FULL PRESCRIBING INFORMATION
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAI NERVOUS SYTEM EFFECTS and EXACERBATION OF MYASTHENIA GRAVIS
• Fluoroquinolones, including moxifloxacin, have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together [see Warnings and Precautions (5.1)], including:
• Tendinitis and tendon rupture [see Warnings and Precautions (5.2)]
• Peripheral neuropathy [see Warnings and Precautions (5.3)]
• Central nervous system effects [see Warnings and Precautions (5.4)]
Discontinue moxifloxacin, immediately and avoid the use of fluoroquinolones, including moxifloxacin, in patients who experience any of these serious adverse reactions [see Warnings and Precautions (5.1)].
• Fluoroquinolones, including moxifloxacin, may exacerbate muscle weakness in patients with myasthenia gravis. Avoid moxifloxacin, in patients with known history of myasthenia gravis [see Warnings and Precautions (5.5)].
• Because fluoroquinolones, including moxifloxacin, have been associated with serious adverse reactions [see Warnings and Precautions(5.1-5.13)], reserve moxifloxacin, for use in patients who have no alternative treatment options for the following indications:
• Acute bacterial sinusitis [see Indications and Usage (1.6)]
• Acute bacterial exacerbation of chronic bronchitis [see Indications and Usage (1.7)]
1 INDICATIONS AND USAGE
1.1 Community Acquired Pneumonia
Moxifloxacin tablets are indicated in adult patients for the treatment of Community Acquired Pneumonia caused by susceptible isolates of Streptococcus pneumoniae (including multi-drug resistant Streptococcus pneumoniae [MDRSP]), Haemophilus influenzae, Moraxella catarrhalis, methicillin-susceptible Staphylococcus aureus, Klebsiella pneumoniae, Mycoplasma pneumoniae, or Chlamydophila pneumoniae [see Clinical Studies (14.3)].
MDRSP isolates are isolates resistant to two or more of the following antibacterial drugs: penicillin (minimum inhibitory concentrations [MIC] ≥ 2 mcg/mL), 2nd generation cephalosporins (for example, cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.
1.2 Uncomplicated Skin and Skin Structure Infections
Moxifloxacin tablets are indicated in adult patients for the treatment of Uncomplicated Skin and Skin Structure Infections caused by susceptible isolates of methicillin-susceptible Staphylococcus aureus or Streptococcus pyogenes [see Clinical Studies (14.4)].
1.3 Complicated Skin and Skin Structure Infections
Moxifloxacin tablets are indicated in adult patients for the treatment of Complicated Skin and Skin Structure Infections caused by susceptible isolates of methicillin-susceptible Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, or Enterobacter cloacae [see Clinical Studies (14.5)].
1.4 Complicated Intra-Abdominal Infections
Moxifloxacin tablets are indicated in adult patients for the treatment of Complicated Intra-Abdominal Infections (cIAI) including polymicrobial infections such as abscess caused by susceptible isolates of Escherichia coli, Bacteroides fragilis, Streptococcus anginosus, Streptococcus constellatus, Enterococcus faecalis, Proteus mirabilis, Clostridium perfringens, Bacteroides thetaiotaomicron, or Peptostreptococcus species [see Clinical Studies (14.6)].
1.5 Plague
Moxifloxacin tablets are indicated in adult patients for the treatment of plague, including pneumonic and septicemic plague, due to susceptible isolates of Yersinia pestis and prophylaxis of plague in adult patients. Efficacy studies of moxifloxacin could not be conducted in humans with plague for feasibility reasons. Therefore, this indication is based on an efficacy study conducted in animals only [see Clinical Studies (14.7)].
1.6 Acute Bacterial Sinusitis
Moxifloxacin tablets are indicated in adult patients for the treatment of acute bacterial Sinusitis caused by susceptible isolates of Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis [see Clinical Studies (14.1)].
Because fluoroquinolones, including moxifloxacin tablets, have been associated with serious adverse reactions [see Warnings and Precautions (5.1 - 5.14)] and for some patients ABS is self-limiting, reserve moxifloxacin tablets for treatment of ABS in patients who have no alternative treatment options.
1.7 Acute Bacterial Exacerbation of Chronic Bronchitis
Moxifloxacin tablets are indicated in adult patients for the treatment of Acute Bacterial Exacerbation of Chronic Bronchitis (ABECB) caused by susceptible isolates of Streptococcus pneumoniae, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella pneumoniae, methicillin-susceptible Staphylococcus aureus, or Moraxella catarrhalis [see Clinical Studies (14.2)].
Because fluoroquinolones, including moxifloxacin tablets, have been associated with serious adverse reactions [see Warnings and Precautions (5.1 -5.14)] and for some patients ABECB is self-limiting, reserve moxifloxacin tablets for treatment of ABECB in patients who have no alternative treatment options.
1.8 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of moxifloxacin tablets and other antibacterial drugs, moxifloxacin tablets should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adult Patients
The dose of moxifloxacin tablet is 400 mg (orally) once every 24 hours. The duration of therapy depends on the type of infection as described in Table 1.
Table 1: Dosage and Duration of Therapy in Adult Patients
| Type of Infectiona | Dose Every 24 hours | Durationb (days) |
| Community Acquired Pneumonia (1.1) | 400 mg | 7 to 14 |
| Uncomplicated Skin and Skin Structure Infections (SSSI ) (1.2) | 400 mg | 7 |
| Complicated SSSI (1.3) | 400 mg | 7 to 21 |
| Complicated Intra-Abdominal Infections (1.4) | 400 mg | 5 to 14 |
| Plague (1.5)c | 400 mg | 10 to 14 |
| Acute Bacterial Sinusitis (ABS) (1.6) | 400 mg | 10 |
| Acute Bacterial Exacerbation of Chronic Bronchitis (ABECB) (1.7) | 400 mg | 5 |
a Due to the designated pathogens [see Indications and Usage (1)]
b Sequential therapy (intravenous to oral) may be instituted at the discretion of the physician
c Drug administration should begin as soon as possible after suspected or confirmed exposure to Yersinia pestis.
Conversion of Intravenous to Oral Dosing in Adults
Intravenous formulation is indicated when it offers a route of administration advantageous to the patient (for example, patient cannot tolerate an oral dosage form). When switching from intravenous to oral formulation, no dosage adjustment is necessary. Patients whose therapy is started with moxifloxacin hydrochloride injection may be switched to moxifloxacin tablets when clinically indicated at the discretion of the physician.
2.2 Important Administration Instructions
Moxifloxacin Tablets
With Multivalent Cations
Administer moxifloxacin tablets at least 4 hours before or 8 hours after products containing magnesium, aluminum, iron or zinc, including antacids, sucralfate, multivitamins and didanosine buffered tablets for oral suspension or the pediatric powder for oral solution [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
With Food
Moxifloxacin tablets can be taken with or without food, drink fluids liberally.
Missed Doses
If a dose is missed, it should be taken anytime but not later than 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next dose, the missed dose should not be taken and treatment should be continued as prescribed with the next scheduled dose. Double doses should not be taken to compensate for a missed dose.
4 CONTRAINDICATIONS
Moxifloxacin tablets are contraindicated in persons with a history of hypersensitivity to moxifloxacin or any member of the quinolone class of antibacterials [see Warnings and Precautions (5.8 )].
5 WARNINGS AND PRECAUTIONS
5.1 Disabling and Potentially Irreversible Serious Adverse Reactions Including Tendinitis and Tendon Rupture, Peripheral Neuropathy, and Central Nervous System Effects
Fluoroquinolones, including moxifloxacin, have been associated with disabling and potentially irreversible serious adverse reactions from different body systems that can occur together in the same patient. Commonly seen adverse reactions include tendinitis, tendon rupture, arthralgia, myalgia, peripheral neuropathy, and central nervous system effects (hallucinations, anxiety, depression, insomnia, severe headaches, and confusion). These reactions can occur within hours to weeks after starting moxifloxacin. Patients of any age or without pre-existing risk factors have experienced these adverse reactions [see Warnings and Precautions (5.2, 5.3, 5.4)].
Discontinue moxifloxacin immediately at the first signs or symptoms of any serious adverse reaction. In addition, avoid the use of fluoroquinolones, including moxifloxacin, in patients who have experienced any of these serious adverse reactions associated with fluoroquinolones.
5.2 Tendinitis and Tendon Rupture
Fluoroquinolones, including moxifloxacin, have been associated with an increased risk of tendinitis and tendon rupture in all ages [see Warnings and Precautions (5.1) and Adverse Reactions (6.2)]. This adverse reaction most frequently involves the Achilles tendon, and has also been reported with the rotator cuff (the shoulder), the hand, the biceps, the thumb, and other tendons. Tendinitis or tendon rupture can occur within hours or days of starting moxifloxacin or as long as several months after completion of therapy. Tendinitis and tendon rupture can occur bilaterally.
The risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants. Other factors that may independently increase the risk of tendon rupture include strenuous physical activity, renal failure, and previous tendon disorders such as rheumatoid arthritis. Tendinitis and tendon rupture have also occurred in patients taking fluoroquinolones who do not have the above risk factors. Discontinue moxifloxacin immediately if the patient experiences pain, swelling, inflammation or rupture of a tendon.
Patients should be advised to rest at the first sign of tendinitis or tendon rupture, and to contact their healthcare provider regarding changing to a non-quinolone antimicrobial drug. Avoid fluoroquinolones, including moxifloxacin, in patients who have a history of tendon disorders or who have experienced tendinitis or tendon rupture [see Adverse Reactions (6.2)].
5.3 Peripheral Neuropathy
Fluoroquinolones, including moxifloxacin, have been associated with an increased risk of peripheral neuropathy. Cases of sensory or sensorimotor axonal polyneuropathy affecting small and/or large axons resulting in paresthesias, hypoesthesias, dysesthesias and weakness have been reported in patients receiving fluoroquinolones including moxifloxacin. Symptoms may occur soon after initiation of moxifloxacin and may be irreversible in some patients [see Warnings and Precautions (5.1) and Adverse Reactions (6.1, 6.2)].
Discontinue moxifloxacin immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation. Avoid fluoroquinolones, including moxifloxacin, in patients who have previously experienced peripheral neuropathy
5.4 Central Nervous System Effects
Psychiatric Adverse Reactions
Fluoroquinolones, including moxifloxacin, have been associated with an increased risk of psychiatric adverse reactions, including: toxic psychosis, hallucinations, or paranoia; depression or suicidal thoughts or acts; anxiety, agitation, or nervousness; confusion, delirium, disorientation, or disturbances in attention; insomnia or nightmares; memory impairment. These adverse reactions may occur following the first dose. If these reactions occur in patients receiving moxifloxacin, discontinue moxifloxacin immediately and institute appropriate measures [see Adverse Reactions (6.1, 6.2)].
Central Nervous System Adverse Reactions
Fluoroquinolones, including moxifloxacin, have been associated with an increased risk of seizures (convulsions), increased intracranial pressure (including pseudotumor cerebri), dizziness, and tremors. As with all fluoroquinolones, use moxifloxacin with caution in patients with known or suspected CNS disorders (for example, severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold. These adverse reactions may occur following the first dose. If these reactions occur in patients receiving moxifloxacin, discontinue moxifloxacin immediately and institute appropriate measures [see Drug Interactions (7.4) Adverse Reactions (6.1, 6.2), and Patient Counseling Information (17)].
5.5 Exacerbation of Myasthenia Gravis
Fluoroquinolones, including moxifloxacin, have neuromuscular blocking activity and may exacerbate muscle weakness in patients with myasthenia gravis. Postmarketing serious adverse reactions, including deaths and requirement for ventilatory support, have been associated with fluoroquinolone use in patients with myasthenia gravis. Avoid moxifloxacin in patients with known history of myasthenia gravis.
5.6 QT Prolongation
Moxifloxacin has been shown to prolong the QT interval of the electrocardiogram in some patients. Following oral dosing with 400 mg of moxifloxacin the mean (± SD) change in QTc from the pre-dose value at the time of maximum drug concentration was 6 msec (± 26) (n = 787). Following a course of daily intravenous dosing (400 mg; 1 hour infusion each day) the mean change in QTc from the Day 1 pre-dose value was 10 msec (±22) on Day 1 (n=667) and 7 msec (± 24) on Day 3 (n = 667).
Avoid moxifloxacin in patients with the following risk factors due to the lack of clinical experience with the drug in these patient populations:
• Known prolongation of the QT interval
• Ventricular arrhythmias including torsade de pointes because QT prolongation may lead to an increased risk for these conditions
• Ongoing proarrhythmic conditions, such as clinically significant bradycardia and acute myocardial ischemia,
• Uncorrected hypokalemia or hypomagnesemia
• Class IA (for example, quinidine, procainamide) or Class III (for example, amiodarone, sotalol) antiarrhythmic agents
• Other drugs that prolong the QT interval such as cisapride, erythromycin, antipsychotics, and tricyclic antidepressants
Elderly patients using intravenous moxifloxacin may be more susceptible to drug-associated QT prolongation [see Use In Specific Populations (8.5)].
In patients with mild, moderate, or severe liver cirrhosis, metabolic disturbances associated with hepatic insufficiency may lead to QT prolongation. Monitor ECG in patients with liver cirrhosis treated with moxifloxacin [see Clinical Pharmacology (12.3)].
The magnitude of QT prolongation may increase with increasing concentrations of the drug or increasing rates of infusion of the intravenous formulation. Therefore, the recommended dose or infusion rate should not be exceeded.
In premarketing clinical trials, the rate of cardiovascular adverse reactions was similar in 798 moxifloxacin and 702 comparator treated patients who received concomitant therapy with drugs known to prolong the QTc interval. No excess in cardiovascular morbidity or mortality attributable to QTc prolongation occurred with moxifloxacin treatment in over 15,500 patients in controlled clinical studies, including 759 patients who were hypokalemic at the start of treatment, and there was no increase in mortality in over 18,000 moxifloxacin tablet treated patients in a postmarketing observational study in which ECGs were not performed.
5.7 Other Serious and Sometimes Fatal Adverse Reactions
Other serious and sometimes fatal adverse reactions, some due to hypersensitivity, and some due to uncertain etiology, have been reported in patients receiving therapy with fluoroquinolones, including moxifloxacin. These reactions may be severe and generally occur following the administration of multiple doses. Clinical manifestations may include one or more of the following:
• Fever, rash, or severe dermatologic reactions (for example, toxic epidermal necrolysis, Stevens-Johnson syndrome)
• Vasculitis; arthralgia; myalgia; serum sickness
• Allergic pneumonitis
• Interstitial nephritis; acute renal insufficiency or failure
• Hepatitis; jaundice; acute hepatic necrosis or failure
• Anemia, including hemolytic and aplastic; thrombocytopenia, including thrombotic thrombocytopenic purpura; leukopenia; agranulocytosis; pancytopenia; and/or other hematologic abnormalities
Discontinue moxifloxacin immediately at the first appearance of a skin rash, jaundice, or any other sign of hypersensitivity and institute supportive measures.
5.8 Hypersensitivity Reactions
Serious anaphylactic reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy, including moxifloxacin. Some reactions were accompanied by cardiovascular collapse, loss of consciousness, tingling, pharyngeal or facial edema, dyspnea, urticaria, and itching. Discontinue moxifloxacin at the first appearance of a skin rash or any other sign of hypersensitivity [see Warnings and Precautions (5.7)]
5.9 Risk of Aortic Aneurysm and Dissection
Epidemiologic studies report an increased rate of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients. The cause for the increased risk has not been identified. In patients with a known aortic aneurysm or patients who are at greater risk for aortic aneurysms, reserve moxifloxacin for use only when there are no alternative antibacterial treatments available.
5.10 Clostridioides difficile -Associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including moxifloxacin, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.11 Arthropathic Effects in Animals
In immature dogs, oral administration of moxifloxacin caused lameness. Histopathological examination of the weight-bearing joints of these dogs revealed permanent lesions of the cartilage. Related quinolone-class drugs also produce erosions of cartilage of weight-bearing joints and other signs of arthropathy in immature animals of various species [see Nonclinical Toxicology (13.2)].
5.12 Blood Glucose Disturbances
As with all fluoroquinolones, disturbances in blood glucose, including both hypoglycemia and hyperglycemia have been reported with moxifloxacin. In moxifloxacin-treated patients, dysglycemia occurred predominantly in elderly diabetic patients receiving concomitant treatment with an oral hypoglycemic agent (for example, sulfonylurea) or with insulin. Severe cases of hypoglycemia resulting in coma or death have been reported. In diabetic patients, careful monitoring of blood glucose is recommended. If a hypoglycemic reaction occurs, discontinue moxifloxacin and initiate appropriate therapy immediately [see Adverse Reactions (6.1), Drug Interactions (7.3) and Patient Counseling Information (17)].
5.13 Photosensitivity/Phototoxicity
Moderate to severe photosensitivity/phototoxicity reactions, the latter of which may manifest as exaggerated sunburn reactions (for example, burning, erythema, exudation, vesicles, blistering, edema) involving areas exposed to light (typically the face, “V” area of the neck, extensor surfaces of the forearms, dorsa of the hands), can be associated with the use of fluoroquinolones, including moxifloxacin, after sun or UV light exposure. Therefore, excessive exposure to these sources of light should be avoided. Moxifloxacin therapy should be discontinued if phototoxicity occurs [see Clinical Pharmacology (12.2)].
6 ADVERSE REACTIONS
The following serious and otherwise important adverse reactions are discussed in greater detail in the warnings and precautions section of the label:
• Disabling and Potentially Irreversible Serious Adverse Reactions Including Tendinitis and Tendon Rupture, Peripheral Neuropathy, and Central Nervous System Effects [see Warnings and Precautions (5.1)]
• Tendinitis and Tendon Rupture[see Warnings and Precautions (5.2)]
• Peripheral Neuropathy [see Warnings and Precautions (5.3)]
• Central Nervous System Effects [see Warnings and Precautions (5.4)]
• Exacerbation of Myasthenia Gravis [see Warnings and Precautions (5.5)]
• QT Prolongation [see Warnings and Precautions (5.6)]
• Other Serious and Sometimes Fatal Adverse Reactions [see Warnings and Precautions (5.7)]
• Hypersensitivity Reactions [see Warnings and Precautions (5.8)]
• Risk of Aortic Aneurysm and Dissection [see Warnings and Precautions (5.9)]
• Clostridioides difficile-Associated Diarrhea [see Warnings and Precautions (5.10)]
• Blood Glucose Disturbances [see Warnings and Precautions (5.12)]
• Photosensitivity/Phototoxicity [see Warnings and Precautions (5.13)]
• Development of Drug Resistant Bacteria [see Warnings and Precautions (5.14)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to moxifloxacin in 14981 patients in 71 active controlled Phase II to IV clinical trials in different indications [see Indications and Usage (1)]. The population studied had a mean age of 50 years (approximately 73% of the population was less than 65 years of age), 50% were male, 63% were Caucasian, 12% were Asian and 9% were Black. Patients received moxifloxacin 400 mg once daily oral, intravenous, or sequentially (intravenous followed by oral). Treatment duration was usually 6 to 10 days, and the mean number of days on therapy was 9 days.
Discontinuation of moxifloxacin due to adverse reactions occurred in 5% of patients overall, 4% of patients treated with 400 mg PO, 4% with 400 mg intravenous and 8% with sequential therapy 400 mg oral/intravenous. The most common adverse reactions (>0.3%) leading to discontinuation with the 400 mg oral doses were nausea, diarrhea, dizziness, and vomiting. The most common adverse reaction leading to discontinuation with the 400 mg intravenous dose was rash. The most common adverse reactions leading to discontinuation with the 400 mg intravenous/oral sequential dose were diarrhea, pyrexia.
Adverse reactions occurring in 1% of moxifloxacin-treated patients and less common adverse reactions, occurring in 0.1 to 1% of moxifloxacin-treated patients, are shown in Tables 2 and Table 3, respectively. The most common adverse drug reactions (3%) are nausea, diarrhea, headache, and dizziness.
Table 2 Common (1% or more) Adverse Reactions Reported in Active-Controlled Clinical Trials with Moxifloxacin
| System Organ Class | Adverse Reactions | % (N=14,981) |
| Blood and Lymphatic System Disorders | Anemia | 1 |
| Gastrointestinal Disorders | Nausea | 7 |
| Diarrhea | 6 | |
| Vomiting | 2 | |
| Constipation | 2 | |
| Abdominal pain | 2 | |
| Dyspepsia | 1 | |
| General Disorders and Administration Site Conditions | Pyrexia | 1 |
| Investigations | Alanine aminotransferase increased | 1 |
| Metabolism and Nutritional Disorder | Hypokalemia | 1 |
| Nervous System Disorders | HeadacheDizziness | 43 |
| Psychiatric Disorders | Insomnia | 2 |
Table 3 Less Common (0.1 to less than 1%) Adverse Reactions Reported in Active-Controlled Clinical Trials with Moxifloxacin (N=14,981)
| System Organ Class | Adverse Reactions |
| Blood and Lymphatic System Disorders | Thrombocythemia Eosinophilia Neutropenia Thrombocytopenia Leukopenia Leukocytosis |
| Cardiac Disorders | Atrial fibrillation Palpitations Tachycardia Angina pectoris Cardiac failure Cardiac arrest Bradycardia |
| Ear and Labyrinth Disorders | Vertigo Tinnitus |
| Eye Disorders | Vision blurred |
| Gastrointestinal Disorders | Dry mouth Abdominal discomfort Flatulence Abdominal distention Gastritis Gastroesophageal reflux disease |
| General Disorders and Administration Site Conditions | Fatigue Chest pain Asthenia Pain Malaise Infusion site extravasation Edema Chills Chest discomfort Facial pain |
| Hepatobiliary disorders | Hepatic function abnormal |
| Infections and Infestations | Candidiasis Vaginal infection Fungal infection Gastroenteritis |
| Investigations | Aspartate aminotransferase increased Gamma-glutamyltransferase increased Blood alkaline phosphatase increased Electrocardiogram QT prolonged Blood lactate dehydrogenase increased Blood amylase increased Lipase increased Blood creatinine increased Blood urea increased Hematocrit decreased Prothrombin time prolonged Eosinophil count increased Activated partial thromboplastin time prolonged Blood triglycerides increased Blood uric acid increased |
| Metabolism and Nutrition Disorders | Hyperglycemia Anorexia HHyperlipidemia Decreased appetite Dehydration |
| Musculoskeletal and Connective Tissue Disorders | Back pain Pain in extremity Arthralgia Muscle spasms Musculoskeletal pain |
| Nervous System Disorders | Dysgeusia Somnolence Tremor Lethargy Paresthesia Hypoesthesia Syncope |
| Psychiatric Disorders | Anxiety Confusional state Agitation Depression Nervousness Restlessness Hallucination Disorientation |
| Renal and Urinary Disorders | Renal failure Dysuria |
| Reproductive System and Breast Disorders | Vulvovaginal pruritus |
| Respiratory, Thoracic, and Mediastinal Disorders | Dyspnea Asthma Wheezing Bronchospasm |
| Skin and Subcutaneous Tissue Disorders | Rash Pruritus Hyperhidrosis Erythema Urticaria Dermatitis allergic Night sweats |
| Vascular Disorders | Hypertension Hypotension Phlebitis * |
Laboratory Changes
Changes in laboratory parameters, which are not listed above and which occurred in 2% or more of patients and at an incidence greater than in controls included: increases in mean corpuscular hemoglobin (MCH), neutrophils, white blood cells (WBCs), prothrombin time (PT) ratio, ionized calcium, chloride, albumin, globulin, bilirubin; decreases in hemoglobin, red blood cells (RBCs), neutrophils, eosinophils, basophils, glucose, oxygen partial pressure (pO2), bilirubin, and amylase. It cannot be determined if any of the above laboratory abnormalities were caused by the drug or the underlying condition being treated.
6.2 Postmarketing Experience
Table 4 below lists adverse reactions that have been identified during post-approval use of moxifloxacin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Table 4: Postmarketing Reports of Adverse Drug Reactions
| System Organ Class | Adverse Reactions |
| Blood and Lymphatic System Disorders | Agranulocytosis Pancytopenia [see Warnings and Precautions (5.7)] |
| Cardiac Disorders | Ventricular tachyarrhythmias (including in very rare cases cardiac arrest and torsade de pointes, and usually in patients with concurrent severe underlying proarrhythmic conditions) |
| Ear and Labyrinth Disorders | Hearing impairment, including deafness (reversible in majority of cases) |
| Eye Disorders | Vision loss (especially in the course of CNS reactions, transient in majority of cases) |
| Hepatobiliary Disorders | Hepatitis (predominantly cholestatic) Hepatic failure (including fatal cases) Jaundice Acute hepatic necrosis [see Warnings and Precautions (5.7)] |
| Immune System Disorders | Anaphylactic reaction Anaphylactic shock Angioedema (including laryngeal edema) [see Warnings and Precautions (5.7, 5.8)] |
| Musculoskeletal and Connective Tissue Disorders | Tendon rupture [see Warnings and Precautions (5.2)] |
| Nervous System Disorders | Altered coordination Abnormal gait [see Warnings and Precautions (5.3)] Myasthenia gravis (exacerbation of) [see Warnings and Precautions (5.5)] Muscle weakness Peripheral neuropathy (that may be irreversible), polyneuropathy [see Warnings and Precautions (5.3)] |
| Psychiatric Disorders | Psychotic reaction (very rarely culminating in self-injurious behavior, such as suicidal ideation/thoughts or suicide attempts [see Warnings and Precautions (5.4)] |
| Renal and Urinary Disorders | Interstitial nephritis [see Warnings and Precautions (5.7)] |
| Respiratory, Thoracic and Mediastinal Disorders | Allergic pneumonitis [see Warnings and Precautions (5.7)] |
| Skin and Subcutaneous Tissue Disorders | Photosensitivity/phototoxicity reaction [see Warnings and Precautions (5.13)] Stevens-Johnson syndrome Toxic epidermal necrolysis [see Warnings and Precautions (5.7)] |
7 DRUG INTERACTIONS
7.1 Antacids, Sucralfate, Multivitamins and Other Products Containing Multivalent Cations
Fluoroquinolones, including moxifloxacin, form chelates with alkaline earth and transition metal cations. Oral administration of moxifloxacin with antacids containing aluminum or magnesium, with sucralfate, with metal cations such as iron, or with multivitamins containing iron or zinc, or with formulations containing divalent and trivalent cations such as didanosine buffered tablets for oral suspension or the pediatric powder for oral solution, may substantially interfere with the absorption of moxifloxacin, resulting in systemic concentrations considerably lower than desired. Therefore, moxifloxacin should be taken at least 4 hours before or 8 hours after these agents [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]
7.2 Warfarin
Fluoroquinolones, including moxifloxacin, have been reported to enhance the anticoagulant effects of warfarin or its derivatives in the patient population. In addition, infectious disease and its accompanying inflammatory process, age, and general status of the patient are risk factors for increased anticoagulant activity. Therefore the prothrombin time, International Normalized Ratio (INR), or other suitable anticoagulation tests should be closely monitored if moxifloxacin is administered concomitantly with warfarin or its derivatives [see Adverse Reactions (6.2) and Clinical Pharmacology (12.3)].
7.3 Antidiabetic Agents
Disturbances of blood glucose, including hyperglycemia and hypoglycemia, have been reported in patients treated concomitantly with fluoroquinolones, including moxifloxacin, and an antidiabetic agent. Therefore, careful monitoring of blood glucose is recommended when these agents are co-administered. If a hypoglycemic reaction occurs, moxifloxacin should be discontinued and appropriate therapy should be initiated immediately [see Warnings and Precautions (5.12) and Adverse Reactions (6.1)]
7.4 Nonsteroidal Anti-Inflammatory Drugs
The concomitant administration of a nonsteroidal anti-inflammatory drug (NSAID) with a fluoroquinolone, including moxifloxacin, may increase the risks of CNS stimulation and convulsions [see Warnings and Precautions (5.4)].
7.5 Drugs that Prolong QT
There is limited information available on the potential for a pharmacodynamic interaction in humans between moxifloxacin and other drugs that prolong the QTc interval of the electrocardiogram. Sotalol, a Class III antiarrhythmic, has been shown to further increase the QTc interval when combined with high doses of intravenous (IV) moxifloxacin in dogs. Therefore, moxifloxacin should be avoided with Class IA and Class III antiarrhythmics [see Warnings and Precautions (5.6), and Nonclinical Toxicology (13.2)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available human data establishing a drug associated risk with the use of moxifloxacin.
Based on animal studies with moxifloxacin, moxifloxacin may cause fetal harm. Moxifloxacin did not cause fetal malformations when administered to pregnant rats (IV and oral), rabbits (IV), and monkeys (oral) at exposures that were 0.24 to 2.5 times of those at the human clinical dose (400 mg/day moxifloxacin). However, when moxifloxacin was administered to rats and rabbits during pregnancy and throughout lactation (rats only) at doses associated with maternal toxicity, decreased neonatal body weights, increased incidence of skeletal variations (rib and vertebra combined), and increased fetal loss were observed (see Data). Advise pregnant women of the potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Animal reproductive and development studies were done in rats, rabbits and cynomolgus macaques. Moxifloxacin did not cause fetal malformations when administered to pregnant rats during organogenesis (gestation days 6 to 17) at oral doses as high as 500 mg/kg/day or 0.24 times the maximum recommended human dose based on systemic exposure (AUC), but decreased fetal body weights and slightly delayed fetal skeletal development were observed. Intravenous administration of 80 mg/kg/day (approximately 2 times the maximum recommended human dose based on body surface area) to pregnant rats resulted in maternal toxicity and a marginal effect on fetal and placental weights and the appearance of the placenta (Gestation days 6 to 17). Fetal malformations were not observed at intravenous doses as high as 80 mg/kg/day (approximately 2 times the maximum recommended human dose based on body surface area) in litters of pregnant rats that received moxifloxacin during organogenesis (Gestation days 6 to 17). Intravenous administration of 20 mg/kg/day (approximately equal to the maximum recommended human oral dose based upon systemic exposure) to pregnant rabbits during organogenesis (gestation days 6 to 20) resulted in decreased fetal body weights and delayed fetal skeletal ossification. When rib and vertebral malformations were combined, there was an increased fetal and litter incidence of these effects in rabbits. Signs of maternal toxicity in rabbits at this dose included mortality, abortions, marked reduction of food consumption, decreased water intake, body weight loss and hypoactivity. Fetal malformations were not observed when pregnant cynomolgus macaques were given oral doses as high as 100 mg/kg/day (2.5 times the maximum recommended human dose based upon systemic exposure) during organogenesis (gestation days 20 to 50). An increased incidence of smaller fetuses was observed at 100 mg/kg/day in macaques. In a pre- and postnatal development study conducted in rats given oral doses from Gestation day 6, throughout gestation and rearing to Postpartum day 21, effects observed at 500 mg/kg/day (0.24 times the maximum recommended human dose based on systemic exposure (AUC)) included slight increases in duration of pregnancy and prenatal loss, reduced pup birth weight and decreased neonatal survival. Treatment-related maternal mortality occurred during gestation at 500 mg/kg/day in this study.
8.2 Lactation
Risk Summary
It is not known if moxifloxacin is present in human milk. Based on animal studies in rats, moxifloxacin may be excreted in human milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for moxifloxacin and any potential adverse effects on the breastfed child from moxifloxacin or from the underlying maternal condition.
Data
In lactating rats given a single oral dose of 4.59 mg/kg moxifloxacin (approximately 9 times less than the recommended human dose based on body surface area) 8 days postpartum, there was very low excretion of substance-related radioactivity into the milk, amounting to approximately 0.03% of the dose.
8.4 Pediatric Use
Effectiveness in pediatric patients and adolescents less than 18 years of age has not been established. Moxifloxacin causes arthropathy in juvenile animals. Limited information on the safety of moxifloxacin in 301 pediatric patients is available from the cIAI trial [see Boxed Warning, Warnings and Precautions (5.11) and Nonclinical Toxicology (13.2)].
Active Controlled Trial in Complicated Intra-Abdominal Infection (cIAI)
The safety and efficacy of moxifloxacin in pediatric patients for the treatment of cIAI has not been demonstrated.
Pediatric patients 3 months to <18 years of age (mean age of 12 ± 4 years) were enrolled in a single randomized, double-blind, active controlled trial in cIAI including appendicitis with perforation, abscesses and peritonitis.
Pediatric patients were randomized (2:1) to receive either moxifloxacin or comparator. This study enrolled 451 patients who received study medication, 301 treated with moxifloxacin, and 150 with comparator. Of the 301 pediatric patients treated with moxifloxacin, 15 were below the age of 6 years and 286 were between the ages of 6 to <18 years.
Patients received sequential intravenous/oral moxifloxacin or comparator (intravenous ertapenem followed by oral amoxicillin/clavulanate) for 5 to 14 days (mean duration was 9 days with a range of 1 to 24 days).
The overall adverse reaction profile in pediatric patients was comparable to that of adult patients. The most frequently occurring adverse reactions in pediatric patients treated with moxifloxacin were QT prolongation 9.3% (28/301), vomiting, 6.6% (20/301) diarrhea 3.7% (11/301), arthralgia 3% (9/301), and phlebitis 2.7% (8/301) (see Table 5). Discontinuation of study drug due to an adverse reaction was reported in 5.3% (16/301) of moxifloxacin-treated patients versus 1.3% (2/150) of comparator-treated patients. The adverse reaction profile of moxifloxacin or comparator was similar across all age groups studied.
Musculoskeletal adverse reactions were monitored and followed up to 5 years after the end of study treatment. The rates of musculoskeletal adverse reactions were 4.3% (13/301) in the moxifloxacin-treated group versus 3.3% (5/150) in the comparator-treated group. The majority of musculoskeletal adverse reactions were reported between 12 and 53 weeks after start of study treatment with complete resolution at the end of the study [see Warnings and Precautions (5.11) and Nonclinical Toxicology (13.2)].
Table 5 Incidence (%) of Selected Adverse Reactions in ≥2% of Pediatric Patients Treated with Moxifloxacin in cIAI Clinical Trial
| System Organ Class | Adverse Reactions | Moxifloxacin
N = 301 (%) | Comparator
N = 150 (%) |
| Gastrointestinal disorders | Abdominal pain | 8 (2.7) | 3 (2.0) |
| Diarrhea | 11 (3.7) | 1 (0.7) | |
| Vomiting | 20 (6.6) | 12 (8.0) | |
| General disorders andadministration site conditions | Pyrexia | 6 (2.0) | 4 (2.7) |
| Investigations | Aspartate aminotransferaseincreased | 2 (0.7) | 3 (2.0) |
| Electrocardiogram QTprolonged | 28 (9.3) | 4 (2.7) | |
| Musculoskeletal andconnective tissue disorders | Arthralgia | 9 (3.0) | 2 (1.3) |
| Nervous system disorders | Headache | 6 (2.0) | 2 (1.3) |
| Vascular disorders | Phlebitis | 8 (2.7) | 0 (0) |
Clinical response was assessed at the test-of-cure visit (28 to 42 days after end of treatment). The clinical response rates observed in the modified intent to treat population were 83.9% (208/248) for moxifloxacin and 95.5% (127/133) for comparator; see Table 6.
Table 6: Clinical Response Rates at 28 to 42 Days After End of Treatment in Pediatric Patients with cIAI
| Moxifloxacin
n (%) | Comparator
n (%) | Difference2
(95% CI) |
|
| mITT Population1 | N=248 | N=133 | |
| Cure | 208 (83.9) | 127 (95.5) | -12.2 (-17.9, -6.4) |
| Failure | 17 (6.9) | 3 (2.3) | |
| Indeterminate | 21 (8.5) | 3 (2.3) | |
| Missing | 2 (0.8) | 0 |
1The modified intent-to-treat (mITT) population is defined as all subjects who were treated with at least one dose of study medication and who have at least one pre-treatment causative organism from the intra-abdominal site of infection or from blood cultures.
2 Difference in clinical cure rates (Moxifloxacin - Comparator) and 95% confidence intervals, presented as percentages, are based on stratified analysis by age group using Mantel-Haenszel methods.
8.5 Geriatric Use
Geriatric patients are at increased risk for developing severe tendon disorders including tendon rupture when being treated with a fluoroquinolone such as moxifloxacin. This risk is further increased in patients receiving concomitant corticosteroid therapy. Tendinitis or tendon rupture can involve the Achilles, hand, shoulder, or other tendon sites and can occur during or after completion of therapy; cases occurring up to several months after fluoroquinolone treatment have been reported. Caution should be used when prescribing moxifloxacin to elderly patients especially those on corticosteroids. Patients should be informed of this potential side effect and advised to discontinue moxifloxacin and contact their healthcare provider if any symptoms of tendinitis or tendon rupture occur [seeBoxed Warning, Warnings and Precautions (5.2)]
Epidemiologic studies report an increased rate of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients [see Warnings and Precautions (5.9)].
In controlled multiple-dose clinical trials, 23% of patients receiving oral moxifloxacin were greater than or equal to 65 years of age and 9% were greater than or equal to 75 years of age. The clinical trial data demonstrate that there is no difference in the safety and efficacy of oral moxifloxacin in patients aged 65 or older compared to younger adults.
In trials of intravenous use, 42% of moxifloxacin patients were greater than or equal to 65 years of age, and 23% were greater than or equal to 75 years of age. The clinical trial data demonstrate that the safety of intravenous moxifloxacin in patients aged 65 or older was similar to that of comparator-treated patients. In general, elderly patients may be more susceptible to drug-associated effects of the QT interval. Therefore, moxifloxacin should be avoided in patients taking drugs that can result in prolongation of the QT interval (for example, class IA or class III antiarrhythmics) or in patients with risk factors for torsade de pointes (for example, known QT prolongation, uncorrected hypokalemia) [see Warnings and Precautions ( 5.6 ), Drug Interactions (7.5), and Clinical Pharmacology (12.3)].
8.6 Renal Impairment
The pharmacokinetic parameters of moxifloxacin are not significantly altered in mild, moderate, severe, or end-stage renal disease. No dosage adjustment is necessary in patients with renal impairment, including those patients requiring hemodialysis (HD) or continuous ambulatory peritoneal dialysis (CAPD) [see Dosage and Administration (2), and Clinical Pharmacology (12.3).]
8.7 Hepatic Impairment
No dosage adjustment is recommended for mild, moderate, or severe hepatic insufficiency (Child-Pugh Classes A, B, or C). However, due to metabolic disturbances associated with hepatic insufficiency, which may lead to QT prolongation, moxifloxacin should be used with caution in these patients [see Warnings and Precautions (5.6), and Clinical Pharmacology, (12.3)].
10 OVERDOSAGE
Single oral overdoses up to 2.8 g were not associated with any serious adverse events. In the event of acute overdose, empty the stomach and maintain adequate hydration. Monitor ECG due to the possibility of QT interval prolongation. Carefully observe the patient and give supportive treatment. The administration of activated charcoal as soon as possible after oral overdose may prevent excessive increase of systemic moxifloxacin exposure. About 3% and 9% of the dose of moxifloxacin, as well as about 2% and 4.5% of its glucuronide metabolite are removed by continuous ambulatory peritoneal dialysis and hemodialysis, respectively.
11 DESCRIPTION
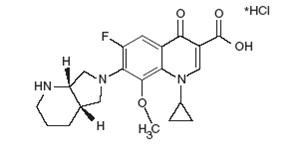
Moxifloxacin hydrochloride USP is a synthetic antibacterial agent for oral administration. Moxifloxacin, a fluoroquinolone, is available as the monohydrochloride salt of 1-cyclopropyl-7-[(S,S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-1,4-dihydro-4-oxo-3 quinoline carboxylic acid. It is a slightly yellow to yellow powder or crystals with a molecular weight of 437.9. Its molecular formula is C21H24FN3O4*HCl and its chemical structure is as follows
11.1 Moxifloxacin Tablets, USP
Moxifloxacin Tablets, USP are available as film-coated tablets containing 400 mg moxifloxacin (equivalent to 436.30 mg moxifloxacin hydrochloride, USP).
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose, iron oxide red, iron oxide yellow, magnesium stearate, microcrystalline cellulose, polyethylene glycol 400, polysorbate 80, sodium stearyl fumarate, talc and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Moxifloxacin is a member of the flouroquinolone class of antibacterial agents [see Microbiology (12.4)].
12.2 Pharmacodynamics
Photosensitivity PotentialA study of the skin response to ultraviolet (UVA and UVB) and visible radiation conducted in 32 healthy volunteers (8 per group) demonstrated that moxifloxacin does not show phototoxicity in comparison to placebo. The minimum erythematous dose (MED) was measured before and after treatment with moxifloxacin (200 mg or 400 mg once daily), lomefloxacin (400 mg once daily), or placebo. In this study, the MED measured for both doses of moxifloxacin were not significantly different from placebo, while lomefloxacin significantly lowered the MED [see Warnings and Precautions (5.13)].
12.3 Pharmacokinetics
Absorption
Moxifloxacin, given as an oral tablet, is well absorbed from the gastrointestinal tract. The absolute bioavailability of moxifloxacin is approximately 90 percent. Co-administration with a high fat meal (that is, 500 calories from fat) does not affect the absorption of moxifloxacin.
Consumption of 1 cup of yogurt with moxifloxacin does not affect the rate or extent of the systemic absorption (that is, area under the plasma concentration time curve (AUC).
Table 7: Mean (± SD) Cmax and AUC values following single and multiple doses of 400 mg moxifloxacin given orally
| Cmax(mg/L) | AUC | Half-life | |
| (mg•h/L) | (hr) | ||
| Single Dose Oral Healthy (n = 372) | 3.1 ± 1 | 36.1 ± 9.1 | 11.5 to 15.6a |
| Multiple Dose Oral Healthy young male/female (n = 15) Healthy elderly male (n = 8) Healthy elderly female (n = 8) Healthy young male (n = 8) Healthy young female (n = 9) | 4.5 ± 0.5 3.8 ± 0.3 4.6 ± 0.6 3.6 ± 0.5 4.2 ± 0.5 | 48 ± 2.7 51.8 ± 6.7 54.6 ± 6.7 48.2 ± 9 49.3 ± 9.5 | 12.7 ± 1.9 |
a Range of means from different studies
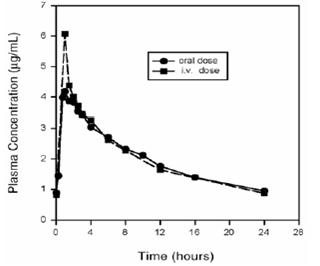
Plasma concentrations increase proportionately with dose up to the highest dose tested (1200 mg single oral dose). The mean (± SD) elimination half-life from plasma is 12 ± 1.3 hours; steady-state is achieved after at least three days with a 400 mg once daily regimen.
Mean Steady-State Plasma Concentrations of Moxifloxacin Obtained With Once Daily Dosing of 400 mg Either Orally (n=10) or by Intravenous Infusion (n=12)
Distribution
Moxifloxacin is approximately 30 to 50% bound to serum proteins, independent of drug concentration. The volume of distribution of moxifloxacin ranges from 1.7 to 2.7 L/kg. Moxifloxacin is widely distributed throughout the body, with tissue concentrations often exceeding plasma concentrations. Moxifloxacin has been detected in the saliva, nasal and bronchial secretions, mucosa of the sinuses, skin blister fluid, subcutaneous tissue, skeletal muscle, and abdominal tissues and fluids following oral administration of 400 mg. Moxifloxacin concentrations measured post-dose in various tissues and fluids following a 400 mg oral dose are summarized in Table 9. The rates of elimination of moxifloxacin from tissues generally parallel the elimination from plasma.
Table 9: Moxifloxacin Concentrations (mean ± SD) in Tissues and the Corresponding Plasma Concentrations After a Single 400 mg Oral Dosea
| Tissue or Fluid | N | Plasma Concentration (mcg/mL) | Tissue or Fluid Concentration (mcg/mL or mcg/g) | Tissue Plasma Ratio |
| Respiratory | ||||
| Alveolar Macrophages Bronchial Mucosa Epithelial Lining Fluid | 5 8 5 | 3.3 ± 0.7 3.3 ± 0.7 .3 ± 0.7 | 61.8 ± 27.3 5.5 ± 1.3 24.4 ± 14.7 | 21.2 ± 10 1.7 ± 0.3 8.7 ± 6.1 |
| Sinus | ||||
| Maxillary Sinus Mucosa Anterior Ethmoid Mucosa Nasal Polyps | 4 3 4 | 3.7 ± 1.1b
3.7 ± 1.1b 3.7 ± 1.1b | 7.6 ± 1.7 8.8 ± 4.3 9.8 ± 4.5 | 2 ± 0.3 2.2 ± 0.6 2.6 ± 0.6 |
| Skin, Musculoskeletal | ||||
| Blister Fluid Subcutaneous Tissue Skeletal Muscle | 5 6 6 | 3 ± 0.5c
2.3 ± 0. 2.3 ± 0.4d | 2.6 ± 0.9 0.9 ± 0.3e 0.9 ± 0.2e | 0.9 ± 0.2 0.4 ± 0.6 0.4 ± 0.1 |
| Intra-Abdominal | ||||
| Abdominal tissue Abdominal exudate Abscess fluid | 8 10 6 | 2.9 ± 0.5 2.3 ± 0.5 2.7 ± 0.7 | 7.6 ± 2 3.5 ±1.2 2.3 ±1.5 | 2.7 ± 0.8 1.6 ± 0.7 0.8±0.4 |
a All moxifloxacin concentrations were measured 3 hours after a single 400 mg dose, except the abdominal tissue and exudate concentrations which were measured at 2 hours post-dose and the sinus concentrations which were measured 3 hours post-dose after 5 days of dosing.
b N = 5
c N = 7
d N = 12
e Reflects only non-protein bound concentrations of drug.
Metabolism
Approximately 52% of an oral or intravenous dose of moxifloxacin is metabolized via glucuronide and sulfate conjugation. The cytochrome P450 system is not involved in moxifloxacin metabolism, and is not affected by moxifloxacin. The sulfate conjugate (M1) accounts for approximately 38% of the dose, and is eliminated primarily in the feces. Approximately 14% of an oral or intravenous dose is converted to a glucuronide conjugate (M2), which is excreted exclusively in the urine. Peak plasma concentrations of M2 are approximately 40% those of the parent drug, while plasma concentrations of M1 are generally less than 10% those of moxifloxacin.
In vitro studies with cytochrome (CYP) P450 enzymes indicate that moxifloxacin does not inhibit CYP3A4, CYP2D6, CYP2C9, CYP2C19, or CYP1A2.
Excretion
Approximately 45% of an oral or intravenous dose of moxifloxacin is excreted as unchanged drug (~20% in urine and ~25% in feces). A total of 96% ± 4% of an oral dose is excreted as either unchanged drug or known metabolites. The mean (± SD) apparent total body clearance and renal clearance are 12 ± 2 L/hr and 2.6 ± 0.5 L/hr, respectively.
Pharmacokinetics in Specific Populations
Geriatric
Following oral administration of 400 mg moxifloxacin for 10 days in 16 elderly (8 male; 8 female) and 17 young (8 male; 9 female) healthy volunteers, there were no age-related changes in moxifloxacin pharmacokinetics. In 16 healthy male volunteers (8 young; 8 elderly) given a single 200 mg dose of oral moxifloxacin, the extent of systemic exposure (AUC and Cmax) was not statistically different between young and elderly males and elimination half-life was unchanged. No dosage adjustment is necessary based on age. In large phase III studies, the concentrations around the time of the end of the infusion in elderly patients following intravenous infusion of 400 mg were similar to those observed in young patients. [see Use In Specific Populations (8.5).]
Gender
Following oral administration of 400 mg moxifloxacin daily for 10 days to 23 healthy males (19 to 75 years) and 24 healthy females (19 to 70 years), the mean AUC and Cmax were 8% and 16% higher, respectively, in females compared to males. There are no significant differences in moxifloxacin pharmacokinetics between male and female subjects when differences in body weight are taken into consideration.
A 400 mg single dose study was conducted in 18 young males and females. The comparison of moxifloxacin pharmacokinetics in this study (9 young females and 9 young males) showed no differences in AUC or Cmax due to gender. Dosage adjustments based on gender are not necessary.
Race
Steady-state moxifloxacin pharmacokinetics in male Japanese subjects were similar to those determined in Caucasians, with a mean Cmax of 4.1 mcg/mL, an AUC24 of 47 mcg•h/mL, and an elimination half-life of 14 hours, following 400 mg p.o. daily.
Renal Insufficiency
The pharmacokinetic parameters of moxifloxacin are not significantly altered in mild, moderate, severe, or end-stage renal disease. No dosage adjustment is necessary in patients with renal impairment, including those patients requiring hemodialysis (HD) or continuous ambulatory peritoneal dialysis (CAPD).
In a single oral dose study of 24 patients with varying degrees of renal function from normal to severely impaired, the mean peak concentrations (Cmax) of moxifloxacin were reduced by 21% and 28% in the patients with moderate (CLCR≥ 30 and ≤ 60 mL/min) and severe (CLCR<30 mL/min) renal impairment, respectively. The mean systemic exposure (AUC) in these patients was increased by 13%. In the moderate and severe renally impaired patients, the mean AUC for the sulfate conjugate (M1) increased by 1.7-fold (ranging up to 2.8-fold) and mean AUC and Cmax for the glucuronide conjugate (M2) increased by 2.8-fold (ranging up to 4.8-fold) and 1.4-fold (ranging up to 2.5-fold), respectively [see Use in Specific Populations( 8.6 ).]
The pharmacokinetics of single dose and multiple dose moxifloxacin were studied in patients with CLCR< 20 mL/min on either hemodialysis or continuous ambulatory peritoneal dialysis (8 HD, 8 CAPD). Following a single 400 mg oral dose, the AUC of moxifloxacin in these HD and CAPD patients did not vary significantly from the AUC generally found in healthy volunteers. Cmax values of moxifloxacin were reduced by about 45% and 33% in HD and CAPD patients, respectively, compared to healthy, historical controls. The exposure (AUC) to the sulfate conjugate (M1) increased by 1.4- to 1.5-fold in these patients. The mean AUC of the glucuronide conjugate (M2) increased by a factor of 7.5, whereas the mean Cmax values of the glucuronide conjugate (M2) increased by a factor of 2.5 to 3, compared to healthy subjects. The sulfate and the glucuronide conjugates of moxifloxacin are not microbiologically active, and the clinical implication of increased exposure to these metabolites in patients with renal disease including those undergoing HD and CAPD has not been studied.
Oral administration of 400 mg QD moxifloxacin for 7 days to patients on HD or CAPD produced mean systemic exposure (AUCss) to moxifloxacin similar to that generally seen in healthy volunteers. Steady-state Cmax values were about 22% lower in HD patients but were comparable between CAPD patients and healthy volunteers. Both HD and CAPD removed only small amounts of moxifloxacin from the body (approximately 9% by HD, and 3% by CAPD). HD and CAPD also removed about 4% and 2% of the glucuronide metabolite (M2), respectively.
Hepatic Insufficiency
No dosage adjustment is recommended for mild, moderate, or severe hepatic insufficiency (Child-Pugh Classes A, B, or C). However, due to metabolic disturbances associated with hepatic insufficiency, which may lead to QT prolongation, moxifloxacin should be used with caution in these patients [see Warnings and Precautions (5.6)and Use in Specific Populations (8.7)].
In 400 mg single oral dose studies in 6 patients with mild (Child-Pugh Class A) and 10 patients with moderate (Child-Pugh Class B) hepatic insufficiency, moxifloxacin mean systemic exposure (AUC) was 78% and 102%, respectively, of 18 healthy controls and mean peak concentration (Cmax) was 79% and 84% of controls.
The mean AUC of the sulfate conjugate of moxifloxacin (M1) increased by 3.9-fold (ranging up to 5.9-fold) and 5.7-fold (ranging up to 8-fold) in the mild and moderate groups, respectively. The mean Cmax of M1 increased by approximately 3 fold in both groups (ranging up to 4.7- and 3.9-fold). The mean AUC of the glucuronide conjugate of moxifloxacin (M2) increased by 1.5-fold (ranging up to 2.5-fold) in both groups. The mean Cmax of M2 increased by 1.6- and 1.3-fold (ranging up to 2.7- and 2.1-fold), respectively. The clinical significance of increased exposure to the sulfate and glucuronide conjugates has not been studied. In a subset of patients participating in a clinical trial, the plasma concentrations of moxifloxacin and metabolites determined approximately at the moxifloxacin Tmax following the first intravenous or oral moxifloxacin dose in the Child-Pugh Class C patients (n=10) were similar to those in the Child-Pugh Class A/B patients (n=5), and also similar to those observed in healthy volunteer studies.
Drug-Drug Interactions
The following drug interactions were studied in healthy volunteers or patients.
Antacids and iron significantly reduced bioavailability of moxifloxacin, as observed with other fluoroquinolones [see Drug Interactions (7.1)].
Calcium, digoxin, itraconazole, morphine, probenecid, ranitidine, theophylline, cyclosporine and warfarin did not significantly affect the pharmacokinetics of moxifloxacin. These results and the data from in vitro studies suggest that moxifloxacin is unlikely to significantly alter the metabolic clearance of drugs metabolized by CYP3A4, CYP2D6, CYP2C9, CYP2C19, or CYP1A2 enzymes.
Moxifloxacin had no clinically significant effect on the pharmacokinetics of atenolol, digoxin, glyburide, itraconazole, oral contraceptives, theophylline, cyclosporine and warfarin. However, fluoroquinolones, including moxifloxacin, have been reported to enhance the anticoagulant effects of warfarin or its derivatives in the patient population [see Drug Interactions (7.2)].
Antacids
When moxifloxacin (single 400 mg tablet dose) was administered two hours before, concomitantly, or 4 hours after an aluminum/magnesium-containing antacid (900 mg aluminum hydroxide and 600 mg magnesium hydroxide as a single oral dose) to 12 healthy volunteers there was a 26%, 60% and 23% reduction in the mean AUC of moxifloxacin, respectively. Moxifloxacin should be taken at least 4 hours before or 8 hours after antacids containing magnesium or aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc, or didanosine buffered tablets for oral suspension or the pediatric powder for oral solution [see Dosage and Administration (2.2) and Drug Interactions (7.1)].
Atenolol
In a crossover study involving 24 healthy volunteers (12 male; 12 female), the mean atenolol AUC following a single oral dose of 50 mg atenolol with placebo was similar to that observed when atenolol was given concomitantly with a single 400 mg oral dose of moxifloxacin. The mean Cmax of single dose atenolol decreased by about 10% following co-administration with a single dose of moxifloxacin.
Calcium
Twelve healthy volunteers were administered concomitant moxifloxacin (single 400 mg dose) and calcium (single dose of 500 mg Ca++ dietary supplement) followed by an additional two doses of calcium 12 and 24 hours after moxifloxacin administration. Calcium had no significant effect on the mean AUC of moxifloxacin. The mean Cmax was slightly reduced and the time to maximum plasma concentration was prolonged when moxifloxacin was given with calcium compared to when moxifloxacin was given alone (2.5 hours versus 0.9 hours). These differences are not considered to be clinically significant.
Digoxin
No significant effect of moxifloxacin (400 mg once daily for two days) on digoxin (0.6 mg as a single dose) AUC was detected in a study involving 12 healthy volunteers. The mean digoxin Cmax increased by about 50% during the distribution phase of digoxin. This transient increase in digoxin Cmax is not viewed to be clinically significant. Moxifloxacin pharmacokinetics were similar in the presence or absence of digoxin. No dosage adjustment for moxifloxacin or digoxin is required when these drugs are administered concomitantly.
Glyburide
In diabetics, glyburide (2.5 mg once daily for two weeks pretreatment and for five days concurrently) mean AUC and Cmax were 12% and 21% lower, respectively, when taken with moxifloxacin (400 mg once daily for five days) in comparison to placebo. Nonetheless, blood glucose levels were decreased slightly in patients taking glyburide and moxifloxacin in comparison to those taking glyburide alone, suggesting no interference by moxifloxacin on the activity of glyburide. These interaction results are not viewed as clinically significant.
Iron
When moxifloxacin tablets were administered concomitantly with iron (ferrous sulfate 100 mg once daily for two days), the mean AUC and Cmax of moxifloxacin was reduced by 39% and 59%, respectively. Moxifloxacin should only be taken more than 4 hours before or 8 hours after iron products. [see Dosage and Administration ( 2.2) and Drug Interactions (7.1)].
Itraconazole
In a study involving 11 healthy volunteers, there was no significant effect of itraconazole (200 mg once daily for 9 days), a potent inhibitor of cytochrome P4503A4, on the pharmacokinetics of moxifloxacin (a single 400 mg dose given on the 7th day of itraconazole dosing). In addition, moxifloxacin was shown not to affect the pharmacokinetics of itraconazole.
Morphine
No significant effect of morphine sulfate (a single 10 mg intramuscular dose) on the mean AUC and Cmax of moxifloxacin (400 mg single dose) was observed in a study of 20 healthy male and female volunteers.
Oral Contraceptives
A placebo-controlled study in 29 healthy female subjects showed that moxifloxacin 400 mg daily for 7 days did not interfere with the hormonal suppression of oral contraception with 0.15 mg levonorgestrel/0.03 mg ethinylestradiol (as measured by serum progesterone, FSH, estradiol, and LH), or with the pharmacokinetics of the administered contraceptive agents.
Probenecid
Probenecid (500 mg twice daily for two days) did not alter the renal clearance and total amount of moxifloxacin (400 mg single dose) excreted renally in a study of 12 healthy volunteers.
Ranitidine
No significant effect of ranitidine (150 mg twice daily for three days as pretreatment) on the pharmacokinetics of moxifloxacin (400 mg single dose) was detected in a study involving 10 healthy volunteers.
Theophylline
No significant effect of moxifloxacin (200 mg every twelve hours for 3 days) on the pharmacokinetics of theophylline (400 mg every twelve hours for 3 days) was detected in a study involving 12 healthy volunteers. In addition, theophylline was not shown to affect the pharmacokinetics of moxifloxacin. The effect of co-administration of 400 mg once daily of moxifloxacin with theophylline has not been studied.
Warfarin
No significant effect of moxifloxacin (400 mg once daily for eight days) on the pharmacokinetics of R- and S-warfarin (25 mg single dose of warfarin sodium on the fifth day) was detected in a study involving 24 healthy volunteers. No significant change in prothrombin time was observed. However, fluoroquinolones, including moxifloxacin, have been reported to enhance the anticoagulant effects of warfarin or its derivatives in the patient population [see Adverse Reactions (6.2) and Drug Interactions (7.2)].
12.4 Microbiology
Mechanism of Action
The bactericidal action of moxifloxacin results from inhibition of the topoisomerase II (DNA gyrase) and topoisomerase IV required for bacterial DNA replication, transcription, repair, and recombination.
Resistance
The mechanism of action for fluoroquinolones, including moxifloxacin, is different from that of macrolides, beta-lactams, aminoglycosides, or tetracyclines; therefore, microorganisms resistant to these classes of drugs may be susceptible to moxifloxacin. Resistance to fluoroquinolones occurs primarily by a mutation in topoisomerase II (DNA gyrase) or topoisomerase IV genes, decreased outer membrane permeability or drug efflux. In vitro resistance to moxifloxacin develops slowly via multiple-step mutations. Resistance to moxifloxacin occurs in vitro at a general frequency of between 1.8 x 10-9to < 1 x 10-11 for Gram-positive bacteria.
Cross Resistance
Cross-resistance has been observed between moxifloxacin and other fluoroquinolones against Gram-negative bacteria. Gram-positive bacteria resistant to other fluoroquinolones may, however, still be susceptible to moxifloxacin. There is no known cross-resistance between moxifloxacin and other classes of antimicrobials.
Antimicrobial Activity
Moxifloxacin has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections . [see Indications and Usage (1)].
Gram-positive bacteria
Enterococcus faecalis
Staphylococcus aureus
Streptococcus anginosus
Streptococcus constellatus
Streptococcus pneumoniae (including multi-drug resistant isolates [MDRSP]**)
Streptococcus pyogenes
**MDRSP, Multi-drug resistant Streptococcus pneumoniae includes isolates previously known as PRSP (Penicillinresistant S. pneumoniae), and are isolates resistant to two or more of the following antibiotics: penicillin (MIC) ≥2 mcg/mL), 2nd generation cephalosporins (for example, cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.
Gram-negative bacteria
Enterobacter cloacae
Escherichia coli
Haemophilus influenzae
Haemophilus parainfluenzae
Klebsiella pneumoniae
Moraxella catarrhalis
Proteus mirabilis
Yersinia pestis
Anaerobic bacteria
Bacteroides fragilis
Bacteroides thetaiotaomicron
Clostridium perfringens
Peptostreptococcus species
Other microorganisms
Chlamydophila pneumoniae
Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for moxifloxacin against isolates of similar genus or organism group. However, the efficacy of moxifloxacin in treating clinical infections due to these bacteria has not been established in adequate and well controlled clinical trials.
Gram-positive bacteria
Staphylococcus epidermidis
Streptococcus agalactiae
Streptococcus viridans group
Gram-negative bacteria
Citrobacter freundii
Klebsiella oxytoca
Legionella pneumophila
Anaerobic bacteria
Fusobacterium species
Prevotella species
Susceptibility Tests Methods
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
Long term studies in animals to determine the carcinogenic potential of moxifloxacin have not been performed.
Moxifloxacin was not mutagenic in 4 bacterial strains (TA 98, TA 100, TA 1535, TA 1537) used in the Ames Salmonella reversion assay. As with other fluoroquinolones, the positive response observed with moxifloxacin in strain TA 102 using the same assay may be due to the inhibition of DNA gyrase. Moxifloxacin was not mutagenic in the CHO/HGPRT mammalian cell gene mutation assay. An equivocal result was obtained in the same assay when v79 cells were used. Moxifloxacin was clastogenic in the v79 chromosome aberration assay, but it did not induce unscheduled DNA synthesis in cultured rat hepatocytes. There was no evidence of genotoxicity in vivo in a micronucleus test or a dominant lethal test in mice.
Moxifloxacin had no effect on fertility in male and female rats at oral doses as high as 500 mg/kg/day, approximately 12 times the maximum recommended human dose based on body surface area, or at intravenous doses as high as 45 mg/kg/day, approximately equal to the maximum recommended human dose based on body surface area. At 500 mg/kg orally there were slight effects on sperm morphology (head-tail separation) in male rats and on the estrous cycle in female rats.
13.2 Animal Toxicology and/or Pharmacology
Fluoroquinolones have been shown to cause arthropathy in immature animals. In studies in juvenile dogs oral doses of moxifloxacin 30 mg/kg/day or more (approximately 1.5 times the maximum recommended human dose based upon systemic exposure) for 28 days resulted in arthropathy. There was no evidence of arthropathy in mature monkeys and rats at oral doses up to 135 and 500 mg/kg/day, respectively.
Moxifloxacin at an oral dose of 300 mg/kg did not show an increase in acute toxicity or potential for CNS toxicity (for example, seizures) in mice when used in combination with NSAIDs such as diclofenac, ibuprofen, or fenbufen. Some fluoroquinolones have been reported to have proconvulsant activity that is exacerbated with concomitant use of NSAIDs.
A QT-prolonging effect of moxifloxacin was found in dog studies, at plasma concentrations about five times the human therapeutic level. The combined infusion of sotalol, a Class III antiarrhythmic agent, with moxifloxacin induced a higher degree of QTc prolongation in dogs than that induced by the same dose (30 mg/kg) of moxifloxacin alone. Electrophysiological in vitro studies suggested an inhibition of the rapid activating component of the delayed rectifier potassium current (IKr) as an underlying mechanism.
No signs of local intolerability were observed in dogs when moxifloxacin was administered intravenously. After intra-arterial injection, inflammatory changes involving the peri-arterial soft tissue were observed suggesting that intra-arterial administration of moxifloxacin should be avoided.
14 CLINICAL STUDIES
14.1 Acute Bacterial Sinusitis
In a controlled double-blind study conducted in the US, moxifloxacin tablets (400 mg once daily for ten days) were compared with cefuroxime axetil (250 mg twice daily for ten days) for the treatment of acute bacterial sinusitis. The trial included 457 patients valid for the efficacy analysis. Clinical success (cure plus improvement) at the 7 to 21 day post therapy test of cure visit was 90% for moxifloxacin and 89% for cefuroxime.
An additional non-comparative study was conducted to gather bacteriological data and to evaluate microbiological eradication in adult patients treated with moxifloxacin 400 mg once daily for seven days. All patients (n = 336) underwent antral puncture in this study. Clinical success rates and eradication/presumed eradication rates at the 21 to 37 day follow up visit were 97% (29 out of 30) for Streptococcus pneumoniae, 83% (15 out of 18) for Moraxella catarrhalis, and 80% (24 out of 30) for Haemophilus influenzae.
14.2 Acute Bacterial Exacerbation of Chronic Bronchitis
Moxifloxacin tablets (400 mg once daily for five days) were evaluated for the treatment of acute bacterial exacerbation of chronic bronchitis in a randomized, double-blind, controlled clinical trial conducted in the US. This study compared moxifloxacin with clarithromycin (500 mg twice daily for 10 days) and enrolled 629 patients. Clinical success was assessed at 7 to 17 days post-therapy. The clinical success for moxifloxacin was 89% (222/250) compared to 89% (224/251) for clarithromycin.
Table 10: Clinical Success Rates at Follow-Up Visit for Clinically Evaluable Patients by Pathogen (Acute Bacterial Exacerbation of Chronic Bronchitis)
| PATHOGEN | Moxifloxacin | Clarithromycin |
| Streptococcus pneumoniae | 16/16 (100%) | 20/23 (87%) |
| Haemophilus influenzae | 33/37 (89%) | 36/41 (88%) |
| Haemophilus parainfluenzae | 16/16 (100%) | 14/14 (100%) |
| Moraxella catarrhalis | 29/34 (85%) | 24/24 (100%) |
| Staphylococcus aureus | 15/16 (94%) | 6/8 (75%) |
| Klebsiella pneumoniae | 17/20 (85%) | 10/11 (91%) |
The microbiological eradication rates (eradication plus presumed eradication) in moxifloxacin treated patients were Streptococcus pneumoniae 100%, Haemophilus influenzae 89%, Haemophilus parainfluenzae 100%, Moraxella catarrhalis 85%, Staphylococcus aureus 94%, and Klebsiella pneumoniae 85%.
14.3 Community Acquired Pneumonia
A randomized, double-blind, controlled clinical trial was conducted in the US to compare the efficacy of moxifloxacin tablets (400 mg once daily) to that of high-dose clarithromycin (500 mg twice daily) in the treatment of patients with clinically and radiologically documented community acquired pneumonia. This study enrolled 474 patients (382 of whom were valid for the efficacy analysis conducted at the 14 to 35 day follow-up visit). Clinical success for clinically evaluable patients was 95% (184/194) for moxifloxacin and 95% (178/188) for high dose clarithromycin.
A randomized, double-blind, controlled trial was conducted in the US and Canada to compare the efficacy of sequential intravenous/oral moxifloxacin 400 mg once a day for 7 to 14 days to an intravenous/oral fluoroquinolone control (trovafloxacin or levofloxacin) in the treatment of patients with clinically and radiologically documented community acquired pneumonia. This study enrolled 516 patients, 362 of whom were valid for the efficacy analysis conducted at the 7 to 30 day post-therapy visit. The clinical success rate was 86% (157/182) for moxifloxacin therapy and 89% (161/180) for the fluoroquinolone comparators.
An open-label ex-US study that enrolled 628 patients compared moxifloxacin to sequential intravenous/oral amoxicillin/clavulanate (1.2 gram intravenously every 8 hours/625 mg orally every 8 hours) with or without high-dose intravenous/oral clarithromycin (500 mg twice a day). The intravenous formulations of the comparators are not FDA approved. The clinical success rate at Day 5 to 7 for moxifloxacin therapy was 93% (241/258) and demonstrated superiority to amoxicillin/clavulanate ± clarithromycin (85%, 239/280) [95% C.I. of difference in success rates between moxifloxacin and comparator (2.9%, 13.2%)]. The clinical success rate at the 21 to 28 days post-therapy visit for moxifloxacin was 84% (216/258), which also demonstrated superiority to the comparators (74%, 208/280) [95% C.I. of difference in success rates between moxifloxacin and comparator (2.6%, 16.3%)].
The clinical success rates by pathogen across four CAP studies are presented in Table 11.
Table 11: Clinical Success Rates By Pathogen (Pooled CAP Studies)
| PATHOGEN | Moxifloxacin | |
| Streptococcus pneumoniae | 80/85 | (94%) |
| Staphylococcus aureus | 17/20 | (85%) |
| Klebsiella pneumoniae | 11/12 | (92%) |
| Haemophilus influenzae | 56/61 | (92%) |
| Chlamydophila pneumoniae | 119/128 | (93%) |
| Mycoplasma pneumoniae | 73/76 | (96%) |
| Moraxella catarrhalis | 11/12 | (92%) |
Community Acquired Pneumonia caused by Multi-Drug Resistant Streptococcus pneumoniae (MDRSP)*
Moxifloxaxin was effective in the treatment of community acquired pneumonia (CAP) caused by multi-drug resistant Streptococcus pneumoniae MDRSP* isolates. Of 37 microbiologically evaluable patients with MDRSP isolates, 35 patients (95%) achieved clinical and bacteriological success post-therapy. The clinical and bacteriological success rates based on the number of patients treated are shown in Table 12.
* MDRSP, Multi-drug resistant Streptococcus pneumoniae includes isolates previously known as PRSP (Penicillin-resistant S.pneumoniae), and are isolates resistant to two or more of the following antibiotics: penicillin (MIC ≥ 2 mcg/mL), 2nd generation cephalosporins (for example, cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.
Table 12: Clinical and Bacteriological Success Rates for Moxifloxacin-Treated MDRSP CAP Patients (Population: Valid for Efficacy)
| Screening Susceptibility | Clinical Success | Bacteriological Success | |||
| n/Na | % | n/Nb | % | ||
| Penicillin-resistant | 21/21 | 100%c | 21/21 | 100%c | |
| 2nd generation cephalosporin-resistant | 25/26 | 96%c | 25/26 | 96%c | |
| Macrolide-resistantd | 22/23 | 96% | 22/23 | 96% | |
| Trimethoprim/sulfamethoxazole-resistant | 28/30 | 93% | 28/30 | 93% | |
| Tetracycline-resistant | 17/18 | 94% | 17/18 | 94% | |
a) n = number of patients successfully treated; N = number of patients with MDRSP (from a total of 37 patients)
b) n = number of patients successfully treated (presumed eradication or eradication); N = number of patients with MDRSP (from a total of 37 patients)
c) One patient had a respiratory isolate that was resistant to penicillin and cefuroxime but a blood isolate that was intermediate to penicillin and cefuroxime. The patient is included in the database based on the respiratory isolate.
d) Azithromycin, clarithromycin, and erythromycin were the macrolide antimicrobials tested.
Not all isolates were resistant to all antimicrobial classes tested. Success and eradication rates are summarized in Table 13.
Table 13: Clinical Success Rates and Microbiological Eradication Rates for Resistant Streptococcus pneumoniae (Community Acquired Pneumonia)
| S. pneumoniaewith MDRSP | Clinical Success | Bacteriological Eradication Rate |
| Resistant to 2 antimicrobials | 12/13 (92.3 %) | 12/13 (92.3 %) |
| Resistant to 3 antimicrobials | 10/11 (90.9 %)a | 10/11 (90.9 %)a |
| Resistant to 4 antimicrobials | 6/6 (100%) | 6/6 (100%) |
| Resistant to 5 antimicrobials | 7/7 (100%)a | 7/7 (100%)a |
| Bacteremia with MDRSP | 9/9 (100%) | 9/9 (100%) |
a) One patient had a respiratory isolate resistant to 5 antimicrobials and a blood isolate resistant to 3 antimicrobials. The patient was included in the category resistant to 5 antimicrobials.
14.4 Uncomplicated Skin and Skin Structure Infections
A randomized, double-blind, controlled clinical trial conducted in the US compared the efficacy of moxifloxacin 400 mg once daily for seven days with cephalexin HCl 500 mg three times daily for seven days. The percentage of patients treated for uncomplicated abscesses was 30%, furuncles 8%, cellulitis 16%, impetigo 20%, and other skin infections 26%. Adjunctive procedures (incision and drainage or debridement) were performed on 17% of the moxifloxacin treated patients and 14% of the comparator treated patients. Clinical success rates in evaluable patients were 89% (108/122) for moxifloxacin and 91% (110/121) for cephalexin HCl.
14.5 Complicated Skin and Skin Structure Infections
Two randomized, active controlled trials of cSSSI were performed. A double-blind trial was conducted primarily in North America to compare the efficacy of sequential intravenous/oral moxifloxacin 400 mg once a day for 7 to 14 days to an intravenous/oral beta-lactam/beta-lactamase inhibitor control in the treatment of patients with cSSSI. This study enrolled 617 patients, 335 of which were valid for the efficacy analysis. A second open-label International study compared moxifloxacin 400 mg once a day for 7 to 21 days to sequential intravenous/oral beta-lactam/beta-lactamase inhibitor control in the treatment of patients with cSSSI. This study enrolled 804 patients, 632 of which were valid for the efficacy analysis. Surgical incision and drainage or debridement was performed on 55% of the moxifloxacin treated and 53% of the comparator treated patients in these studies and formed an integral part of therapy for this indication. Success rates varied with the type of diagnosis ranging from 61% in patients with infected ulcers to 90% in patients with complicated erysipelas. These rates were similar to those seen with comparator drugs. The overall success rates in the evaluable patients and the clinical success by pathogen are shown in Tables 14 and 15.
Table 14: Overall Clinical Success Rates in Patients with Complicated Skin and Skin Structure Infections
| Study | Moxifloxacin n/ N (%) | Comparator n/N (%) | 95% Confidence Intervala |
| North America | 125/162 (77.2%) | 141/173 (81.5%) | (-14.4%, 2%) |
| International | 254/315 (80.6%) | 268/317 (84.5%) | (-9.4%, 2.2%) |
a) of difference in success rates between Moxifloxacin and comparator (Moxifloxacin – comparator)
Table 15: Clinical Success Rates by Pathogen in Patients with Complicated Skin and Skin Structure Infections
| Pathogen | Moxifloxacin
n/ N (%) | Comparator
n/N (%) |
| Staphylococcus aureus
(methicillin-susceptible isolates)a | 106/129 (82.2%) | 120/137 (87.6%) |
| Escherichia coli | 31/38 (81.6 %) | 28/33 (84.8 %) |
| Klebsiella pneumoniae | 11/12 (91.7 % ) | 7/10 (70%) |
| Enterobacter cloacae | 9/11 (81.8%) | 4/7 (57.1%) |
a) methicillin susceptibility was only determined in the North American Study
14.6 Complicated Intra-Abdominal Infections
Two randomized, active controlled trials of cIAI were performed. A double-blind trial was conducted primarily in North America to compare the efficacy of sequential intravenous/oral moxifloxacin 400 mg once a day for 5 to 14 days to intravenous/piperacillin/tazobactam followed by oral amoxicillin/clavulanic acid in the treatment of patients with cIAI, including peritonitis, abscesses, appendicitis with perforation, and bowel perforation. This study enrolled 681 patients, 379 of which were considered clinically evaluable. A second open-label international study compared moxifloxacin 400 mg once a day for 5 to 14 days to intravenous ceftriaxone plus intravenous metronidazole followed by oral amoxicillin/clavulanic acid in the treatment of patients with cIAI. This study enrolled 595 patients, 511 of which were considered clinically evaluable. The clinically evaluable population consisted of subjects with a surgically confirmed complicated infection, at least 5 days of treatment and a 25 to 50 day follow-up assessment for patients at the Test of Cure visit. The overall clinical success rates in the clinically evaluable patients are shown in Table 16.
Table 16: Clinical Success Rates in Patients with Complicated Intra-Abdominal Infections
| Study | Moxifloxacin n/ N (%) | Comparator n/N (%) | 95% Confidence Intervala |
| North America (overall) | 146/183 (79.8 %) | 153/196 (78.1 %) | (-7.4%, 9.3%) |
| Abscess | 40/57 (70.2 %) | 49/63 (77.8 %)b | NAc |
| Non-abscess | 106/126 (84.1 %) | 104/133 (78.2 %) | NA |
| International (overall) | 199/246 (80.9 %) | 218/265 (82.3 %) | (-8.9 %, 4.2%) |
| Abscess | 73/93 (78.5 %) | 86/99 (86.9 %) | NA |
| Non-abscess | 126/153 (82.4 %) | 132/166 (79.5 %) | NA |
a) of difference in success rates between moxifloxacin and comparator
b) Excludes 2 patients who required additional surgery within the first 48 hours.
c) NA - not applicable
14.7 Plague
Efficacy studies of moxifloxacin could not be conducted in humans with pneumonic plague for ethical and feasibility reasons. Therefore, approval of this indication was based on an efficacy study conducted in animals and supportive pharmacokinetic data in adult humans and animals.
A randomized, blinded, placebo-controlled study was conducted in an African Green Monkey (AGM) animal model of pneumonic plague. Twenty AGM (10 males and 10 females) were exposed to an inhaled mean (± SD) dose of 100 ± 50 LD50 (range 92 to 127 LD50) of Yersinia pestis (CO92 strain) aerosol. The minimal inhibitory concentration (MIC) of moxifloxacin for the Y. pestis strain used in this study was 0.06 mcg/mL. Development of sustained fever for at least 4 hours duration was used as the trigger for the initiation of 10 days of treatment with either a humanized regimen of moxifloxacin or placebo. All study animals were febrile and bacteremic with Y. pestis prior to the initiation of study treatment. Ten of 10 (100%) of the animals receiving the placebo succumbed to disease between 83 to 139 h (mean 115 ± 19 hours) post treatment. Ten of 10 (100%) moxifloxacin-treated animals survived for the 30-day period after completion of the study treatment. Compared to the placebo group, mortality in the moxifloxacin group was significantly lower (difference in survival: 100% with a two-sided 95% exact confidence interval [66.3%, 100%], p-value<0.0001).
The mean plasma concentrations of moxifloxacin associated with a statistically significant improvement in survival over placebo in an AGM model of pneumonic plague are reached or exceeded in human adults receiving the recommended oral and intravenous dosage regimens. The mean (± SD) peak plasma concentration (Cmax) and total plasma exposure defined as the area under the plasma concentration-time curve (AUC) in human adults receiving 400 mg intravenously were 3.9 ± 0.9 mcg/mL and 39.3 ± 8.6 mcg•h/mL, respectively [see Clinical Pharmacology (12.3)]. The mean (± SD) peak plasma concentration and AUC0-24 in AGM following one- day administration of a humanized dosing regimen simulating the human AUC0-24 at a 400 mg dose were 4.4 ± 1.5 mcg/mL and 22 ± 8.0 mcg·h/mL, respectively.
15 REFERENCES
1.Clinical and Laboratory Standards Institute (CLSI), Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically Approved Standard – Tenth Edition. CLSI Document M7-A10 [2015], CLSI, 950 West Valley Rd., Suite 2500, Wayne, PA 19087, USA.
2. Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-sixth Informational Supplement, CLSI document M100-S26 [2016], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA. .
3. Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Disk Diffusion Susceptibility Tests; Approved Standard – Twelfth Edition. CLSI document M02-A12 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
4. Clinical and Laboratory Standards Institute (CLSI). Methods for Antimicrobial Dilution and Disk Susceptibility Testing for Infrequently Isolated or Fastidious Bacteria: Approved Guidelines—Third Edition CLSI document M45-A3 [2016], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
5. Clinical and Laboratory Standards Institute (CLSI). Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard -Eighth Edition. CLSI document M11-A8 [2012]. Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Moxifloxacin Tablets, USP
Moxifloxacin Tablets USP, 400 mg are available as Beige colored, capsule shaped, biconvex tablets debossed ‘112’ on one side and plain on other side. They are supplied in bottles of 30, 100, 500, and unit dose package of 50’s (5 x 10’s).
Bottles of 30 NDC 55111-112-30
Bottles of 100 NDC 55111-112-01
Bottles of 500 NDC 55111-112-05
Unit dose package of 50 (5 x 10’s) NDC 55111-112-66
Store at 20 to 25°C (68 to 77°F); [see USP Controlled Room Temperature]. Avoid high humidity.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Serious Adverse Reactions Advise patients to stop taking moxifloxacin if they experience an adverse reaction and to call their healthcare provider for advice on completing the full course of treatment with another antibacterial drug.
Inform patients of the following serious adverse reactions that have been associated with moxifloxacin or other fluoroquinolone use:
• Disabling and potentially irreversible serious adverse reactions that may occur together: Inform patients that disabling and potentially irreversible serious adverse reactions, including tendinitis and tendon rupture, peripheral neuropathies, and central nervous system effects, have been associated with use of moxifloxacin and may occur together in the same patient. Inform patients to stop taking moxifloxacin immediately if they experience an adverse reaction and to call their healthcare provider.
• Tendinitis and Tendon Rupture: Instruct patients to contact their healthcare provider if they experience pain, swelling, or inflammation of a tendon, or weakness or inability to use one of their joints; rest and refrain from exercise; and discontinue moxifloxacin treatment. Symptoms may be irreversible. The risk of severe tendon disorder with fluoroquinolones is higher in older patients usually over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants.
• Peripheral Neuropathies: Inform patients that peripheral neuropathies have been associated with moxifloxacin use, symptoms may occur soon after initiation of therapy and may be irreversible. If symptoms of peripheral neuropathy including pain, burning, tingling, numbness and/or weakness develop, immediately discontinue moxifloxacin and tell them to contact their physician.
• Central nervous system effects (for example, convulsions, dizziness, lightheadedness, increased intracranial pressure): Inform patients that convulsions have been reported in patients receiving fluoroquinolones, including moxifloxacin. Instruct patients to notify their physician before taking this drug if they have a history of convulsions. Inform patients that they should know how they react to moxifloxacin before they operate an automobile or machinery or engage in other activities requiring mental alertness and coordination. Instruct patients to notify their physician if persistent headache with or without blurred vision occurs.
• Exacerbation of Myasthenia Gravis: Instruct patients to inform their physician of any history of myasthenia gravis. Instruct patients to notify their physician if they experience any symptoms of muscle weakness, including respiratory difficulties.
• Hypersensitivity Reactions: Inform patients that moxifloxacin can cause hypersensitivity reactions, even following a single dose, and to discontinue the drug at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
•Hepatotoxicity: Inform patients that severe hepatotoxicity (including acute hepatitis and fatal events) has been reported in patients taking moxifloxacin. Instruct patients to inform their physician if they experience any signs or symptoms of liver injury including: loss of appetite, nausea, vomiting, fever, weakness, tiredness, right upper quadrant tenderness, itching, yellowing of the skin and eyes, light colored bowel movements or dark colored urine.
• Aortic aneurysm and dissection: Inform patients to seek emergency medical care if they experience sudden chest, stomach, or back pain.
• Diarrhea: Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, instruct patients to contact their physician as soon as possible.
• Prolongation of the QT Interval: Instruct patients to inform their physician of any personal or family history of QT prolongation or proarrhythmic conditions such as hypokalemia, bradycardia, or recent myocardial ischemia; if they are taking any Class IA (quinidine, procainamide), or Class III (amiodarone, sotalol) antiarrhythmic agents. Instruct patients to notify their physician if they have any symptoms of prolongation of the QT interval, including prolonged heart palpitations or a loss of consciousness.
• Blood Glucose Disturbances: Inform the patients that if they are diabetic and are being treated with insulin or an oral hypoglycemic agent and a hypoglycemic reaction occurs, they should discontinue moxifloxacin and consult a physician.
• Photosensitivity/Phototoxicity: Inform patients that photosensitivity/phototoxicity has been reported in patients receiving fluoroquinolones. Inform patients to minimize or avoid exposure to natural or artificial sunlight (tanning beds or UVA/B treatment) while taking quinolones. If patients need to be outdoors while using quinolones, instruct them to wear loose-fitting clothes that protect skin from sun exposure and discuss other sun protection measures with their physician. If a sunburn-like reaction or skin eruption occurs, instruct patients to contact their physician.
Antibacterial Resistance
Inform patients that antibacterial drugs including moxifloxacin should only be used to treat bacterial infections. They do not treat viral infections (for example, the common cold). When moxifloxacin is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by moxifloxacin or other antibacterial drugs in the future.
Administration Instructions
- Inform patients that moxifloxacin tablets may be taken with or without food. Advise patients drink fluids liberally.
- Inform patients that moxifloxacin tablets should be taken at least 4 hours before or 8 hours after multivitamins (containing iron or zinc), antacids (containing magnesium or aluminum), sucralfate, or didanosine buffered tablets for oral suspension or the pediatric powder for oral solution.
- Advise patients that if a dose is missed, it should be taken anytime but not later than 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next dose, the missed dose should not be taken and treatment should be continued as prescribed with the next scheduled dose. Double doses should not be taken to compensate for a missed dose.
Plague Studies
Inform patients given moxifloxacin for plague that efficacy studies could not be conducted in humans for feasibility reasons. Therefore, approval for plague was based on efficacy studies conducted in animals.
FDA-Approved Medication Guide
MEDICATION GUIDE
Moxifloxacin Tablets, USP
(mox'' i flox' a sin hye'' droe klor' ide)
Read the Medication Guide that comes with moxifloxacin tablets before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about moxifloxacin tablets?
Moxifloxacin tablet is in a class of antibiotics called fluoroquinolones. Moxifloxacin tablets can cause serious side effects that can happen at the same time and could result in death. If you get any of the following serious side effects, you should stop taking moxifloxacin tablets and get medical help right away. Talk with your healthcare provider about whether you should continue to take moxifloxacin tablets.
1. Tendon rupture or swelling of the tendon (tendinitis).
-
Tendon problems can happen in people of all ages who take moxifloxacin tablets. Tendons are tough cords of tissue that connect muscles to bones. Symptoms of tendon problems may include:
- Pain, swelling, tears and inflammation of tendons including the back of the ankle (Achilles), shoulder, hand, or other tendon sites.
-
The risk of getting tendon problems while you take moxifloxacintablets is higher if you:
- Are over 60 years of age .
- Are taking steroids (corticosteroids) .
- Have had a kidney, heart or lung transplant .
- Tendon problems can happen in people who do not have the above risk factors when they take moxifloxacin tablets.
-
Other reasons that can increase your risk of tendon problems can include:
- Physical activity or exercise .
- Kidney failure .
- Tendon problems in the past, such as in people with rheumatoid arthritis (RA).
-
Stop taking moxifloxacin tablets immediately and call your healthcare provider right away at the first sign of tendon pain, swelling or inflammation. Stop taking moxifloxacin tablets until tendinitis or tendon rupture has been ruled out by your healthcare provider.
Avoid exercise and using the affected area. The most common area of pain and swelling is in the Achilles tendon at the back of your ankle. This can also happen with other tendons.
- Talk to your healthcare provider about the risk of tendon rupture with continued use of moxifloxacin tablets. You may need a different antibiotic that is not a fluoroquinolone to treat your infection.
- Tendon rupture can happen while you are taking or after you have stopped taking moxifloxacin tablets. Tendon ruptures can happen within hours or days after taking moxifloxacin tablets and have happened up to several months after people have stopped taking their fluoroquinolone.
-
Stop taking moxifloxacin tablets immediately and get medical help right away if you get any of the following signs or symptoms of a tendon rupture:
- Hear or feel a snap or pop in a tendon area.
- Bruising right after an injury in a tendon area.
- Unable to move the affected area or put weight on the area.
2.Changes in sensation and possible nerve damage (Peripheral Neuropathy). Damage to the nerves in arms, hands, legs, or feet can happen in people who take fluoroquinolones, including moxifloxacin tablets. Stop taking moxifloxacin tablets immediately and talk to your healthcare provider right away if you get any of the following symptoms of peripheral neuropathy in your arms, hands, legs, or feet:
• pain
• burning
• tingling
• numbness
• weakness
Moxifloxacin tablets may need to be stopped to prevent permanent nerve damage.
3. Central Nervous System (CNS) effects. Seizures have been reported in people who take fluoroquinolone antibiotic medicines, including moxifloxacin tablets. Tell your healthcare provider if you have a history of seizures before you start taking moxifloxacin tablets. CNS side effects may happen as soon as after taking the first dose of moxifloxacin tablets. Stop taking moxifloxacin tablets immediately and talk to your healthcare provider right away if you get any of these side effects, or other changes in mood or behavior:
• seizures
• hear voices, see things, or sense things that are not there (hallucinations)
• feel restless
• tremors
• feel anxious or nervous
• confusion
• depression
• trouble sleeping
• nightmares
• feel lightheaded or dizzy
• feel more suspicious (paranoia)
• suicidal thoughts or acts
• headaches that will not go away, with or without blurred vision.
4. Worsening of myasthenia gravis (a disease which causes muscle weakness). Fluoroquinolones like moxifloxacin tablets may cause worsening of myasthenia gravis symptoms, including muscle weakness and breathing problems. Tell your healthcare provider if you have a history of myasthenia gravis. Moxifloxacin tablets should not be used in people who have a history of myasthenia gravis. Call your healthcare provider right away if you have any worsening muscle weakness or breathing problems.
See the section “What are the possible side effects of moxifloxacin tablets?” for more information about side effects.
What are moxifloxacin tablets?
Moxifloxacin tablets are a fluoroquinolone antibiotic medicine used to treat certain types of infections caused by certain germs called bacteria in adults 18 years or older. These bacterial infections include:
• Community Acquired Pneumonia
• Uncomplicated Skin and Skin Structure Infections
• Complicated Skin and Skin Structure Infections
• Complicated Intra-Abdominal Infections
• Plague
• Acute Bacterial Sinusitis
• Acute Bacterial Exacerbation of Chronic Bronchitis
Moxifloxacin tablets should not be used in people with acute bacterial sinusitis or acute bacterial exacerbation of chronic bronchitis if there are other treatment options available.
Studies of moxifloxacin for use in the treatment of plague were done in animals only, because plague could not be studied in people.
It is not known if moxifloxacin tablets are safe and works in people under 18 years of age. Children have a higher chance of getting bone, joint, and tendon (musculoskeletal) problems while taking fluoroquinolone antibiotic medicines.
Sometimes infections are caused by viruses rather than by bacteria. Examples include viral infections in the sinuses and lungs, such as the common cold or flu. Antibiotics, including moxifloxacin tablets, do not kill viruses.
Call your healthcare provider if you think your condition is not getting better while you are taking moxifloxacin tablets.
Who should not take moxifloxacin tablets?
Do not take moxifloxacin tablets if you have ever had an allergic reaction to moxifloxacin, other fluoroquinolone antibiotics, or any of the ingredients in moxifloxacin tablets. Ask your healthcare provider if you are not sure. See the list of ingredients in moxifloxacin tablets at the end of this Medication Guide for a complete list of ingredients in moxifloxacin tablets.
What should I tell my healthcare provider before taking moxifloxacin tablets?
- See “What is the most important information I should know about moxifloxacin tablets?”
Tell your healthcare provider about all your medical conditions, including if you:
- Have tendon problems. Moxifloxacin tablets should not be used in people who have a history of tendon problems.
- Have a disease that causes muscle weakness (myasthenia gravis). Moxifloxacin tablets should not used in people who have a history of myasthenia gravis.
- Have central nervous system problems (such as epilepsy) .
- Have nerve problems. Moxifloxacin tablets should not be used in patients who have a history of a nerve problem called peripheral neuropathy.
- Have or anyone in your family has an irregular heartbeat, especially a condition called “QT prolongation” .
- Have low blood potassium (hypokalemia) .
- Have a slow heartbeat (bradycardia) .
- Have a history of seizures .
- Have kidney problems .
- Have rheumatoid arthritis (RA) or other history of joint problems .
- Are pregnant or planning to become pregnant. It is not known if moxifloxacin tablets will harm your unborn child .
- Are breastfeeding or plan to breastfeed. It is not known if moxifloxacin tablets passes into breast milk. You and your healthcare provider should decide whether you will take moxifloxacin tablets or breastfeed.
- Have diabetes or problems with low blood sugar (hypoglycemia).
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, herbal and dietary supplements. Moxifloxacin tablets and other medicines can affect each other causing side effects.
Especially tell your healthcare provider if you take:
- A Non-Steroidal Anti-Inflammatory Drug (NSAID). Many common medicines for pain relief are NSAIDs. Taking an NSAID while you take moxifloxacin tablets or other fluoroquinolones may increase your risk of central nervous system effects and seizures.
- A blood thinner (warfarin, Coumadin, Jantoven).
- A medicine to control your heart rate or rhythm (antiarrhythmic) See “What are the possible side effects of moxifloxacin tablets?”
- An anti-psychotic medicine.
- A tricyclic antidepressant.
- An oral anti-diabetes medicine or insulin.
- Erythromycin.
- A water pill (diuretic).
- A steroid medicine. Corticosteroids taken by mouth or by injection may increase the chance of tendon injury. See “What is the most important information I should know about moxifloxacin tablets?”
- Certain medicines may keep moxifloxacin tablets from working correctly. Take moxifloxacin tablets either 4 hours before or 8 hours after taking these products:
- An antacid, multivitamin, or other product that has magnesium, aluminum, iron, or zinc
- Sucralfate (Carafate®)
- Didanosine oral suspension or solution
Ask your healthcare provider if you are not sure if any of your medicines are listed above. Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take moxifloxacin tablets?
- Take moxifloxacin tablets 1 time each day exactly as prescribed by your healthcare provider.
- Take moxifloxacin tablets at about the same time each day.
- Moxifloxacin tablets can be taken with or without food.
- If you miss a dose of moxifloxacin tablets and it is:
- 8 hours or more until your next scheduled dose, take your missed dose right away. Then take the next dose at your regular time.
- less than 8 hours until your next scheduled dose, do not take the missed dose. Take the next dose at your regular time.
- Do not take 2 doses of moxifloxacin tablets to make up for a missed dose. If you are not sure about when to take moxifloxacin tablets after a missed dose, ask your doctor or pharmacist.
- Drink plenty of fluids while taking moxifloxacin tablets.
- Do not skip any doses, or stop taking moxifloxacin tablets even if you begin to feel better, until you finish your prescribed treatment, unless:
- You have tendon problems. (See “What is the most important information I should know about moxifloxacin tablets?”).
- You have nerve problems. See " What is the most important information I should know about moxifloxacin tablets?”.
- You have central nervous system problems. See " What is the most important information I should know about moxifloxacin tablets?”.
- You have a serious allergic reaction (see “What are the possible side effects of moxifloxacin tablets?”), or your healthcare provider tells you to stop taking moxifloxacin tablets.
- This will help make sure that all of the bacteria are killed and lower the chance that the bacteria will become resistant to moxifloxacin tablets. If this happens, moxifloxacin tablets and other antibiotic medicines may not work in the future.
- If you miss a dose of moxifloxacin tablets, take it as soon as you remember.
- Do not take more than 1 dose of moxifloxacin tablets in one day.
- If you take too much moxifloxacin tablets, call your healthcare provider or get medical help immediately.
What should I avoid while taking moxifloxacin tablets?
- Moxifloxacin tablets can make you feel dizzy and lightheaded. Do not drive, operate machinery, or do other activities that require mental alertness or coordination until you know how moxifloxacin tablets affects you.
- Avoid sunlamps, tanning beds, and try to limit your time in the sun. Moxifloxacin tablets can make your skin sensitive to the sun (photosensitivity) and the light from sunlamps and tanning beds. You could get severe sunburn, blisters or swelling of your skin. If you get any of these symptoms while taking moxifloxacin tablets, call your healthcare provider right away. You should use a sunscreen and wear a hat and clothes that cover your skin if you have to be in sunlight.
What are the possible side effects of moxifloxacin tablets?
Moxifloxacin tablets can cause side effects that may be serious or even cause death including.
- See “What is the most important information I should know about moxifloxacin tablets?”
- Serious heart rhythm changes (QT prolongation and torsade de pointes). Tell your healthcare provider right away if you have a change in your heartbeat (a fast or irregular heartbeat), or if you faint. Moxifloxacin tablets may cause a rare heart problem known as prolongation of the QT interval. This condition can cause an abnormal heartbeat and can be very dangerous. The chances of this event are higher in people:
- Who are elderly
- With a family history of prolonged QT interval
- With low blood potassium (hypokalemia)
- Who take certain medicines to control heart rhythm (antiarrhythmics)
-
Serious allergic reactions. Allergic reactions can happen in people taking fluoroquinolones, including moxifloxacin tablets, even after only 1 dose. Stop taking moxifloxacin tablets and get emergency medical help right away if you get any of the following symptoms of a severe allergic reaction:
- Hives
- Trouble breathing or swallowing
- Swelling of the lips, tongue, face
- Throat tightness, hoarseness
- Fast heartbeat
- Faint
- Yellowing of the skin or eyes. Stop taking moxifloxacin tablets and tell your healthcare provider right away if you get yellowing of your skin or white part of your eyes, or if you have dark urine. These can be signs of a serious reaction to moxifloxacin tablets (a liver problem).
- Skin rash. Skin rash may happen in people taking moxifloxacin tablets even after only 1 dose. Stop taking moxifloxacin tablets at the first sign of a skin rash and call your healthcare provider. Skin rash may be a sign of a more serious reaction to moxifloxacin tablets.
- Aortic aneurysm and dissection. Tell your healthcare provider if you have ever been told that you have a swelling of the large artery that carries blood from the heart to the body (aortic aneurysm). Get emergency medical help right away if you have sudden chest, stomach, or back pain.
- Intestine infection (Pseudomembranous colitis). Pseudomembranous colitis can happen with most antibiotics, including moxifloxacin tablets. Call your healthcare provider right away if you get watery diarrhea, diarrhea that does not go away, or bloody stools. You may have stomach cramps and a fever. Pseudomembranous colitis can happen 2 or more months after you have stopped taking moxifloxacin tablets.
- Changes in blood sugar. People who take moxifloxacin tablets and other fluoroquinolone medicines with oral anti-diabetes medicines or with insulin can get low blood sugar (hypoglycemia) and high blood sugar (hyperglycemia). Follow your healthcare provider’s instructions for how often to check your blood sugar. If you have diabetes and you get low blood sugar while taking moxifloxacin tablets, stop taking moxifloxacin tablets and call your healthcare provider right away. Your antibiotic medicine may need to be changed.
- Sensitivity to sunlight (photosensitivity). See “What should I avoid while taking moxifloxacin tablets?”
The most common side effects of moxifloxacin tablets include:
- Nausea
- Diarrhea
- Headache
- dizziness
These are not all the possible side effects of moxifloxacin tablets. Tell your healthcare provider about any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store moxifloxacin tablets?
Moxifloxacin Tablets
- Store moxifloxacin tablets at 20 to 25°C (68 to 77°F).
- Keep moxifloxacin tablets away from moisture (humidity).
Keep moxifloxacin tablets and all medicines out of the reach of children.
General Information about the safe and effective use of moxifloxacin tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use moxifloxacin tablets for a condition for which it is not prescribed. Do not give moxifloxacin tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about moxifloxacin tablets. If you would like more information about moxifloxacin tablets, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about moxifloxacin tablets that is written for healthcare professionals. For more information call 1-888-375-3784 (toll free).
What are the ingredients in moxifloxacin tablets?
Moxifloxacin Tablets:
Active ingredient: moxifloxacin hydrochloride
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose, iron oxide red, iron oxide yellow, magnesium stearate, microcrystalline cellulose, polyethylene glycol 400, polysorbate 80, sodium stearyl fumarate, talc and titanium dioxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Rx only
Distributed by:
Dr. Reddy's Laboratories, Inc.
Princeton, NJ 08540
Made in India
Revised: 1220
Dispense with Medication Guide available at:
www.drreddys.com/medguide/moxifloxacintabs.pdf

