FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Hypertension
Nebivolol tablets are indicated for the treatment of hypertension, to lower blood pressure [see Clinical Studies (14.1)]. Nebivolol tablets may be used alone or in combination with other antihypertensive agents [see Drug Interactions (7)].
Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes, including the class to which this drug principally belongs. There are no controlled trials demonstrating risk reduction with nebivolol tablets.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
The dose of nebivolol tablets must be individualized to the needs of the patient. For most patients, the recommended starting dose is 5 mg once daily, with or without food, as monotherapy or in combination with other agents. For patients requiring further reduction in blood pressure, the dose can be increased at 2-week intervals up to 40 mg. A more frequent dosing regimen is unlikely to be beneficial.
Renal Impairment
In patients with severe renal impairment (ClCr less than 30 mL/min) the recommended initial dose is 2.5 mg once daily; titrate up slowly if needed. Nebivolol tablets have not been studied in patients receiving dialysis [see Clinical Pharmacology (12.4)].
Hepatic Impairment
In patients with moderate hepatic impairment, the recommended initial dose is 2.5 mg once daily; titrate up slowly if needed. Nebivolol tablets have not been studied in patients with severe hepatic impairment and therefore it is not recommended in that population [see Clinical Pharmacology (12.4)].
2.2 Subpopulations
Geriatric Patients
It is not necessary to adjust the dose in the elderly [see use in Specific Populations (8.5)].
CYP2D6 Polymorphism
No dose adjustments are necessary for patients who are CYP2D6 poor metabolizers. The clinical effect and safety profile observed in poor metabolizers were similar to those of extensive metabolizers [see Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Nebivolol is available as tablets for oral administration containing nebivolol hydrochloride equivalent to 2.5 mg, 5 mg, 10 mg, and 20 mg of nebivolol.
Nebivolol tablets 2.5 mg are light blue colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘78’ on the other side.
Nebivolol tablets 5 mg are beige colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘79’ on the other side.
Nebivolol tablets 10 mg are pink colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘80’ on the other side.
Nebivolol tablets 20 mg are light blue colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘85’ on the other side.
4 CONTRAINDICATIONS
Nebivolol tablets are contraindicated in the following conditions:
- Severe bradycardia
- Heart block greater than first degree
- Patients with cardiogenic shock
- Decompensated cardiac failure
- Sick sinus syndrome (unless a permanent pacemaker is in place)
- Patients with severe hepatic impairment (Child-Pugh >B)
- Patients who are hypersensitive to any component of this product.
5 WARNINGS AND PRECAUTIONS
5.1 Abrupt Cessation of Therapy
Do not abruptly discontinue nebivolol therapy in patients with coronary artery disease. Severe exacerbation of angina, myocardial infarction and ventricular arrhythmias have been reported in patients with coronary artery disease following the abrupt discontinuation of therapy with β-blockers. Myocardial infarction and ventricular arrhythmias may occur with or without preceding exacerbation of the angina pectoris. Caution patients without overt coronary artery disease against interruption or abrupt discontinuation of therapy. As with other β-blockers, when discontinuation of nebivolol is planned, carefully observe and advise patients to minimize physical activity. Taper nebivolol over 1 to 2 weeks when possible. If the angina worsens or acute coronary insufficiency develops, re-start nebivolol promptly, at least temporarily.
5.2 Angina and Acute Myocardial Infarction
Nebivolol was not studied in patients with angina pectoris or who had a recent MI.
5.3 Bronchospastic Diseases
In general, patients with bronchospastic diseases should not receive β-blockers.
5.4 Anesthesia and Major Surgery
Because beta-blocker withdrawal has been associated with an increased risk of MI and chest pain, patients already on beta-blockers should generally continue treatment throughout the perioperative period. If nebivolol is to be continued perioperatively, monitor patients closely when anesthetic agents which depress myocardial function, such as ether, cyclopropane, and trichloroethylene, are used. If β-blocking therapy is withdrawn prior to major surgery, the impaired ability of the heart to respond to reflex adrenergic stimuli may augment the risks of general anesthesia and surgical procedures.
The β-blocking effects of nebivolol can be reversed by β-agonists, e.g., dobutamine or isoproterenol. However, such patients may be subject to protracted severe hypotension. Additionally, difficulty in restarting and maintaining the heartbeat has been reported with β-blockers.
5.5 Hypoglycemia
Beta-blockers may prevent early warning signs of hypoglycemia, such as tachycardia, and increase the risk for severe or prolonged hypoglycemia at anytime during treatment, especially in patients with diabetes mellitus or children and patients who are fasting (i.e., surgery, not eating regularly, or are vomiting). If severe hypoglycemia occurs, patients should be instructed to seek emergency treatment.
5.6 Thyrotoxicosis
β-blockers may mask clinical signs of hyperthyroidism, such as tachycardia. Abrupt withdrawal of β-blockers may be followed by an exacerbation of the symptoms of hyperthyroidism or may precipitate a thyroid storm.
5.7 Peripheral Vascular Disease
β-blockers can precipitate or aggravate symptoms of arterial insufficiency in patients with peripheral vascular disease.
5.8 Non-dihydropyridine Calcium Channel Blockers
Because of significant negative inotropic and chronotropic effects in patients treated with β-blockers and calcium channel blockers of the verapamil and diltiazem type, monitor the ECG and blood pressure in patients treated concomitantly with these agents.
5.9 Use with CYP2D6 Inhibitors
Nebivolol exposure increases with inhibition of CYP2D6 [see Drug Interactions (7)]. The dose of nebivolol may need to be reduced.
5.10 Impaired Renal Function
Renal clearance of nebivolol is decreased in patients with severe renal impairment. Nebivolol has not been studied in patients receiving dialysis [see Clinical Pharmacology (12.4) and Dosage and Administration (2.1)].
5.11 Impaired Hepatic Function
Metabolism of nebivolol is decreased in patients with moderate hepatic impairment. Nebivolol has not been studied in patients with severe hepatic impairment [see Clinical Pharmacology (12.4) and Dosage and Administration (2.1)].
5.12 Risk of Anaphylactic Reactions
While taking β-blockers, patients with a history of severe anaphylactic reactions to a variety of allergens may be more reactive to repeated accidental, diagnostic, or therapeutic challenge. Such patients may be unresponsive to the usual doses of epinephrine used to treat allergic reactions.
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Nebivolol has been evaluated for safety in patients with hypertension and in patients with heart failure. The observed adverse reaction profile was consistent with the pharmacology of the drug and the health status of the patients in the clinical trials. Adverse reactions reported for each of these patient populations are provided below. Excluded are adverse reactions considered too general to be informative and those not reasonably associated with the use of the drug because they were associated with the condition being treated or are very common in the treated population.
The data described below reflect worldwide clinical trial exposure to nebivolol in 6545 patients, including 5038 patients treated for hypertension and the remaining 1507 subjects treated for other cardiovascular diseases. Doses ranged from 0.5 mg to 40 mg. Patients received nebivolol for up to 24 months, with over 1900 patients treated for at least 6 months, and approximately 1300 patients for more than one year.
HYPERTENSION: In placebo-controlled clinical trials comparing nebivolol with placebo, discontinuation of therapy due to adverse reactions was reported in 2.8% of patients treated with nebivolol and 2.2% of patients given placebo. The most common adverse reactions that led to discontinuation of nebivolol were headache (0.4%), nausea (0.2%) and bradycardia (0.2%).
Table 1 lists treatment-emergent adverse reactions that were reported in three 12-week, placebo-controlled monotherapy trials involving 1597 hypertensive patients treated with either 5 mg, 10 mg, or 20 mg to 40 mg of nebivolol and 205 patients given placebo and for which the rate of occurrence was at least 1% of patients treated with nebivolol and greater than the rate for those treated with placebo in at least one dose group.
Table 1. Treatment-Emergent Adverse Reactions with an Incidence (over 6 weeks) ≥ 1% in Nebivolol-Treated Patients and at a Higher Frequency than Placebo-Treated Patients
| System Organ Class – Preferred Term | Placebo (n = 205) (%) | Nebivolol 5 mg (n = 459) (%) | Nebivolol 10 mg (n = 461) (%) | Nebivolol 20 mg to 40 mg (n = 677) (%) |
| Cardiac Disorders
| | | | |
| Bradycardia | 0 | 0 | 0 | 1 |
| Gastrointestinal Disorders
| | | | |
| Diarrhea | 2 | 2 | 2 | 3 |
| Nausea | 0 | 1 | 3 | 2 |
| General Disorders
| | | | |
| Fatigue | 1 | 2 | 2 | 5 |
| Chest pain | 0 | 0 | 1 | 1 |
| Peripheral edema | 0 | 1 | 1 | 1 |
| Nervous System Disorders
| | | | |
| Headache | 6 | 9 | 6 | 7 |
| Dizziness | 2 | 2 | 3 | 4 |
| Psychiatric Disorders
| | | | |
| Insomnia | 0 | 1 | 1 | 1 |
| Respiratory Disorders
| | | | |
| Dyspnea | 0 | 0 | 1 | 1 |
| Skin and subcutaneous
Tissue Disorders | | | | |
| Rash | 0 | 0 | 1 | 1 |
Listed below are other reported adverse reactions with an incidence of at least 1% in the more than 4300 patients treated with nebivolol in controlled or open-label trials except for those already appearing in Table 1, terms too general to be informative, minor symptoms, or adverse reactions unlikely to be attributable to drug because they are common in the population. These adverse reactions were in most cases observed at a similar frequency in placebo-treated patients in the controlled studies.
Body as a Whole: asthenia.
Gastrointestinal System Disorders: abdominal pain
Metabolic and Nutritional Disorders: hypercholesterolemia
Nervous System Disorders: paraesthesia
6.2 Laboratory Abnormalities
In controlled monotherapy trials of hypertensive patients, nebivolol was associated with an increase in BUN, uric acid, triglycerides and a decrease in HDL cholesterol and platelet count.
6.3 Postmarketing Experience
The following adverse reactions have been identified from spontaneous reports of nebivolol received worldwide and have not been listed elsewhere. These adverse reactions have been chosen for inclusion due to a combination of seriousness, frequency of reporting or potential causal connection to nebivolol. Adverse reactions common in the population have generally been omitted. Because these adverse reactions were reported voluntarily from a population of uncertain size, it is not possible to estimate their frequency or establish a causal relationship to nebivolol exposure: abnormal hepatic function (including increased AST, ALT and bilirubin), acute pulmonary edema, acute renal failure, atrioventricular block (both second and third degree), bronchospasm, erectile dysfunction, hypersensitivity (including urticaria, allergic vasculitis and rare reports of angioedema), hypotension, myocardial infarction, pruritus, psoriasis, Raynaud’s phenomenon, peripheral ischemia/claudication, somnolence, syncope, thrombocytopenia, various rashes and skin disorders, vertigo, and vomiting.
7 DRUG INTERACTIONS
7.1 CYP2D6 Inhibitors
Use caution when nebivolol is co-administered with CYP2D6 inhibitors (quinidine, propafenone, fluoxetine, paroxetine, etc.) [see Clinical Pharmacology (12.5)].
7.2 Hypotensive Agents
Do not use nebivolol with other β-blockers. Closely monitor patients receiving catecholamine-depleting drugs, such as reserpine or guanethidine, because the added β-blocking action of nebivolol may produce excessive reduction of sympathetic activity. In patients who are receiving nebivolol and clonidine, discontinue nebivolol for several days before the gradual tapering of clonidine.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data regarding use of nebivolol in pregnant women are insufficient to determine whether there are drug-associated risks of adverse developmental outcomes. There are risks to the mother and fetus associated with poorly controlled hypertension in pregnancy. The use of beta blockers during the third trimester of pregnancy may increase the risk of hypotension, bradycardia, hypoglycemia, and respiratory depression in the neonate [see Clinical Considerations]. Oral administration of nebivolol to pregnant rats during organogenesis resulted in embryofetal and perinatal lethality at doses approximately equivalent to the maximum recommended human dose (MRHD).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal adverse reactions
Neonates of women with hypertension, who are treated with beta-blockers during the third trimester of pregnancy, may be at increased risk for hypotension, bradycardia, hypoglycemia, and respiratory depression. Observe newborns for symptoms of hypotension, bradycardia, hypoglycemia and respiratory depression and manage accordingly.
Data
Animal Data
Nebivolol was shown to increase embryo-fetal and perinatal lethality in rats at approximately 1.2 times the MRHD or 40 mg/day on a mg/m2 basis. Decreased pup body weights occurred at 1.25 and 2.5 mg/kg in rats, when exposed during the perinatal period (late gestation, parturition and lactation). At 5 mg/kg and higher doses (1.2 times the MRHD), prolonged gestation, dystocia and reduced maternal care were produced with corresponding increases in late fetal deaths and stillbirths and decreased birth weight, live litter size and pup survival. These events occurred only when nebivolol was given during the perinatal period (late gestation, parturition and lactation). Insufficient numbers of pups survived at 5 mg/kg to evaluate the offspring for reproductive performance.
In studies in which pregnant rats were given nebivolol during organogenesis, reduced fetal body weights were observed at maternally toxic doses of 20 and 40 mg/kg/day (5 and 10 times the MRHD), and small reversible delays in sternal and thoracic ossification associated with the reduced fetal body weights and a small increase in resorption occurred at 40 mg/kg/day (10 times the MRHD).
No adverse effects on embryo-fetal viability, sex, weight or morphology were observed in studies in which nebivolol was given to pregnant rabbits at doses as high as 20 mg/kg/day (10 times the MRHD).
8.2 Lactation
Risk Summary
There is no information regarding the presence of nebivolol in human milk, the effects on the breastfed infant, or the effects on milk production. Nebivolol is present in rat milk [see Data]. Because of the potential for β-blockers to produce serious adverse reactions in nursing infants, especially bradycardia, nebivolol is not recommended during nursing.
Data
In lactating rats, maximum milk levels of unchanged nebivolol were observed at 4 hours after single and repeat doses of 2.5 mg/kg/day. The daily dose (mg/kg body weight) ingested by a rat pup is 0.3% of the dam dose for unchanged nebivolol.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Pediatric studies in ages newborn to 18 years old have not been conducted because of incomplete characterization of developmental toxicity and possible adverse effects on long-term fertility [see Nonclinical Toxicology (13.1)].
Juvenile Animal Toxicity Data
Daily oral doses of nebivolol to juvenile rats from post-natal day 14 to post-natal day 27 showed sudden unexplained death at exposures equal to those in human poor metabolizers given a single dose of 10 mg. No mortality was seen at half the adult human exposure.
In surviving rats, cardiomyopathy was seen at exposures greater than or equal to the human exposure. Male rat pups exposed to twice the human exposure showed decreases in total sperm count as well as decreases in the total and percentage of motile sperm.
8.5 Geriatric Use
Of the 2800 patients in the U.S. sponsored placebo-controlled clinical hypertension studies, 478 patients were 65 years of age or older. No overall differences in efficacy or in the incidence of adverse events were observed between older and younger patients.
8.6 Heart Failure
In a placebo-controlled trial of 2128 patients (1067 nebivolol, 1061 placebo) over 70 years of age with chronic heart failure receiving a maximum dose of 10 mg per day for a median of 20 months, no worsening of heart failure was reported with nebivolol compared to placebo. However, if heart failure worsens consider discontinuation of nebivolol.
10 OVERDOSAGE
In clinical trials and worldwide postmarketing experience there were reports of nebivolol overdose. The most common signs and symptoms associated with nebivolol overdosage are bradycardia and hypotension. Other important adverse reactions reported with nebivolol overdose include cardiac failure, dizziness, hypoglycemia, fatigue and vomiting. Other adverse reactions associated with β-blocker overdose include bronchospasm and heart block.
The largest known ingestion of nebivolol worldwide involved a patient who ingested up to 500 mg of nebivolol along with several 100 mg tablets of acetylsalicylic acid in a suicide attempt. The patient experienced hyperhydrosis, pallor, depressed level of consciousness, hypokinesia, hypotension, sinus bradycardia, hypoglycemia, hypokalemia, respiratory failure and vomiting. The patient recovered.
Because of extensive drug binding to plasma proteins, hemodialysis is not expected to enhance nebivolol clearance.
If overdose occurs, provide general supportive and specific symptomatic treatment. Based on expected pharmacologic actions and recommendations for other β-blockers, consider the following general measures, including stopping nebivolol, when clinically warranted:
Bradycardia: Administer IV atropine. If the response is inadequate, isoproterenol or another agent with positive chronotropic properties may be given cautiously. Under some circumstances, transthoracic or transvenous pacemaker placement may be necessary.
Hypotension: Administer IV fluids and vasopressors. Intravenous glucagon may be useful.
Heart Block (second or third degree): Monitor and treat with isoproterenol infusion. Under some circumstances, transthoracic or transvenous pacemaker placement may be necessary.
Congestive Heart Failure: Initiate therapy with digitalis glycoside and diuretics. In certain cases, consider the use of inotropic and vasodilating agents.
Bronchospasm: Administer bronchodilator therapy such as a short acting inhaled β2-agonist and/or aminophylline.
Hypoglycemia: Administer IV glucose. Repeated doses of IV glucose or possibly glucagon may be required.
Supportive measures should continue until clinical stability is achieved. The half-life of low doses of nebivolol is 12 to 19 hours.
Call the National Poison Control Center (800-222-1222) for the most current information on β-blocker overdose treatment.
11 DESCRIPTION
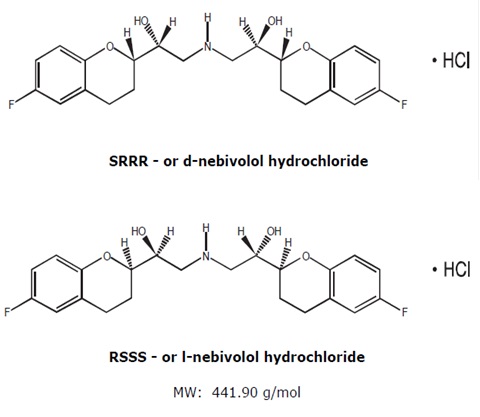
The chemical name for the active ingredient in nebivolol tablets is (1RS,1’RS)-1,1’-[(2RS,2’SR)-bis(6-fluoro-3,4-dihydro-2H-1-benzopyran-2-yl)]-2,2’-iminodiethanol hydrochloride. Nebivolol is a racemate composed of d-Nebivolol and l-Nebivolol with the stereochemical designations of [SRRR]-nebivolol and [RSSS]-nebivolol, respectively. Nebivolol’s molecular formula is (C22H25F2NO4•HCl) with the following structural formula:
Nebivolol hydrochloride is a white to off-white crystalline powder that is sparingly soluble in dimethyl formamide, slightly soluble in methanol, very slightly soluble in water and practically insoluble in 0.1 M hydrochloric acid.
Nebivolol as tablets for oral administration contains nebivolol hydrochloride equivalent to 2.5 mg, 5 mg, 10 mg, and 20 mg of nebivolol base. In addition, nebivolol tablets contain the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80 and pregelatinized starch (maize). In addition 2.5 mg and 20 mg tablets contain FD&C Blue No.2 Lake, 5 mg tablets contain FD&C Yellow No.6 Lake and 10 mg tablets contain phloxine B (D&C RED No. 27 Lake).
12 CLINICAL PHARMACOLOGY
Nebivolol is a β-adrenergic receptor blocking agent. In extensive metabolizers (most of the population) and at doses less than or equal to 10 mg, nebivolol is preferentially β1 selective. In poor metabolizers and at higher doses, nebivolol inhibits both β1 - and β2 - adrenergic receptors. Nebivolol lacks intrinsic sympathomimetic and membrane stabilizing activity at therapeutically relevant concentrations. At clinically relevant doses, nebivolol does not demonstrate α1-adrenergic receptor blockade activity. Various metabolites, including glucuronides, contribute to β-blocking activity.
12.1 Mechanism of Action
The mechanism of action of the antihypertensive response of nebivolol has not been definitively established. Possible factors that may be involved include: (1) decreased heart rate, (2) decreased myocardial contractility, (3) diminution of tonic sympathetic outflow to the periphery from cerebral vasomotor centers, (4) suppression of renin activity and (5) vasodilation and decreased peripheral vascular resistance.
12.3 Pharmacokinetics
Nebivolol is metabolized by a number of routes, including glucuronidation and hydroxylation by CYP2D6. The active isomer (d-nebivolol) has an effective half-life of about 12 hours in CYP2D6 extensive metabolizers (most people), and 19 hours in poor metabolizers and exposure to d-nebivolol is substantially increased in poor metabolizers. This has less importance than usual, however, because the metabolites, including the hydroxyl metabolite and glucuronides (the predominant circulating metabolites), contribute to β-blocking activity.
Plasma levels of d–nebivolol increase in proportion to dose in EMs and PMs for doses up to 20mg. Exposure to l-nebivolol is higher than to d-nebivolol but l-nebivolol contributes little to the drug’s activity as d-nebivolol’s beta receptor affinity is > 1000-fold higher than l-nebivolol. For the same dose, PMs attain a 5-fold higher Cmax and 10-fold higher AUC of d-nebivolol than do EMs. d-Nebivolol accumulates about 1.5-fold with repeated once-daily dosing in EMs.
Absorption
Absorption of nebivolol is similar to an oral solution. The absolute bioavailability has not been determined.
Mean peak plasma nebivolol concentrations occur approximately 1.5 to 4 hours post-dosing in EMs and PMs.
Food does not alter the pharmacokinetics of nebivolol. Under fed conditions, nebivolol glucuronides are slightly reduced. Nebivolol may be administered without regard to meals.
Distribution
The in vitro human plasma protein binding of nebivolol is approximately 98%, mostly to albumin, and is independent of nebivolol concentrations.
Metabolism
Nebivolol is predominantly metabolized via direct glucuronidation of parent and to a lesser extent via N-dealkylation and oxidation via cytochrome P450 2D6. Its stereospecific metabolites contribute to the pharmacologic activity [see Drug Interactions (7)].
Elimination
After a single oral administration of 14C-nebivolol, 38% of the dose was recovered in urine and 44% in feces for EMs and 67% in urine and 13% in feces for PMs. Essentially all nebivolol was excreted as multiple oxidative metabolites or their corresponding glucuronide conjugates.
12.4 Pharmacokinetics in Special Populations
Hepatic Disease
d-Nebivolol peak plasma concentration increased 3-fold, exposure (AUC) increased 10-fold, and the apparent clearance decreased by 86% in patients with moderate hepatic impairment (Child-Pugh Class B). No formal studies have been performed in patients with severe hepatic impairment and nebivolol should be contraindicated for these patients [see Dosage and Administration (2.1)].
Renal Disease
The apparent clearance of nebivolol was unchanged following a single 5 mg dose of nebivolol in patients with mild renal impairment (ClCr 50 to 80 mL/min, n=7), and it was reduced negligibly in patients with moderate (ClCr 30 to 50 mL/min, n=9), but clearance was reduced by 53% in patients with severe renal impairment (ClCr <30 mL/min, n=5). No studies have been conducted in patients on dialysis [see Dosage and Administration (2.1)].
12.5 Drug-Drug Interactions
Drugs that inhibit CYP2D6 can be expected to increase plasma levels of nebivolol. When nebivolol is co-administered with an inhibitor or an inducer of this enzyme, monitor patients closely and adjust the nebivolol dose according to blood pressure response. In vitro studies have demonstrated that at therapeutically relevant concentrations, d- and l-nebivolol do not inhibit any cytochrome P450 pathways.
Digoxin: Concomitant administration of nebivolol (10 mg once daily) and digoxin (0.25 mg once daily) for 10 days in 14 healthy adult individuals resulted in no significant changes in the pharmacokinetics of digoxin or nebivolol [see Drug Interactions (7)].
Warfarin: Administration of nebivolol (10 mg once daily for 10 days) led to no significant changes in the pharmacokinetics of nebivolol or R- or S-warfarin following a single 10 mg dose of warfarin. Similarly, nebivolol has no significant effects on the anticoagulant activity of warfarin, as assessed by Prothrombin time and INR profiles from 0 to 144 hours after a single 10 mg warfarin dose in 12 healthy adult volunteers.
Diuretics: No pharmacokinetic interactions were observed in healthy adults between nebivolol (10 mg daily for 10 days) and furosemide (40 mg single dose), hydrochlorothiazide (25 mg once daily for 10 days), or spironolactone (25 mg once daily for 10 days).
Ramipril: Concomitant administration of nebivolol (10 mg once daily) and ramipril (5 mg once daily) for 10 days in 15 healthy adult volunteers produced no pharmacokinetic interactions.
Losartan: Concomitant administration of nebivolol (10 mg single dose) and losartan (50 mg single dose) in 20 healthy adult volunteers did not result in pharmacokinetic interactions.
Fluoxetine: Fluoxetine, a CYP2D6 inhibitor, administered at 20 mg per day for 21 days prior to a single 10 mg dose of nebivolol to 10 healthy adults, led to an 8-fold increase in the AUC and 3-fold increase in Cmax for d-nebivolol [see Drug Interactions (7)].
Histamine-2 Receptor Antagonists: The pharmacokinetics of nebivolol (5 mg single dose) were not affected by the co-administration of ranitidine (150 mg twice daily). Cimetidine (400 mg twice daily) causes a 23% increase in the plasma levels of d-nebivolol.
Charcoal: The pharmacokinetics of nebivolol (10 mg single dose) were not affected by repeated co-administration (4, 8, 12, 16, 22, 28, 36, and 48 hours after nebivolol administration) of activated charcoal (Actidose®-Aqua).
Sildenafil: The co-administration of nebivolol and sildenafil decreased AUC and Cmax of sildenafil by 21 and 23% respectively. The effect on the Cmax and AUC for d-nebivolol was also small (< 20%). The effect on vital signs (e.g., pulse and blood pressure) was approximately the sum of the effects of sildenafil and nebivolol.
Other Concomitant Medications: Utilizing population pharmacokinetic analyses, derived from hypertensive patients, the following drugs were observed not to have an effect on the pharmacokinetics of nebivolol: acetaminophen, acetylsalicylic acid, atorvastatin, esomeprazole, ibuprofen, levothyroxine sodium, metformin, sildenafil, simvastatin, or tocopherol.
Protein Binding: No meaningful changes in the extent of in vitro binding of nebivolol to human plasma proteins were noted in the presence of high concentrations of diazepam, digoxin, diphenylhydantoin, enalapril, hydrochlorothiazide, imipramine, indomethacin, propranolol, sulfamethazine, tolbutamide, or warfarin. Additionally, nebivolol did not significantly alter the protein binding of the following drugs: diazepam, digoxin, diphenylhydantoin, hydrochlorothiazide, imipramine, or warfarin at their therapeutic concentrations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a two-year study of nebivolol in mice, a statistically significant increase in the incidence of testicular Leydig cell hyperplasia and adenomas was observed at 40 mg/kg/day (5 times the maximally recommended human dose of 40 mg on a mg/m2 basis). Similar findings were not reported in mice administered doses equal to approximately 0.3 or 1.2 times the maximum recommended human dose. No evidence of a tumorigenic effect was observed in a 24-month study in Wistar rats receiving doses of nebivolol 2.5, 10 and 40 mg/kg/day (equivalent to 0.6, 2.4, and 10 times the maximally recommended human dose). Co-administration of dihydrotestosterone reduced blood LH levels and prevented the Leydig cell hyperplasia, consistent with an indirect LH-mediated effect of nebivolol in mice and not thought to be clinically relevant in man.
A randomized, double-blind, placebo- and active-controlled, parallel-group study in healthy male volunteers was conducted to determine the effects of nebivolol on adrenal function, luteinizing hormone, and testosterone levels. This study demonstrated that 6 weeks of daily dosing with 10 mg of nebivolol had no significant effect on ACTH-stimulated mean serum cortisol AUC0-120 min, serum LH, or serum total testosterone.
Effects on spermatogenesis were seen in male rats and mice at ≥ 40 mg/kg/day (10 and 5 times the MRHD, respectively). For rats the effects on spermatogenesis were not reversed and may have worsened during a four week recovery period. The effects of nebivolol on sperm in mice, however, were partially reversible.
Mutagenesis: Nebivolol was not genotoxic when tested in a battery of assays (Ames, in vitro mouse lymphoma TK+/-, in vitro human peripheral lymphocyte chromosome aberration, in vivo Drosophila melanogaster sex-linked recessive lethal, and in vivo mouse bone marrow micronucleus tests).
14 CLINICAL STUDIES
14.1 Hypertension
The antihypertensive effectiveness of nebivolol as monotherapy has been demonstrated in three randomized, double-blind, multi-center, placebo-controlled trials at doses ranging from 1.25 to 40 mg for 12 weeks (Studies 1, 2, and 3). A fourth placebo-controlled trial demonstrated additional antihypertensive effects of nebivolol at doses ranging from 5 to 20 mg when administered concomitantly with up to two other antihypertensive agents (ACE inhibitors, angiotensin II receptor antagonists, and thiazide diuretics) in patients with inadequate blood pressure control.
The three monotherapy trials included a total of 2016 patients (1811 nebivolol, 205 placebo) with mild to moderate hypertension who had baseline diastolic blood pressures (DBP) of 95 to 109 mmHg. Patients received either nebivolol or placebo once daily for twelve weeks. Two of these monotherapy trials (Studies 1 and 2) studied 1716 patients in the general hypertensive population with a mean age of 54 years, 55% males, 26% non-Caucasians, 7% diabetics and 6% genotyped as PMs. The third monotherapy trial (Study 3) studied 300 Black patients with a mean age of 51 years, 45% males, 14% diabetics, and 3% as PMs.
Placebo-subtracted blood pressure reductions by dose for each study are presented in Table 2. Most studies showed increasing response to doses above 5 mg.
Table 2. Placebo-Subtracted Least-Square Mean Reductions in Trough Sitting Systolic/Diastolic Blood Pressure (SiSBP/SiDBP mmHg) by Dose in Studies with Once Daily Nebivolol
| | Nebivolol dose (mg)
|
|||||
| | 1.25 | 2.5 | 5 | 10 | 20 | 30 to 40 |
| Study 1 |
-6.6*/-5.1* |
-8.5*/-5.6* |
-8.1*/-5.5* |
-9.2*/-6.3* |
-8.7*/-6.9* |
-11.7*/-8.3* |
| Study 2 |
|
| -3.8/-3.2* | -3.1/-3.9* | -6.3*/-4.5* |
|
| Study 3¶
|
| -1.5/-2.9 | -2.6/-4.9* | -6.0*/-6.1* | -7.2*/-6.1* | -6.8*/-5.5* |
| Study 4^
|
|
| -5.7*/-3.3* | -3.7*/-3.5* | -6.2*/-4.6* |
|
* p<0.05 based on pair-wise comparison vs. placebo
¶ Study enrolled only African Americans.
^ Study on top of one or two other antihypertensive medications.
Study 4 enrolled 669 patients with a mean age of 54 years, 55% males, 54% Caucasians, 29% Blacks, 15% Hispanics, 1% Asians, 14% diabetics, and 5% PMs. Nebivolol, 5 mg to 20 mg, administered once daily concomitantly with stable doses of up to two other antihypertensive agents (ACE inhibitors, angiotensin II receptor antagonists, and thiazide diuretics) resulted in significant additional antihypertensive effects over placebo compared to baseline blood pressure.
Effectiveness was similar in subgroups analyzed by age and sex. Effectiveness was established in Blacks, but as monotherapy the magnitude of effect was somewhat less than in Caucasians.
The blood pressure lowering effect of nebivolol was seen within two weeks of treatment and was maintained over the 24-hour dosing interval.
There are no trials of nebivolol demonstrating reductions in cardiovascular risk in patients with hypertension, but at least one pharmacologically similar drug has demonstrated such benefits.
16 HOW SUPPLIED/STORAGE AND HANDLING
Nebivolol is available as tablets for oral administration containing nebivolol hydrochloride equivalent to 2.5 mg, 5 mg, 10 mg, and 20 mg of nebivolol.
Nebivolol Tablets 2.5 mg are light blue colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘78’ on the other side. They are supplied as follows:
Bottles of 30 NDC 59651-137-30
Bottles of 90 NDC 59651-137-90
Nebivolol Tablets 5 mg are beige colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘79’ on the other side. They are supplied as follows:
Bottles of 30 NDC 59651-138-30
Bottles of 90 NDC 59651-138-90
Nebivolol Tablets 10 mg are pink colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘80’ on the other side. They are supplied as follows:
Bottles of 30 NDC 59651-139-30
Bottles of 90 NDC 59651-139-90
Nebivolol Tablets 20 mg are light blue colored, round shaped, biconvex uncoated tablets debossed with ‘L’ on one side and ‘85’ on the other side. They are supplied as follows:
Bottles of 30 NDC 59651-140-30
Bottles of 90 NDC 59651-140-90
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information).
- Patient Advice
Advise patients to take nebivolol tablets regularly and continuously, as directed. Nebivolol tablets can be taken with or without food. If a dose is missed, take the next scheduled dose only (without doubling it). Do not interrupt or discontinue nebivolol tablets without consulting the physician.
Patients should know how they react to this medicine before they operate automobiles, use machinery, or engage in other tasks requiring alertness.
Advise patients to consult a physician if any difficulty in breathing occurs, or if they develop signs or symptoms of worsening congestive heart failure such as weight gain or increasing shortness of breath, or excessive bradycardia.
Caution patients subject to spontaneous hypoglycemia, or diabetic patients receiving insulin or oral hypoglycemic agents, that β-blockers may mask some of the manifestations of hypoglycemia, particularly tachycardia.
Inform patients or caregivers that there is a risk of hypoglycemia when nebivolol tablets are given to patients who are fasting or who are vomiting. Instruct patients or caregivers how to monitor for signs of hypoglycemia [see Warnings and Precautions (5.5)].
PATIENT INFORMATION
Nebivolol Tablets
(ne-BIV-oh-lol)
Read the Patient Information that comes with nebivolol tablets before you start taking it and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. If you have any questions about nebivolol tablets, ask your doctor or pharmacist.
WHAT ARE NEBIVOLOL TABLETS?
Nebivolol tablets are a kind of prescription medicine called a “beta-blocker”. Nebivolol tablets treat:
- High blood pressure (hypertension)
Nebivolol tablets can lower blood pressure when used by itself and with other medicines.
Nebivolol tablets are not approved for children less than 18 years of age.
WHO SHOULD NOT TAKE NEBIVOLOL TABLETS?
Do not take nebivolol tablets if you:
- Have heart failure and are in the ICU or need medicines to keep up your blood circulation
- Have a slow heartbeat or your heart skips beats (irregular heartbeat)
- Have severe liver damage
- Are allergic to any ingredient in nebivolol tablets. The active ingredient is nebivolol. See the end of this leaflet for a list of ingredients.
WHAT SHOULD I TELL MY DOCTOR BEFORE TAKING NEBIVOLOL TABLETS?
Tell your doctor about all of your medical problems, including if you:
- Have asthma or other lung problems (such as bronchitis or emphysema)
- Have problems with blood flow in your feet and legs (peripheral vascular disease) nebivolol tablets can make symptoms of blood flow problems worse.
- Have diabetes and take medicine to control blood sugar
- Have thyroid problems
- Have liver or kidney problems
- Had allergic reactions to medications or have allergies
- Have a condition called pheochromocytoma
- Are pregnant or trying to become pregnant. It is not known if nebivolol tablets are safe for your unborn baby. Talk with your doctor about the best way to treat high blood pressure while you are pregnant.
- Are breastfeeding. It is not known if nebivolol passes into your breast milk. You should not breastfeed while using nebivolol tablets.
- Are scheduled for surgery and will be given anesthetic agents
Tell your doctor about all the medicines you take. Include prescription and non-prescription medicines, vitamins, and herbal products. Nebivolol tablets and certain other medicines can affect each other and cause serious side effects.
Keep a list of all the medicines you take. Show this list to your doctor and pharmacist before you start a new medicine.
HOW SHOULD I TAKE NEBIVOLOL TABLETS?
- Do not suddenly stop taking nebivolol tablets. You could have chest pain or a heart attack. If your doctor decides to stop nebivolol tablets, your doctor may slowly lower your dose over time before stopping it completely.
- Take nebivolol tablets every day exactly as your doctor tells you. Your doctor will tell you how much nebivolol to take and how often. Your doctor may start with a low dose and raise it over time.
- Do not stop taking nebivolol tablets or change your dose without talking with your doctor.
- Take nebivolol tablets with or without food.
- If you miss a dose, take your dose as soon as you remember, unless it is close to the time to take your next dose. Do not take 2 doses at the same time. Take your next dose at the usual time.
- If you take too much nebivolol, call your doctor or poison control center right away.
WHAT ARE POSSIBLE SIDE EFFECTS OF NEBIVOLOL TABLETS?
- Low blood pressure and feeling dizzy. If you feel dizzy, sit or lie down and tell your doctor right away.
- Tiredness
- Slow heartbeat
- Headache
- Leg swelling due to fluid retention (edema). Tell your doctor if you gain weight or have trouble breathing while taking nebivolol tablets.
- If you are diabetic or take medicine for high blood sugar or if you have a tendency to have low blood sugar, nebivolol tablets can mask/cover some of the signs and symptoms that would tell you that your blood sugar may be low, like heart palpitations or rapid heart beating. Ask your doctor for other signs that may alert you when having low blood sugar.
Tell your doctor if you have any side effects that bother you or don’t go away.
HOW SHOULD I STORE NEBIVOLOL TABLETS?
- Store nebivolol tablets between 20° to 25°C (68° to 77°F).
- Safely throw away nebivolol tablets that are out of date or no longer needed.
- Keep nebivolol tablets and all medicines out of the reach of children.
GENERAL INFORMATION ABOUT NEBIVOLOL TABLETS
Doctors sometimes prescribe medicines for conditions not included in the patient information leaflets.
- Only use nebivolol tablets for the medical problem it was prescribed for.
- Do not give nebivolol tablets to other people, even if they have the same symptoms. It may harm them.
This leaflet summarizes the most important information about nebivolol tablets. For more information:
- Talk with your doctor.
- Ask your doctor or pharmacist for information about nebivolol tablets that is written for healthcare professionals.
- call Aurobindo Pharma USA, Inc. at 1-866-850-2876.
WHAT ARE IN NEBIVOLOL TABLETS?
Active Ingredient: Nebivolol
Inactive Ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80 and pregelatinized starch (maize). In addition 2.5 mg and 20 mg tablets contain FD&C Blue No.2 Lake, 5 mg tablets contain FD&C Yellow No.6 Lake and 10 mg tablets contain phloxine B (D&C RED No. 27 Lake).
WHAT IS HIGH BLOOD PRESSURE (HYPERTENSION)?
Blood pressure is the force in your blood vessels when your heart beats and when your heart rests. You have high blood pressure when the force is too great.
High blood pressure makes the heart work harder to pump blood through the body and causes damage to the blood vessels. Nebivolol tablets can help your blood vessels relax so your blood pressure is lower. Medicines that lower your blood pressure lower your chance of having a stroke or heart attack.
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revised: 08/2023
The brands listed are trademarks of their respective owners and are not trademarks of the Aurobindo Pharma Limited.
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 2.5 mg (30 Tablets Bottle)
NDC 59651-137-30
Rx only
Nebivolol Tablets
2.5 mg
AUROBINDO 30 Tablets

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL – 5 mg (30 Tablets Bottle)
NDC 59651-138-30
Rx only
Nebivolol Tablets
5 mg
AUROBINDO 30 Tablets


