FULL PRESCRIBING INFORMATION
WARNING: QT PROLONGATION and SUDDEN DEATHS
- Tasigna prolongs the QT interval. Prior to Tasigna administration and periodically, monitor for hypokalemia or hypomagnesemia and correct deficiencies [see Warnings and Precautions (5.2)]. Obtain ECGs to monitor the QTc at baseline, seven days after initiation, and periodically thereafter, and following any dose adjustments [see Warnings and Precautions (5.2, 5.3, 5.7, 5.12)].
- Sudden deaths have been reported in patients receiving Tasigna [see Warnings and Precautions (5.3)]. Do not administer Tasigna to patients with hypokalemia, hypomagnesemia, or long QT syndrome [see Contraindications (4), Warnings and Precautions (5.2)].
- Avoid use of concomitant drugs known to prolong the QT interval and strong CYP3A4 inhibitors [see Drug Interactions (7.1, 7.2)].
- Avoid food 2 hours before and 1 hour after taking the dose [see Dosage and Administration (2.1)].
1 INDICATIONS AND USAGE
1.1 Adult and Pediatric Patients With Newly Diagnosed Ph+ CML-CP
Tasigna is indicated for the treatment of adult and pediatric patients greater than or equal to 1 year of age with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase.
1.2 Adult Patients With Resistant or Intolerant Ph+ CML-CP and CML-AP
Tasigna is indicated for the treatment of adult patients with chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) resistant or intolerant to prior therapy that included imatinib.
1.3 Pediatric Patients With Resistant or Intolerant Ph+ CML-CP and CML-AP
Tasigna is indicated for the treatment of pediatric patients greater than or equal to 1 year of age with chronic phase and accelerated phase Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) with resistance or intolerance to prior tyrosine-kinase inhibitor (TKI) therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Dose Tasigna twice daily at approximately 12-hour intervals on an empty stomach. No food should be consumed for at least 2 hours before the dose is taken and for at least 1 hour after the dose is taken. Advise patients to swallow the capsules whole with water [see Boxed Warning, Clinical Pharmacology (12.3)].
For patients who are unable to swallow capsules, the contents of each capsule may be dispersed in 1 teaspoon of applesauce (puréed apple). The mixture should be taken immediately (within 15 minutes) and should not be stored for future use [see Clinical Pharmacology (12.3)].
Tasigna may be given in combination with hematopoietic growth factors, such as erythropoietin or G-CSF if clinically indicated. Tasigna may be given with hydroxyurea or anagrelide if clinically indicated.
Dosage in Adult Patients with Newly Diagnosed Ph+ CML-CP
The recommended dosage of Tasigna is 300 mg orally twice daily.
Dosage in Adult Patients with Resistant or Intolerant Ph+ CML-CP and CML-AP
The recommended dosage of Tasigna is 400 mg orally twice daily.
Dosage in Pediatric Patients with Newly Diagnosed Ph+ CML-CP or Resistant or Intolerant Ph+ CML-CP and CML-AP
The recommended dosage of Tasigna for pediatric patients is 230 mg/m2 orally twice daily, rounded to the nearest 50 mg dose (to a maximum single dose of 400 mg) (see Table 1). If needed, attain the desired dose by combining different strengths of Tasigna capsules. Continue treatment as long as clinical benefit is observed or until unacceptable toxicity occurs.
| Body surface area | Single dose | Total daily dose |
| Up to 0.32 m2 | 50 mg | 100 mg |
| 0.33 – 0.54 m2 | 100 mg | 200 mg |
| 0.55 – 0.76 m2 | 150 mg | 300 mg |
| 0.77 – 0.97 m2 | 200 mg | 400 mg |
| 0.98 – 1.19 m2 | 250 mg | 500 mg |
| 1.20 – 1.41 m2 | 300 mg | 600 mg |
| 1.42 – 1.63 m2 | 350 mg | 700 mg |
| ≥ 1.64 m2 | 400 mg | 800 mg |
2.2 Discontinuation of Treatment After a Sustained Molecular Response (MR4.5) on Tasigna
Patient Selection
Eligibility for Discontinuation of Treatment
Ph+ CML-CP patients with typical BCR-ABL transcripts, who have been taking Tasigna for a minimum of 3 years and have achieved a sustained molecular response (MR4.5, corresponding to = BCR-ABL/ABL ≤ 0.0032% IS), may be eligible for treatment discontinuation [see Clinical Studies (14.3, 14.4)]. Information on FDA authorized tests for the detection and quantitation of BCR-ABL transcripts to determine eligibility for treatment discontinuation is available at http://www.fda.gov/CompanionDiagnostics.
Patients with typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2), who achieve the sustained MR4.5 criteria, are eligible for discontinuation of Tasigna. Patients must continue to be monitored for possible loss of molecular remission after treatment discontinuation. Use the same FDA-authorized test to consistently monitor molecular response levels while on and off treatment.
Consider discontinuation in patients with newly diagnosed Ph+ CML-CP who have:
- been treated with Tasigna for at least 3 years
- maintained a molecular response of at least MR4.0 (corresponding to = BCR-ABL/ABL ≤ 0.01% IS) for one year prior to discontinuation of therapy
- achieved an MR4.5 for the last assessment taken immediately prior to discontinuation of therapy
- been confirmed to express the typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2)
- no history of accelerated phase or blast crisis
- no history of prior attempts of treatment-free remission discontinuation that resulted in relapse.
Consider discontinuation in patients with Ph+ CML-CP that are resistant or intolerant to imatinib who have achieved a sustained molecular response (MR4.5) on Tasigna who have:
- been treated with Tasigna for a minimum of 3 years
- been treated with imatinib only prior to treatment with Tasigna
- achieved a molecular response of MR4.5 (corresponding to = BCR-ABL/ABL ≤ 0.0032% IS)
- sustained an MR4.5 for a minimum of one year immediately prior to discontinuation of therapy
- been confirmed to express the typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2)
- no history of accelerated phase or blast crisis
- no history of prior attempts of treatment-free remission discontinuation that resulted in relapse.
Monitor BCR-ABL transcript levels and complete blood count (CBC) with differential in patients who have discontinued Tasigna therapy monthly for one year, then every 6 weeks for the second year, and every 12 weeks thereafter [see Warnings and Precautions (5.16)].
Upon the loss of MR4.0 (corresponding to = BCR-ABL/ABL ≤ 0.01% IS) during the treatment-free phase, monitor BCR-ABL transcript levels every 2 weeks until BCR-ABL levels remain lower than major molecular response [(MMR), corresponding to MR3.0 or = BCR-ABL/ABL ≤ 0.1% IS] for 4 consecutive measurements. The patient can then proceed to the original monitoring schedule.
2.3 Reinitiation of Treatment in Patients Who Lose Molecular Response After Discontinuation of Therapy With Tasigna
- Newly diagnosed patients who lose MMR must reinitiate treatment within 4 weeks at the dose level prior to discontinuation of therapy [see Warnings and Precautions (5.16)]. Patients who reinitiate Tasigna therapy should have their BCR-ABL transcript levels monitored monthly until major molecular response is re-established and every 12 weeks thereafter.
- Patients resistant or intolerant to prior treatment that included imatinib with confirmed loss of MR4.0 (2 consecutive measures separated by at least 4 weeks showing loss of MR4.0) or loss of MMR must reinitiate treatment within 4 weeks at the dose level prior to discontinuation of therapy [see Warnings and Precautions (5.16)]. Patients who reinitiate Tasigna therapy should have their BCR-ABL transcript levels monitored monthly until previous major molecular response or MR4.0 is re-established and every 12 weeks thereafter.
2.4 Dosage Modification for QT Interval Prolongation
See Table 2 for dose adjustments for QT interval prolongation [see Warnings and Precautions (5.2), Clinical Pharmacology (12.2)].
| Abbreviation: ECG, electrocardiogram. | |
| Degree of QTc prolongation | Dosage adjustment |
| ECGs with a QTc greater than 480 msec | 1. Withhold Tasigna, and perform an analysis of serum potassium and magnesium, and if below lower limit of normal, correct with supplements to within normal limits. Concomitant medication usage must be reviewed. 2. Resume within 2 weeks at prior dose if QTcF returns to less than 450 msec and to within 20 msec of baseline. 3. If QTcF is between 450 msec and 480 msec after 2 weeks, reduce the dose to 400 mg once daily in adults and 230 mg/m2 once daily in pediatric patients. 4. Discontinue Tasigna if, following dose-reduction to 400 mg once daily in adults and 230 mg/m2 once daily in pediatric patients, QTcF returns to greater than 480 msec. 5. An ECG should be repeated approximately 7 days after any dose adjustment. |
2.5 Dosage Modifications for Myelosuppression
Withhold or reduce Tasigna dosage for hematological toxicities (neutropenia, thrombocytopenia) that are not related to underlying leukemia (Table 3) [see Warnings and Precautions (5.1)].
| Abbreviations: ANC, absolute neutrophil count; Ph+ CML, Philadelphia chromosome positive chronic myeloid leukemia. | ||
| Diagnosis | Degree of myelosuppression | Dosage adjustment |
Adult patients with:
| ANC less than 1.0 x 109/L and/or platelet counts less than 50 x 109/L | 1. Stop Tasigna, and monitor blood counts. 2. Resume within 2 weeks at prior dose if ANC greater than 1.0 x 109/L and platelets greater than 50 x 109/L. 3. If blood counts remain low for greater than 2 weeks, reduce the dose to 400 mg once daily. |
Pediatric patients with:
| ANC less than 1.0 x 109/L and/or platelet counts less than 50 x 109/L | 1. Stop Tasigna and monitor blood counts. 2. Resume within 2 weeks at prior dose if ANC greater than 1.5 x 109/L and/or platelets greater than 75 x 109/L. 3. If blood counts remain low for greater than 2 weeks, a dose reduction to 230 mg/m2 once daily may be required. 4. If event occurs after dose reduction, consider discontinuing treatment. |
2.6 Dosage Modifications for Selected Non-Hematologic Laboratory Abnormalities and Other Toxicities
See Table 4 for dosage adjustments for elevations of lipase, amylase, bilirubin, and/or hepatic transaminases [see Warnings and Precautions (5.5, 5.6), Adverse Reactions (6.1)].
| Degree of non-hematologic laboratory abnormality | Dosage adjustment |
| Elevated serum lipase or amylase greater than or equal to Grade 3 | Adult patients: 1. Withhold Tasigna, and monitor serum lipase or amylase. 2. Resume treatment at 400 mg once daily if serum lipase or amylase returns to less than or equal to Grade 1. |
| Pediatric patients: 1. Interrupt Tasigna until the event returns to less than or equal to Grade 1. 2. Resume treatment at 230 mg/m2 once daily if prior dose was 230 mg/m2 twice daily; discontinue treatment if prior dose was 230 mg/m2 once daily. |
|
| Elevated bilirubin greater than or equal to Grade 3 in adult patients and greater than or equal to Grade 2 in pediatric patients | Adult patients: 1. Withhold Tasigna, and monitor bilirubin. 2. Resume treatment at 400 mg once daily if bilirubin returns to less than or equal to Grade 1. |
| Pediatric patients: 1. Interrupt Tasigna until the event returns to less than or equal to Grade 1. 2. Resume treatment at 230 mg/m2 once daily if prior dose was 230 mg/m2 twice daily; discontinue treatment if prior dose was 230 mg/m2 once daily, and recovery to less than or equal to Grade 1 takes longer than 28 days. |
|
| Elevated hepatic transaminases greater than or equal to Grade 3 | Adult patients: 1. Withhold Tasigna, and monitor hepatic transaminases. 2. Resume treatment at 400 mg once daily if hepatic transaminases returns to less than or equal to Grade 1. |
| Pediatric patients: 1. Interrupt Tasigna until the event returns to less than or equal to Grade 1. 2. Resume treatment at 230 mg/m2 once daily if prior dose was 230 mg/m2 twice daily; discontinue treatment if prior dose was 230 mg/m2 once daily, and recovery to less than or equal to Grade 1 takes longer than 28 days. |
If clinically significant moderate or severe non-hematologic toxicity develops (including medically severe fluid retention), see Table 5 for dosage adjustments [see Adverse Reactions (6.1)].
| Abbreviations: CML-AP, chronic myeloid leukemia-accelerated phase; CML-CP, chronic myeloid leukemia-chronic phase; Ph+, Philadelphia chromosome positive. | |
| Degree of “other Non-hematologic toxicity” | Dosage adjustment |
| Other clinically moderate or severe non-hematologic toxicity | Adult patients: 1. Withhold Tasigna until toxicity has resolved. 2. Resume treatment at 400 mg once daily if previous dose was 300 mg twice daily in adult patients newly diagnosed with CML-CP or 400 mg twice daily in adult patients with resistant or intolerant CML-CP and CML-AP. 3. Discontinue treatment if the prior dose was 400 mg once daily in adult patients. 4. If clinically appropriate, consider re-escalation of the dose to 300 mg (newly diagnosed Ph+ CML-CP) or 400 mg (resistant or intolerant Ph+ CML-CP and CML-AP) twice daily. |
| Pediatric patients: 1. Interrupt Tasigna until toxicity has resolved. 2. Resume treatment at 230 mg/m2 once daily if previous dose was 230 mg/m2 twice daily; discontinue treatment if prior dose was 230 mg/m2 once daily. 3. If clinically appropriate, consider re-escalation of the dose to 230 mg/m2 twice daily. |
|
2.7 Dosage Modification for Hepatic Impairment
If possible, consider alternative therapies. If Tasigna must be administered to patients with hepatic impairment, consider the following dose reduction [see Use in Specific Populations (8.7)]:
| Diagnosis | Degree of hepatic impairment | Dosage adjustment |
| Newly diagnosed Ph+ CML in chronic phase | Mild (Child-Pugh A), Moderate (Child-Pugh B), or Severe (Child-Pugh C) | Reduce dosage to 200 mg twice daily. Increase dosage to 300 mg twice daily based on tolerability. |
|
Resistant or intolerant Ph+ CML in chronic phase or accelerated phase | Mild or Moderate | Reduce dosage to 300 mg twice daily. Increase dosage to 400 mg twice daily based on tolerability. |
| Severe | Reduce dosage to 200 mg twice daily. Increase dosage to 300 mg twice daily and then to 400 mg twice daily based on tolerability. |
2.8 Dosage Modification With Concomitant Strong CYP3A4 Inhibitors
Avoid the concomitant use of strong CYP3A4 inhibitors. Should treatment with any of these agents be required, interrupt therapy with Tasigna. If patients must be coadministered a strong CYP3A4 inhibitor, reduce dosage to 300 mg once daily in patients with resistant or intolerant Ph+ CML or to 200 mg once daily in patients with newly diagnosed Ph+ CML-CP. However, there are no clinical data with this dose adjustment in patients receiving strong CYP3A4 inhibitors. If the strong inhibitor is discontinued, allow a washout period before adjusting Tasigna dose upward to the indicated dose. For patients who cannot avoid use of strong CYP3A4 inhibitors, monitor closely for prolongation of the QT interval [see Boxed Warning, Warnings and Precautions (5.2), Drug Interactions (7.1, 7.2), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Capsules:
- 50 mg red opaque cap and light-yellow opaque body hard gelatin capsules with black radial imprint “NVR/ABL.”
- 150 mg red opaque hard gelatin capsules with black axial imprint “NVR/BCR.”
- 200 mg light-yellow opaque hard gelatin capsules with a red axial imprint “NVR/TKI.”
4 CONTRAINDICATIONS
Tasigna is contraindicated in patients with hypokalemia, hypomagnesemia, or long QT syndrome [see Boxed Warning].
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Treatment with Tasigna can cause Grade 3/4 thrombocytopenia, neutropenia, and anemia. Perform CBCs every 2 weeks for the first 2 months and then monthly thereafter, or as clinically indicated. Myelosuppression was generally reversible and usually managed by withholding Tasigna temporarily or dose reduction [see Dosage and Administration (2.5)].
5.2 QT Prolongation
Tasigna has been shown to prolong cardiac ventricular repolarization as measured by the QT interval on the surface electrocardiogram (ECG) in a concentration-dependent manner [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)]. Prolongation of the QT interval can result in a type of ventricular tachycardia called torsade de pointes, which may result in syncope, seizure, and/or death. Electrocardiograms should be performed at baseline, 7 days after initiation of Tasigna, and periodically as clinically indicated and following dose adjustments [see Dosage and Administration (2.4), Warnings and Precautions (5.12)].
Tasigna should not be used in patients who have hypokalemia, hypomagnesemia, or long QT syndrome. Before initiating Tasigna and periodically, test electrolyte, calcium, and magnesium blood levels. Hypokalemia or hypomagnesemia must be corrected prior to initiating Tasigna and these electrolytes should be monitored periodically during therapy [see Warnings and Precautions (5.12)].
Significant prolongation of the QT interval may occur when Tasigna is inappropriately taken with food and/or strong CYP3A4 inhibitors and/or medicinal products with a known potential to prolong QT. Therefore, coadministration with food must be avoided and concomitant use with strong CYP3A4 inhibitors and/or medicinal products with a known potential to prolong QT should be avoided [see Dosage and Administration (2.1), Drug Interactions (7.1, 7.2)]. The presence of hypokalemia and hypomagnesemia may further prolong the QT interval [see Warnings and Precautions (5.7, 5.12)].
5.3 Sudden Deaths
Sudden deaths have been reported in 0.3% of patients with CML treated with Tasigna in clinical studies of 5661 patients. The relative early occurrence of some of these deaths relative to the initiation of Tasigna suggests the possibility that ventricular repolarization abnormalities may have contributed to their occurrence.
5.4 Cardiac and Arterial Vascular Occlusive Events
Cardiovascular events, including arterial vascular occlusive events, were reported in a randomized, clinical trial in newly diagnosed CML patients and observed in the postmarketing reports of patients receiving Tasigna therapy [see Adverse Reactions (6.1)]. With a median time on therapy of 60 months in the clinical trial, cardiovascular events, including arterial vascular occlusive events, occurred in 9% and 15% of patients in the Tasigna 300 and 400 mg twice daily arms, respectively, and in 3.2% in the imatinib arm. These included cases of cardiovascular events, including ischemic heart disease-related cardiac events (5% and 9% in the Tasigna 300 mg and 400 mg twice daily arms, respectively, and 2.5% in the imatinib arm), peripheral arterial occlusive disease (3.6% and 2.9% in the Tasigna 300 mg and 400 mg twice daily arms, respectively, and 0% in the imatinib arm), and ischemic cerebrovascular events (1.4% and 3.2% in the Tasigna 300 mg and 400 mg twice daily arms, respectively, and 0.7% in the imatinib arm). If acute signs or symptoms of cardiovascular events occur, advise patients to seek immediate medical attention. The cardiovascular status of patients should be evaluated and cardiovascular risk factors should be monitored and actively managed during Tasigna therapy according to standard guidelines [see Dosage and Administration (2.4)].
5.5 Pancreatitis and Elevated Serum Lipase
Tasigna can cause increases in serum lipase [see Adverse Reactions (6.1)]. Patients with a previous history of pancreatitis may be at greater risk of elevated serum lipase. If lipase elevations are accompanied by abdominal symptoms, interrupt dosing and consider appropriate diagnostics to exclude pancreatitis [see Dosage and Administration (2.6)]. Test serum lipase levels monthly or as clinically indicated.
5.6 Hepatotoxicity
Tasigna may result in hepatotoxicity as measured by elevations in bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase. Grade 3-4 elevations of bilirubin, AST, and ALT were reported at a higher frequency in pediatric than in adult patients. Monitor hepatic function tests monthly or as clinically indicated [see Warnings and Precautions (5.12)] and following dose adjustments. [see Dosage and Administration (2.6)].
5.7 Electrolyte Abnormalities
The use of Tasigna can cause hypophosphatemia, hypokalemia, hyperkalemia, hypocalcemia, and hyponatremia. Correct electrolyte abnormalities prior to initiating Tasigna and during therapy. Monitor these electrolytes periodically during therapy [see Warnings and Precautions (5.12)].
5.8 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS) cases have been reported in Tasigna treated patients with resistant or intolerant CML. Malignant disease progression, high white blood cell (WBC) counts and/or dehydration were present in the majority of these cases. Due to potential for TLS, maintain adequate hydration and correct uric acid levels prior to initiating therapy with Tasigna.
5.9 Hemorrhage
Serious hemorrhagic events, including fatal events, have occurred in patients with CML treated with Tasigna. In a randomized trial in patients with newly diagnosed Ph+ CML in chronic phase comparing Tasigna and imatinib, Grade 3 or 4 hemorrhage occurred in 1.1% of patients in the Tasigna 300 mg twice daily arm, in 1.8% of patients in the Tasigna 400 mg twice daily arm, and 0.4% of patients in the imatinib arm. GI hemorrhage occurred in 2.9% and 5% of patients in the Tasigna 300 mg twice daily and 400 mg twice daily arms and in 1.4% of patients in the imatinib arm, respectively. Grade 3 or 4 events occurred in 0.7% and 1.4% of patients in the Tasigna 300 mg twice daily and 400 mg twice daily arms, respectively, and in no patients in the imatinib arm. Monitor for signs and symptoms of bleeding and medically manage as needed.
5.10 Total Gastrectomy
Since the exposure of Tasigna is reduced in patients with total gastrectomy, perform more frequent monitoring of these patients. Consider dose increase or alternative therapy in patients with total gastrectomy [see Clinical Pharmacology (12.3)].
5.11 Lactose
Since the capsules contain lactose, Tasigna is not recommended for patients with rare hereditary problems of galactose intolerance, severe lactase deficiency with a severe degree of intolerance to lactose-containing products, or of glucose-galactose malabsorption.
5.12 Monitoring Laboratory Tests
Complete blood counts should be performed every 2 weeks for the first 2 months and then monthly thereafter. Perform chemistry panels, including electrolytes, calcium, magnesium, liver enzymes, lipid profile, and glucose prior to therapy and periodically. Electrocardiograms should be obtained at baseline, 7 days after initiation and periodically thereafter, as well as following dose adjustments [see Warnings and Precautions (5.2)]. Monitor lipid profiles and glucose periodically during the first year of Tasigna therapy and at least yearly during chronic therapy. Should treatment with any HMG-CoA reductase inhibitor (a lipid lowering agent) be needed to treat lipid elevations, evaluate the potential for a drug-drug interaction before initiating therapy as certain HMG-CoA reductase inhibitors are metabolized by the CYP3A4 pathway [see Drug Interactions (7.1)]. Assess glucose levels before initiating treatment with Tasigna and monitor during treatment as clinically indicated. If test results warrant therapy, physicians should follow their local standards of practice and treatment guidelines.
5.13 Fluid Retention
In the randomized trial in patients with newly diagnosed Ph+ CML in chronic phase, severe (Grade 3 or 4) fluid retention occurred in 3.9% and 2.9% of patients receiving Tasigna 300 mg twice daily and 400 mg twice daily, respectively, and in 2.5% of patients receiving imatinib. Effusions (including pleural effusion, pericardial effusion, ascites) or pulmonary edema, were observed in 2.2% and 1.1% of patients receiving Tasigna 300 mg twice daily and 400 mg twice daily, respectively, and in 2.1% of patients receiving imatinib. Effusions were severe (Grade 3 or 4) in 0.7% and 0.4% of patients receiving Tasigna 300 mg twice daily and 400 mg twice daily, respectively, and in no patients receiving imatinib. Similar events were also observed in postmarketing reports. Monitor patients for signs of severe fluid retention (e.g., unexpected rapid weight gain or swelling) and for symptoms of respiratory or cardiac compromise (e.g., shortness of breath) during Tasigna treatment; evaluate etiology and treat patients accordingly.
5.14 Effects on Growth and Development in Pediatric Patients
Growth retardation has been reported in pediatric patients with Ph+ CML in chronic phase treated with Tasigna. In a pediatric trial with 58 patients with Ph+ CML in chronic phase with a median exposure of 56.7 months, growth deceleration (crossing at least two main height percentile lines from baseline) was observed in eight patients: five (9%) crossed two main percentile lines from baseline and three (5%) crossed three main percentile lines from baseline (percentile lines: 5th, 10th, 25th, 50th, 75th, 90th, and 95th). Growth deceleration was more pronounced in children who were less than age 12 at baseline. Adverse reactions associated with growth retardation were reported in 3 patients (5%). Monitor growth and development in pediatric patients receiving Tasigna treatment.
5.15 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, Tasigna can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of nilotinib to pregnant rats and rabbits during organogenesis caused adverse developmental outcomes, including embryo-fetal lethality/fetal effects (small renal papilla, fetal edema, and skeletal variations) in rats and increased resorptions of fetuses and fetal skeletal variations in rabbits at maternal area under the curve (AUCs) approximately 2 and 0.5 times, respectively, the AUC in patients receiving the recommended dose. Advise pregnant women of the potential risk to a fetus.
Advise females of reproductive potential to use effective contraception during treatment and for 14 days after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
5.16 Monitoring of BCR-ABL Transcript Levels
Monitoring of BCR-ABL Transcript Levels in Patients Who Discontinued Tasigna
Monitor BCR-ABL transcript levels in patients eligible for treatment discontinuation using an FDA authorized test validated to measure molecular response levels with a sensitivity of at least MR4.5 (BCR-ABL/ABL ≤ 0.0032% IS). In patients who discontinue Tasigna therapy, assess BCR-ABL transcript levels monthly for one year, then every 6 weeks for the second year, and every 12 weeks thereafter during treatment discontinuation [see Clinical Studies (14.3,14.4), Dosage and Administration (2.2)].
Newly diagnosed patients must reinitiate Tasigna therapy within 4 weeks of a loss of major molecular response [(MMR), corresponding to MR3.0 or = BCR-ABL/ABL ≤ 0.1% IS].
Patients resistant or intolerant to prior treatment which included imatinib must reinitiate Tasigna therapy within 4 weeks of a loss of MMR or confirmed loss of MR4.0 (two consecutive measures separated by at least 4 weeks showing loss of MR4.0, corresponding to = BCR-ABL/ABL ≤ 0.01% IS).
For patients who fail to achieve MMR after three months of treatment reinitiation, BCR-ABL kinase domain mutation testing should be performed.
Monitoring of BCR-ABL Transcript Levels in Patients Who Have Reinitiated Therapy After Loss of Molecular Response
Monitor CBC and BCR-ABL transcripts in patients who reinitiate treatment with Tasigna due to loss of molecular response quantitation every 4 weeks until a major molecular response is re-established, then every 12 weeks.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions can occur with Tasigna and are discussed in greater detail in other sections of labeling:
- Myelosuppression [see Warnings and Precautions (5.1)]
- QT Prolongation [see Boxed Warning, Warnings and Precautions (5.2)]
- Sudden Deaths [see Boxed Warning, Warnings and Precautions (5.3)]
- Cardiac and Arterial Vascular Occlusive Events [see Warnings and Precautions (5.4)]
- Pancreatitis and Elevated Serum Lipase [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
- Electrolyte Abnormalities [see Boxed Warning, Warnings and Precautions (5.7)]
- Hemorrhage [see Warnings and Precautions (5.9)]
- Fluid Retention [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In Adult Patients With Newly Diagnosed Ph+ CML-CP
The data below reflect exposure to Tasigna from a randomized trial in patients with newly diagnosed Ph+ CML in chronic phase treated at the recommended dose of 300 mg twice daily (n = 279). The median time on treatment in the Tasigna 300 mg twice daily group was 61 months (range, 0.1 to 71 months). The median actual dose intensity was 593 mg/day in the Tasigna 300 mg twice daily group.
The most common (greater than 10%) non-hematologic adverse drug reactions were rash, pruritus, headache, nausea, fatigue, alopecia, myalgia, and upper abdominal pain. Constipation, diarrhea, dry skin, muscle spasms, arthralgia, abdominal pain, peripheral edema, vomiting, and asthenia were observed less commonly (less than or equal to 10% and greater than 5%) and have been of mild-to-moderate severity, manageable and generally did not require dose reduction.
Increase in QTcF greater than 60 msec from baseline was observed in 1 patient (0.4%) in the 300 mg twice daily treatment group. No patient had an absolute QTcF of greater than 500 msec while on study drug.
The most common hematologic adverse drug reactions (all Grades) were myelosuppression, including: thrombocytopenia (18%), neutropenia (15%), and anemia (8%). See Table 9 for Grade 3/4 laboratory abnormalities.
Discontinuation due to adverse reactions, regardless of relationship to study drug, was observed in 10% of patients.
In Adult Patients With Resistant or Intolerant Ph+ CML-CP and CML-AP
In the single-arm, open-label multicenter clinical trial, a total of 458 patients with Ph+ CML-CP and CML-AP resistant to or intolerant to at least one prior therapy, including imatinib were treated (CML-CP = 321; CML-AP = 137) at the recommended dose of 400 mg twice daily.
The median duration of exposure in days for CML-CP and CML-AP patients is 561 (range, 1 to 1096) and 264 (range, 2 to 1160), respectively. The median dose intensity for patients with CML-CP and CML-AP is 789 mg/day (range, 151 to 1110) and 780 mg/day (range, 150 to 1149), respectively, and corresponded to the planned 400 mg twice daily dosing.
The median cumulative duration in days of dose interruptions for the CML-CP patients was 20 (range, 1 to 345), and the median duration in days of dose interruptions for the CML-AP patients was 23 (range, 1 to 234).
In patients with CML-CP, the most commonly reported non-hematologic adverse drug reactions (greater than or equal to 10%) were rash, pruritus, nausea, fatigue, headache, constipation, diarrhea, vomiting, and myalgia. The common serious drug-related adverse reactions (greater than or equal to 1% and less than 10%) were thrombocytopenia, neutropenia, and anemia.
In patients with CML-AP, the most commonly reported non-hematologic adverse drug reactions (greater than or equal to 10%) were rash, pruritus and fatigue. The common serious adverse drug reactions (greater than or equal to 1% and less than 10%) were thrombocytopenia, neutropenia, febrile neutropenia, pneumonia, leukopenia, intracranial hemorrhage, elevated lipase, and pyrexia.
Sudden deaths and QT prolongation were reported. The maximum mean QTcF change from baseline at steady-state was 10 msec. Increase in QTcF greater than 60 msec from baseline was observed in 4.1% of the patients and QTcF of greater than 500 msec was observed in 4 patients (less than 1%) [see Boxed Warning, Warnings and Precautions (5.2, 5.3), Clinical Pharmacology (12.2)].
Discontinuation due to adverse drug reactions was observed in 16% of CML-CP and 10% of CML-AP patients.
Most Frequently Reported Adverse Reactions
Tables 7 and 8 show the percentage of adult patients experiencing non-hematologic adverse reactions (excluding laboratory abnormalities) regardless of relationship to study drug. Adverse reactions reported in greater than 10% of adult patients who received at least 1 dose of Tasigna are listed.
| Abbreviations: CML-CP, chronic myeloid leukemia-chronic phase; Ph+, Philadelphia chromosome positive. aExcluding laboratory abnormalities. bNCI Common Terminology Criteria (CTC) for Adverse Events, version 3.0. |
|||||
| Patients With Newly Diagnosed Ph+ CML-CP | |||||
| Tasigna
300 mg twice daily | imatinib
400 mg once daily | Tasigna
300 mg twice daily | imatinib
400 mg once daily |
||
| N = 279 | N = 280 | N = 279 | N = 280 | ||
| Body System and Adverse Reaction | All Grades (%) | CTC Gradesb 3/4 (%) | |||
| Skin and subcutaneous tissue disorders | Rash | 38 | 19 | < 1 | 2 |
| Pruritus | 21 | 7 | < 1 | 0 | |
| Alopecia | 13 | 7 | 0 | 0 | |
| Dry skin | 12 | 6 | 0 | 0 | |
| Gastrointestinal disorders | Nausea | 22 | 41 | 2 | 2 |
| Constipation | 20 | 8 | < 1 | 0 | |
| Diarrhea | 19 | 46 | 1 | 4 | |
| Vomiting | 15 | 27 | < 1 | < 1 | |
| Abdominal pain upper | 18 | 14 | 1 | < 1 | |
| Abdominal pain | 15 | 12 | 2 | 0 | |
| Dyspepsia | 10 | 12 | 0 | 0 | |
| Nervous system disorders | Headache | 32 | 23 | 3 | < 1 |
| Dizziness | 12 | 11 | < 1 | < 1 | |
| General disorders and administration-site conditions | Fatigue | 23 | 20 | 1 | 1 |
| Pyrexia | 14 | 13 | < 1 | 0 | |
| Asthenia | 14 | 12 | < 1 | 0 | |
| Peripheral edema | 9 | 20 | < 1 | 0 | |
| Face edema | < 1 | 14 | 0 | < 1 | |
| Musculoskeletal and connective tissue disorders | Myalgia | 19 | 19 | < 1 | < 1 |
| Arthralgia | 22 | 17 | < 1 | < 1 | |
| Muscle spasms | 12 | 34 | 0 | 1 | |
| Pain in extremity | 15 | 16 | < 1 | < 1 | |
| Back pain | 19 | 17 | 1 | 1 | |
| Respiratory, thoracic, and mediastinal disorders | Cough | 17 | 13 | 0 | 0 |
| Oropharyngeal pain | 12 | 6 | 0 | 0 | |
| Dyspnea | 11 | 6 | 2 | < 1 | |
| Infections and infestations | Nasopharyngitis | 27 | 21 | 0 | 0 |
| Upper respiratory tract infection | 17 | 14 | < 1 | 0 | |
| Influenza | 13 | 9 | 0 | 0 | |
| Gastroenteritis | 7 | 10 | 0 | < 1 | |
| Eye disorders | Eyelid edema | 1 | 19 | 0 | < 1 |
| Periorbital edema | < 1 | 15 | 0 | 0 | |
| Psychiatric disorders | Insomnia | 11 | 9 | 0 | 0 |
| Vascular disorder | Hypertension | 10 | 4 | 1 | < 1 |
| Abbreviations: CML-AP, chronic myeloid leukemia-accelerated phase; CML-CP, chronic myeloid leukemia-chronic phase; Ph+, Philadelphia chromosome positive. aExcluding laboratory abnormalities. bNCI Common Terminology Criteria for Adverse Events, version 3.0. cAlso includes preferred term anorexia. |
|||||
| Body System and Adverse Reaction | CML-CP | CML-AP | |||
| N = 321 | N = 137 | ||||
| All Grades (%) | CTC Gradesb 3/4 (%) | All Grades (%) | CTC Gradesb 3/4 (%) | ||
| Skin and subcutaneous tissue disorders | Rash | 36 | 2 | 29 | 0 |
| Pruritus | 32 | < 1 | 20 | 0 | |
| Night sweat | 12 | < 1 | 27 | 0 | |
| Alopecia | 11 | 0 | 12 | 0 | |
| Gastrointestinal disorders | Nausea | 37 | 1 | 22 | < 1 |
| Constipation | 26 | < 1 | 19 | 0 | |
| Diarrhea | 28 | 3 | 24 | 2 | |
| Vomiting | 29 | < 1 | 13 | 0 | |
| Abdominal pain | 15 | 2 | 16 | 3 | |
| Abdominal pain upper | 14 | < 1 | 12 | < 1 | |
| Dyspepsia | 10 | < 1 | 4 | 0 | |
| Nervous system disorders | Headache | 35 | 2 | 20 | 1 |
| General disorders and administration-site conditions | Fatigue | 32 | 3 | 23 | < 1 |
| Pyrexia | 22 | < 1 | 28 | 2 | |
| Asthenia | 16 | 0 | 14 | 1 | |
| Peripheral edema | 15 | < 1 | 12 | 0 | |
| Musculoskeletal and connective tissue disorders | Myalgia | 19 | 2 | 16 | < 1 |
| Arthralgia | 26 | 2 | 16 | 0 | |
| Muscle spasms | 13 | < 1 | 15 | 0 | |
| Bone pain | 14 | < 1 | 15 | 2 | |
| Pain in extremity | 20 | 2 | 18 | 1 | |
| Back pain | 17 | 2 | 15 | < 1 | |
| Musculoskeletal pain | 11 | < 1 | 12 | 1 | |
| Respiratory, thoracic, and mediastinal disorders | Cough | 27 | < 1 | 18 | 0 |
| Dyspnea | 15 | 2 | 9 | 2 | |
| Oropharyngeal pain | 11 | 0 | 7 | 0 | |
| Infections and infestations | Nasopharyngitis | 24 | < 1 | 15 | 0 |
| Upper respiratory tract infection | 12 | 0 | 10 | 0 | |
| Metabolism and nutrition disorders | Decreased appetitec | 15 | < 1 | 17 | < 1 |
| Psychiatric disorders | Insomnia | 12 | 1 | 7 | 0 |
| Vascular disorders | Hypertension | 10 | 2 | 11 | < 1 |
Laboratory Abnormalities
Table 9 shows the percentage of adult patients experiencing treatment-emergent Grade 3/4 laboratory abnormalities in patients who received at least one dose of Tasigna.
| Abbreviations: ALT alanine aminotransferase; AST, aspartate aminotransferase; CML-AP, chronic myeloid leukemia-accelerated phase; CML-CP, chronic myeloid leukemia-chronic phase; Ph+, Philadelphia chromosome positive. *NCI Common Terminology Criteria for Adverse Events, version 3.0. 1CML-CP: Thrombocytopenia: 12% were Grade 3, 18% were Grade 4. 2CML-CP: Neutropenia: 16% were Grade 3, 15% were Grade 4. 3CML-AP: Thrombocytopenia: 11% were Grade 3, 32% were Grade 4. 4CML-AP: Neutropenia: 16% were Grade 3, 26% were Grade 4. |
||||
| Patient population | ||||
| Newly diagnosed adult Ph+ CML-CP | Resistant or intolerant adult Ph+ | |||
| CML-CP | CML-AP | |||
| Tasigna 300 mg
twice daily N = 279 (%) | imatinib 400 mg
once daily N = 280 (%) | Tasigna 400 mg
twice daily N = 321 (%) | Tasigna 400 mg
twice daily N = 137 (%) |
|
| Hematologic parameters | ||||
| Thrombocytopenia | 10 | 9 | 301 | 423 |
| Neutropenia | 12 | 22 | 312 | 424 |
| Anemia | 4 | 6 | 11 | 27 |
| Biochemistry parameters | ||||
| Elevated lipase | 9 | 4 | 18 | 18 |
| Hyperglycemia | 7 | < 1 | 12 | 6 |
| Hypophosphatemia | 8 | 10 | 17 | 15 |
| Elevated bilirubin (total) | 4 | < 1 | 7 | 9 |
| Elevated SGPT (ALT) | 4 | 3 | 4 | 4 |
| Hyperkalemia | 2 | 1 | 6 | 4 |
| Hyponatremia | 1 | < 1 | 7 | 7 |
| Hypokalemia | < 1 | 2 | 2 | 9 |
| Elevated SGOT (AST) | 1 | 1 | 3 | 2 |
| Decreased albumin | 0 | < 1 | 4 | 3 |
| Hypocalcemia | < 1 | < 1 | 2 | 5 |
| Elevated alkaline phosphatase | 0 | < 1 | < 1 | 1 |
| Elevated creatinine | 0 | < 1 | < 1 | < 1 |
Elevated total cholesterol (all Grades) occurred in 28% (Tasigna 300 mg twice daily) and 4% (imatinib). Elevated triglycerides (all Grades) occurred in 12% and 8% of patients in the Tasigna and imatinib arms, respectively. Hyperglycemia (all Grades) occurred in 50% and 31% of patients in the Tasigna and imatinib arms, respectively.
Most common biochemistry laboratory abnormalities (all Grades) were alanine aminotransferase increased (72%), blood bilirubin increased (59%), aspartate aminotransferase increased (47%), lipase increased (28%), blood glucose increased (50%), blood cholesterol increased (28%), and blood triglyceride increased (12%).
Treatment Discontinuation in Patients With Ph+ CML-CP Who Have Achieved a Sustained Molecular Response (MR4.5)
In eligible patients who discontinued Tasigna therapy after attaining a sustained molecular response (MR4.5), musculoskeletal symptoms (e.g., myalgia, pain in extremity, arthralgia, bone pain, spinal pain, or musculoskeletal pain), were reported more frequently than before treatment discontinuation in the first year, as noted in Table 10. The rate of new musculoskeletal symptoms generally decreased in the second year after treatment discontinuation.
In the newly diagnosed population in whom musculoskeletal symptoms occurred at any time during the TFR phase, 23/53 (43%) had not resolved by the TFR end date or data cut-off date. In the population previously treated with imatinib in whom musculoskeletal events occurred at any time during the TFR phase, 32/57 (56%) had not resolved by the data cut-off date.
The rate of musculoskeletal symptoms decreased in patients who entered the Tasigna treatment reinitiation (NTRI) phase, at 11/88 (13%) in the newly diagnosed population and 14/56 (25%) in the population previously treated with imatinib. Other adverse reactions observed in the Tasigna re-treatment phase were similar to those observed during Tasigna use in patients with newly diagnosed Ph+ CML-CP and resistant or intolerant Ph+ CML-CP and CML-AP.
| Abbreviations: CML-CP, chronic myeloid leukemia-chronic phase; Ph+, Philadelphia chromosome positive ; TFR, treatment-free remission. | |||||||||||
| Entire TFR period in all TFR patients | By time interval, in subset of patients in TFR greater than 48 weeks | ||||||||||
| Ph+ CML-CP patients |
N | Median follow-up in TFR | Patients with musculoskeletal symptoms |
N | Year prior to Tasigna discontinuation | 1st year after Tasigna discontinuation | 2nd year after Tasigna discontinuation |
||||
| All Grades | Grade 3/4 | All Grades | Grade 3/4 | All Grades | Grade 3/4 | All Grades | Grade 3/4 | ||||
| Newly Diagnosed |
190 | 76 weeks |
28% |
1% |
100 |
17% |
0% |
34% |
2% |
9% |
0% |
| Previously treated with imatinib |
126 |
99 weeks |
45% |
2% |
73 |
14% |
0% |
48% |
3% |
15% |
1% |
Additional Data From Clinical Trials
The following adverse drug reactions were reported in adult patients in the Tasigna clinical studies at the recommended doses. These adverse drug reactions are ranked under a heading of frequency, the most frequent first using the following convention: common (greater than or equal to 1% and less than 10%), uncommon (greater than or equal to 0.1% and less than 1%), and unknown frequency (single events). For laboratory abnormalities, very common events (greater than or equal to 10%), which were not included in Tables 7 and 8, are also reported. These adverse reactions are included based on clinical relevance and ranked in order of decreasing seriousness within each category, obtained from 2 clinical studies:
1. Adult patients with newly diagnosed Ph+ CML-CP 60 month analysis and,
2. Adult patients with resistant or intolerant Ph+ CML-CP and CMP-AP 24 months’ analysis.
Infections and Infestations: Common: folliculitis. Uncommon: pneumonia, bronchitis, urinary tract infection, candidiasis (including oral candidiasis). Unknown frequency: hepatitis B reactivation, sepsis, subcutaneous abscess, anal abscess, furuncle, tinea pedis.
Neoplasms Benign, Malignant, and Unspecified: Common: skin papilloma. Unknown frequency: oral papilloma, paraproteinemia.
Blood and Lymphatic System Disorders: Common: leukopenia, eosinophilia, febrile neutropenia, pancytopenia, lymphopenia. Unknown frequency: thrombocythemia, leukocytosis.
Immune System Disorders: Unknown frequency: hypersensitivity.
Endocrine Disorders: Uncommon: hyperthyroidism, hypothyroidism. Unknown frequency: hyperparathyroidism secondary, thyroiditis.
Metabolism and Nutrition Disorders: Very Common: hypophosphatemia. Common: electrolyte imbalance (including hypomagnesemia, hyperkalemia, hypokalemia, hyponatremia, hypocalcemia, hypercalcemia, hyperphosphatemia), diabetes mellitus, hyperglycemia, hypercholesterolemia, hyperlipidemia, hypertriglyceridemia. Uncommon: gout, dehydration, increased appetite. Unknown frequency: hyperuricemia, hypoglycemia.
Psychiatric Disorders: Common: depression, anxiety. Unknown frequency: disorientation, confusional state, amnesia, dysphoria.
Nervous System Disorders: Common: peripheral neuropathy, hypoesthesia, paresthesia. Uncommon: intracranial hemorrhage, ischemic stroke, transient ischemic attack, cerebral infarction, migraine, loss of consciousness (including syncope), tremor, disturbance in attention, hyperesthesia, facial paralysis. Unknown frequency: basilar artery stenosis, brain edema, optic neuritis, lethargy, dysesthesia, restless legs syndrome.
Eye Disorders: Common: eye hemorrhage, eye pruritus, conjunctivitis, dry eye (including xerophthalmia). Uncommon: vision impairment, vision blurred, visual acuity reduced, photopsia, hyperemia (scleral, conjunctival, ocular), eye irritation, conjunctival hemorrhage. Unknown frequency: papilledema, diplopia, photophobia, eye swelling, blepharitis, eye pain, chorioretinopathy, conjunctivitis allergic, ocular surface disease.
Ear and Labyrinth Disorders: Common: vertigo. Unknown frequency: hearing impaired, ear pain, tinnitus.
Cardiac Disorders: Common: angina pectoris, arrhythmia (including atrioventricular block, cardiac flutter, extrasystoles, atrial fibrillation, tachycardia, bradycardia), palpitations, electrocardiogram QT prolonged. Uncommon: cardiac failure, myocardial infarction, coronary artery disease, cardiac murmur, coronary artery stenosis, myocardial ischemia, pericardial effusion, cyanosis. Unknown frequency: ventricular dysfunction, pericarditis, ejection fraction decrease.
Vascular Disorders: Common: flushing. Uncommon: hypertensive crisis, peripheral arterial occlusive disease, intermittent claudication, arterial stenosis limb, hematoma, arteriosclerosis. Unknown frequency: shock hemorrhagic, hypotension, thrombosis, peripheral artery stenosis.
Respiratory, Thoracic and Mediastinal Disorders: Common: dyspnea exertional, epistaxis, dysphonia. Uncommon: pulmonary edema, pleural effusion, interstitial lung disease, pleuritic pain, pleurisy, pharyngolaryngeal pain, throat irritation. Unknown frequency: pulmonary hypertension, wheezing.
Gastrointestinal Disorders: Common: pancreatitis, abdominal discomfort, abdominal distension, dysgeusia, flatulence. Uncommon: gastrointestinal hemorrhage, melena, mouth ulceration, gastroesophageal reflux, stomatitis, esophageal pain, dry mouth, gastritis, sensitivity of teeth. Unknown frequency: gastrointestinal ulcer perforation, retroperitoneal hemorrhage, hematemesis, gastric ulcer, esophagitis ulcerative, subileus, enterocolitis, hemorrhoids, hiatus hernia, rectal hemorrhage, gingivitis.
Hepatobiliary Disorders: Very common: hyperbilirubinemia. Common: hepatic function abnormal. Uncommon: hepatotoxicity, toxic hepatitis, jaundice. Unknown frequency: cholestasis, hepatomegaly.
Skin and Subcutaneous Tissue Disorders: Common: eczema, urticaria, erythema, hyperhidrosis, contusion, acne, dermatitis (including allergic, exfoliative and acneiform). Uncommon: exfoliative rash, drug eruption, pain of skin, ecchymosis. Unknown frequency: psoriasis, erythema multiforme, erythema nodosum, skin ulcer, palmar-plantar erythrodysesthesia syndrome, petechiae, photosensitivity, blister, dermal cyst, sebaceous hyperplasia, skin atrophy, skin discoloration, skin exfoliation, skin hyperpigmentation, skin hypertrophy, hyperkeratosis.
Musculoskeletal and Connective Tissue Disorders: Common: bone pain, musculoskeletal chest pain, musculoskeletal pain, back pain, neck pain, flank pain, muscular weakness. Uncommon: musculoskeletal stiffness, joint swelling. Unknown frequency: arthritis.
Renal and Urinary Disorders: Common: pollakiuria. Uncommon: dysuria, micturition urgency, nocturia. Unknown frequency: renal failure, hematuria, urinary incontinence, chromaturia.
Reproductive System and Breast Disorders: Uncommon: breast pain, gynecomastia, erectile dysfunction. Unknown frequency: breast induration, menorrhagia, nipple swelling.
General Disorders and Administration Site Conditions: Common: pyrexia, chest pain (including non-cardiac chest pain), pain, chest discomfort, malaise. Uncommon: gravitational edema, influenza-like illness, chills, feeling body temperature change (including feeling hot, feeling cold). Unknown frequency: localized edema.
Investigations: Very Common: alanine aminotransferase increased, aspartate aminotransferase increased, lipase increased, lipoprotein cholesterol (including very low density and high density) increased, total cholesterol increased, blood triglycerides increased. Common: hemoglobin decreased, blood amylase increased, gamma-glutamyltransferase increased, blood creatinine phosphokinase increased, blood alkaline phosphatase increased, weight decreased, weight increased, globulins decreased. Uncommon: blood lactate dehydrogenase increased, blood urea increased. Unknown frequency: troponin increased, blood bilirubin unconjugated increased, insulin C-peptide decreased, blood parathyroid hormone increased.
In Pediatric Patients With Newly Diagnosed Ph+ CML-CP or Resistant or Intolerant Ph+ CML-CP
The data below reflect exposure to Tasigna from two studies in pediatric patients from 2 to less than 18 years of age with either newly diagnosed Ph+ CML-CP or imatinib/dasatinib resistant or intolerant Ph+ CML-CP treated at the recommended dose of 230 mg/m2 twice daily (n = 69) [see Clinical Studies (14.5)]. The median time on treatment with Tasigna was 39.6 months (range, 0.7 to 63.5 months). The median actual dose intensity was 427.7 mg/m2/day (range 149.1 to 492.8 mg/m2/day), and the median relative dose intensity was 93% (range, 32.4 to 107.1%). Thirty-nine patients (57%) had relative dose intensity superior to 90%.
In pediatric patients with Ph+ CML-CP, the most common (greater than 20%) non-hematologic adverse reactions were hyperbilirubinemia, headache, alanine aminotransferase increased, rash, pyrexia, nausea, aspartate aminotransferase increased, pain in extremity, upper respiratory tract infection, vomiting, diarrhea, and nasopharyngitis. The most common (greater than 5%) Grade 3/4 non-hematologic adverse reactions were hyperbilirubinemia, rash, alanine aminotransferase increased, and neutropenia.
Laboratory abnormalities of hyperbilirubinemia (Grade 3/4: 16%) and transaminase elevation (AST Grade 3/4: 2.9%, ALT Grade 3/4: 10%), were reported at a higher frequency than in adult patients.
The most common hematological laboratory abnormalities (greater than or equal to 30% of patients, of all Grades) were decreases in total white blood cells (54%), platelet count (44%), absolute neutrophils (44%), hemoglobin (38%), and absolute lymphocytes (36%).
Discontinuation of study treatment due to adverse reactions occurred in 15 patients (22%). The most frequent adverse reactions leading to discontinuation were hyperbilirubinemia (9%) and rash (6%).
Increase in QTcF greater than 30 msec from baseline was observed in 19 patients (28%). No patient had an absolute QTcF of greater than 500 msec or QTcF increase of greater than 60 msec from baseline.
Growth Retardation in Pediatric Population
In a multicenter, open-label, single-arm study of 58 pediatric patients with newly diagnosed or resistant Ph+ CML-CP treated with Tasigna, with a median exposure of 56.7 months, adverse reactions associated with growth and deceleration of growth in regard to height were reported in 3 patients (5%). The adverse reactions include growth retardation in 2 adolescent patients and growth hormone deficiency with short stature in the remaining patient (age category: child). Of the 58 pediatric patients, five (9%) crossed two main percentile lines from baseline and three (5%) crossed three main percentile lines from baseline (percentile lines: 5th, 10th, 25th, 50th, 75th, 90th, and 95th). Close monitoring of growth in pediatric patients under Tasigna treatment is recommended [see Warnings and Precautions (5.14)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Tasigna. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: thrombotic microangiopathy
Nervous System Disorders: facial paralysis
Musculoskeletal and Connective Tissue Disorders: osteonecrosis
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Tasigna
Strong CYP3A Inhibitors
Concomitant use with a strong CYP3A inhibitor increased nilotinib concentrations compared to Tasigna alone [see Clinical Pharmacology (12.3)], which may increase the risk of Tasigna toxicities. Avoid concomitant use of strong CYP3A inhibitors with Tasigna. If patients must be coadministered a strong CYP3A4 inhibitor, reduce Tasigna dose [see Dosage and Administration (2.8)].
Strong CYP3A Inducers
Concomitant use with a strong CYP3A inducer decreased nilotinib concentrations compared to Tasigna alone [see Clinical Pharmacology (12.3)], which may reduce Tasigna efficacy. Avoid concomitant use of strong CYP3A inducers with Tasigna.
Proton Pump Inhibitors
Concomitant use with a proton pump inhibitor (PPI) decreased nilotinib concentrations compared to Tasigna alone [see Clinical Pharmacology (12.3)], which may reduce Tasigna efficacy. Avoid concomitant use of PPI with Tasigna. As an alternative to PPIs, use H2 blockers approximately 10 hours before or approximately 2 hours after the dose of Tasigna, or use antacids approximately 2 hours before or approximately 2 hours after the dose of Tasigna.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, Tasigna can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of nilotinib to pregnant rats and rabbits during organogenesis caused adverse developmental outcomes, including embryo-fetal lethality, fetal effects, and fetal variations in rats and rabbits at maternal exposures (AUC) approximately 2 and 0.5 times, respectively, the exposures in patients at the recommended dose (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of nilotinib up to 100 mg/kg/day and 300 mg/kg/day, respectively, during the period of organogenesis.
In rats, oral administration of nilotinib produced embryo-lethality/fetal effects at doses ≥ 30 mg/kg/day. At ≥ 30 mg/kg/day, skeletal variations of incomplete ossification of the frontals and misshapen sternebra were noted, and there was an increased incidence of small renal papilla and fetal edema. At 100 mg/kg/day, nilotinib was associated with maternal toxicity (decreased gestation weight, gravid uterine weight, net weight gain, and food consumption) and resulted in a single incidence of cleft palate and two incidences of pale skin were noted in the fetuses. A single incidence of dilated ureters was noted in a fetus also displaying small renal papilla at 100 mg/kg/day. Additional variations of forepaw and hindpaw phalanx unossified, fused sternebra, bipartite sternebra ossification, and incomplete ossification of the cervical vertebra were noted at 100 mg/kg/day.
In rabbits, oral administration of nilotinib resulted in the early sacrifice of two females, maternal toxicity and increased resorption of fetuses at 300 mg/kg/day. Fetal skeletal variations (incomplete ossification of the hyoid, bent hyoid, supernumerary short detached ribs and the presence of additional ossification sites near the nasals, frontals and in the sternebral column) were also increased at this dose in the presence of maternal toxicity. Slight maternal toxicity was evident at 100 mg/kg/day but there were no reproductive or embryo-fetal effects at this dose.
At 30 mg/kg/day in rats and 300 mg/kg/day in rabbits, the maternal systemic exposure (AUC) were 72700 ng*hr/mL and 17100 ng*hr/mL, respectively, representing approximately 2 and 0.5 times the exposure in humans at the highest recommended dose 400 mg twice daily.
When pregnant rats were dosed with nilotinib during organogenesis and through lactation, the adverse effects included a longer gestational period, lower pup body weights until weaning and decreased fertility indices in the pups when they reached maturity, all at a maternal dose of 60 mg/kg (i.e., 360 mg/m2, approximately 0.7 times the clinical dose of 400 mg twice daily based on body surface area). At doses up to 20 mg/kg (i.e., 120 mg/m2, approximately 0.25 times the clinical dose of 400 mg twice daily based on body surface area) no adverse effects were seen in the maternal animals or the pups.
8.2 Lactation
Risk Summary
There are no data on the presence of nilotinib or its metabolites in human milk or its effects on a breastfed child or on milk production. However, nilotinib is present in the milk of lactating rats. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with Tasigna and for 14 days after the last dose.
Animal Data
After a single 20 mg/kg of [14C] nilotinib dose to lactating rats, the transfer of parent drug and its metabolites into milk was observed. The overall milk-to-plasma exposure ratio of total radioactivity was approximately 2, based on the AUC0-24h or AUC0-INF values. No rat metabolites of nilotinib were detected that were unique to milk.
8.3 Females and Males of Reproductive Potential
Based on animal studies, Tasigna can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Females of reproductive potential should have a pregnancy test prior to starting treatment with Tasigna.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with Tasigna and for 14 days after the last dose.
Infertility
The risk of infertility in females or males of reproductive potential has not been studied in humans. In studies in rats and rabbits, the fertility in males and females was not affected [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of Tasigna have been established in pediatric patients greater than or equal to 1 year of age with newly diagnosed and resistant or intolerant Ph+ CML in chronic phase [see Clinical Studies (14.5)]. There are no data for pediatric patients under 2 years of age. Use of Tasigna in pediatric patients 1 year to less than 2 years of age with newly diagnosed or resistant or intolerant Ph+ CML in chronic phase is supported by efficacy in pediatric patients 2 to 6 years of age for these indications. The safety and effectiveness of Tasigna have been established in pediatric patients greater than or equal to 1 year of age with resistant or intolerant Ph+ CML in accelerated phase based on evidence of effectiveness from an adequate and well-controlled single-arm study in adults [see Clinical Studies (14.2)] with safety data from two pediatric studies as described in the next paragraph.
Use of Tasigna in pediatric patients 1 to less than 18 years of age is supported by evidence from two clinical trials [see Clinical Studies (14.5)]. The 25 patients with newly diagnosed Ph+ CML-CP were in the following age groups: 6 children (age 2 to less than 12 years) and 19 adolescents (age 12 to less than 18 years). The 44 patients with resistant or intolerant Ph+ CML-CP included 18 children (age 2 to less than 12 years) and 26 adolescents (age 12 to less than 18 years). All pediatric patients received Tasigna treatment at a dose of 230 mg/m2 twice daily, rounded to the nearest 50 mg dose (to a maximum single dose of 400 mg). No differences in efficacy or safety were observed between the different age subgroups in the two trials.
The frequency, type, and severity of adverse reactions observed were generally consistent with those observed in adults, with the exception of the laboratory abnormalities of hyperbilirubinemia (Grade 3/4: 16%) and transaminase elevation (AST Grade 3/4: 2.9%, ALT Grade 3/4: 10%), which were reported at a higher frequency in pediatric patients than in adults [see Adverse Reactions (6.1)]. For pediatric growth and development, growth retardation has been reported in pediatric patients with Ph+ CML-CP treated with Tasigna [see Warnings and Precautions (5.14), Adverse Reactions (6.1)].
The safety and effectiveness of Tasigna in pediatric patients below the age of 1 year with newly diagnosed, or resistant or intolerant Ph+ CML in chronic phase and accelerated phase, have not been established.
8.5 Geriatric Use
In the clinical trials of Tasigna (patients with newly diagnosed Ph+ CML-CP and resistant or intolerant Ph+ CML-CP and CML-AP), approximately 12% and 30% of patients were 65 years or over, respectively.
- Patients with newly diagnosed Ph+ CML-CP: There was no difference in major molecular response between patients aged less than 65 years and those greater than or equal to 65 years.
- Patients with resistant or intolerant CML-CP: There was no difference in major cytogenetic response rate between patients aged less than 65 years and those greater than or equal to 65 years.
- Patients with resistant or intolerant CML-AP: The hematologic response rate was 44% in patients less than 65 years of age and 29% in patients greater than or equal to 65 years.
No major differences for safety were observed in patients greater than or equal to 65 years of age as compared to patients less than 65 years.
8.6 Cardiac Disorders
In the clinical trials, patients with a history of uncontrolled or significant cardiovascular disease, including recent myocardial infarction, congestive heart failure, unstable angina or clinically significant bradycardia, were excluded. Caution should be exercised in patients with relevant cardiac disorders [see Boxed Warning, Warnings and Precautions (5.2)].
10 OVERDOSAGE
Overdose with nilotinib has been reported, where an unspecified number of Tasigna capsules were ingested in combination with alcohol and other drugs. Events included neutropenia, vomiting, and drowsiness. In the event of overdose, observe the patient and provide appropriate supportive treatment.
11 DESCRIPTION
Tasigna contains nilotinib, which belongs to a pharmacologic class of drugs known as kinase inhibitors.
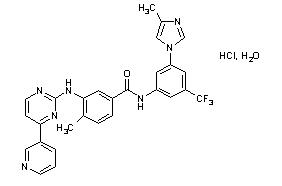
Nilotinib drug substance, in the form of monohydrochloride monohydrate, is a white to slightly yellowish to slightly greenish yellow powder with the molecular formula and weight, respectively, of C28H22F3N7O•HCl • H2O and 584 g/mol (corresponding molecular formula and weight of nilotinib base, anhydrous are C28H22F3N7O and 529 g/mol, respectively). The solubility of nilotinib in aqueous solutions decreases with increasing pH. Nilotinib is not optically active. The pKa1 was determined to be 2.1; pKa2 was estimated to be 5.4.
The chemical name of nilotinib monohydrochloride monohydrate is 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-benzamide, monohydrochloride, monohydrate. Its structure is shown below:
Tasigna (nilotinib) capsules, for oral use, contain 50 mg, 150 mg, or 200 mg nilotinib base, anhydrous (equivalent to 55 mg, 166 mg, and 221 mg nilotinib monohydrochloride monohydrate respectively) with the following inactive ingredients: colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate, and poloxamer 188. The capsules contain gelatin, iron oxide (red), iron oxide (yellow), iron oxide (black), and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nilotinib is an inhibitor of the BCR-ABL kinase. Nilotinib binds to and stabilizes the inactive conformation of the kinase domain of ABL protein. In vitro, nilotinib inhibited BCR-ABL mediated proliferation of murine leukemic cell lines and human cell lines derived from patients with Ph+ CML. Under the conditions of the assays, nilotinib was able to overcome imatinib resistance resulting from BCR-ABL kinase mutations, in 32 out of 33 mutations tested. Nilotinib inhibited the autophosphorylation of the following kinases at IC50 values as indicated: BCR-ABL (20 to 60 nM), PDGFR (69 nM), c-KIT (210 nM), CSF-1R (125 to 250 nM), and DDR1 (3.7 nM).
12.2 Pharmacodynamics
Based on exposure-response analyses for efficacy, a relationship between drug exposure and a greater likelihood of response was observed in clinical studies. Based on exposure-response analyses for safety, a relationship between exposure and a greater likelihood of safety events, including a higher occurrence of total bilirubin elevations, was observed in clinical studies.
Cardiac Electrophysiology
Tasigna is associated with concentration-dependent QT prolongation. At a dose of Tasigna 400 mg twice daily given without food in healthy subjects, the maximum mean placebo-adjusted QTcF changes were 10.4 msec (90% CI: 2.85, 18.0). After a single dose of Tasigna 800 mg (two times the maximum approved recommended dosage) given with a high fat meal to healthy subjects, the maximum mean placebo-adjusted QTcF changes were) 18.0 msec (90% CI: 9.65, 25.8). Peak plasma concentrations in the QT study were 26% lower than or comparable with those observed in patients enrolled in the single-arm study [see Boxed Warning, Warnings and Precautions (5.2), Adverse Reactions (6.1)].
12.3 Pharmacokinetics
Steady-state nilotinib exposure was dose-dependent with less than dose-proportional increases in systemic exposure at dose levels higher than 400 mg given as once or twice daily dosing. In adult patients with resistant or intolerant Ph+ CML given Tasigna 400 mg twice daily, the steady-state mean (% CV) Cmax and AUC0-12h were 2260 ng/mL (35%) and 18000 ng∙h/mL (33%), respectively. In adult patients with newly diagnosed Ph+ CML given Tasigna 300 mg twice daily, the steady-state mean (% CV) Cmax and AUC0-12h were 1540 ng/mL (48%) and 13337 ng∙h/mL (46%), respectively.
Steady state conditions were achieved by Day 8. An increase in serum exposure to nilotinib between the first dose and steady state was approximately 2-fold for daily dosing and 3.8-fold for twice daily dosing. The average steady state nilotinib trough and peak concentrations did not change over 12 months.
Absorption
Relative bioavailability of nilotinib capsule is approximately 50%, as compared to an oral drink solution (pH of 1.2 to 1.3). Peak concentrations of nilotinib are reached 3 hours after oral administration. Nilotinib is a substrate of P-gp in vitro.
Median steady-state trough concentration of nilotinib was decreased by 53% in patients with total gastrectomy compared to patients who had not undergone surgeries [see Warnings and Precautions (5.10)].
Effect of Food
Compared to the fasted state, the systemic exposure (AUC) increased by 82% when the dose was given 30 minutes after a high fat meal (meal of 800 to 1000 calories with fat being 50% of total caloric content; approximately: 150 calories from protein, 250 calories from carbohydrates, and 500-600 calories from fat).
Single dose administration of two 200 mg nilotinib capsules each dispersed in 1 teaspoon of applesauce and administered within 15 minutes was shown to be bioequivalent to a single dose administration of two 200 mg intact capsules.
Distribution
The blood-to-serum ratio of nilotinib is 0.68. Serum protein binding is approximately 98%.
Elimination
The mean (CV%) apparent elimination half-life is estimated to be approximately 17 hours (69%) and the mean (CV%) apparent clearance approximates 29 L/h (61%).
Metabolism
Nilotinib is primarily metabolized via CYP3A4-mediated oxidation and to a minor extent by CYP2C8. Nilotinib is the main circulating component in the serum. None of the metabolites contribute significantly to the pharmacological activity of nilotinib.
Excretion
After a single dose of radiolabeled nilotinib, more than 90% of the administered dose was eliminated within 7 days: 93% of the dose in feces. Parent drug accounted for 69% of the dose.
Specific Populations
Age, sex, race/ethnicity, or body weight did not significantly affect the pharmacokinetics of nilotinib. The effect of renal impairment on nilotinib pharmacokinetics is unknown.
Pediatric Patients
Following administration of the approved recommend pediatric dosage of nilotinib, steady-state exposure of nilotinib were within 2-fold to adult patients treated with 400 mg twice daily. Steady-state Cmin was comparable across all age groups (pediatric patients from ages 2 to less than 18 years), diseases (patients with newly diagnosed and resistant or intolerant Ph+ CML) and studies.
Body surface area correlated with nilotinib clearance and was the primary factor responsible for the PK differences between pediatrics and adults.
Patients with Hepatic Impairment
Following a single dose of Tasigna 200 mg (0.5 times the maximum approved recommended dosage), the mean AUC of nilotinib increased 1.4-fold, 1.4-fold, and 1.6-fold in subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B) and severe (Child-Pugh class C) hepatic impairment, respectively, compared to subjects with normal hepatic function.
Drug Interaction Studies
Clinical Studies
Strong CYP3A Inhibitors: Coadministration of ketoconazole (a strong CYP3A inhibitor) 400 mg once daily for 6 days increased nilotinib AUC by approximately 3-fold. A single concurrent intake of double-strength grapefruit juice increased the nilotinib AUC by 1.3-fold.
Strong CYP3A Inducers: Coadministration of rifampicin (a strong CYP3A inducer) 600 mg daily for 12 days decreased nilotinib AUC by approximately 80%.
Proton Pump Inhibitors (PPIs): Tasigna displays pH-dependent aqueous solubility. Coadministration of multiple doses of esomeprazole (a PPI) at 40 mg daily decreased the nilotinib AUC by 34%. No significant change in nilotinib pharmacokinetics was observed when a single 400 mg dose of Tasigna was administered 10 hours after and 2 hours before famotidine (an H2 blocker), or administered 2 hours after and 2 hours before an antacid (e.g., aluminum hydroxide, magnesium hydroxide, simethicone).
Moderate CYP3A Inhibitors: Following coadministration of nilotinib 400 mg twice daily with imatinib (a moderate CYP3A inhibitor) 400 mg daily or 400 mg twice daily, the AUC increased 30% to 50% for nilotinib and approximately 20% for imatinib.
CYP3A4 Substrates: Multiple doses of Tasigna increased the systemic exposure of oral midazolam (a substrate of CYP3A4) 2.6-fold.
CYP2C9 Substrates: Single-dose of Tasigna did not change the pharmacokinetics and pharmacodynamics of warfarin (a CYP2C9 substrate).
In Vitro Studies Where Drug Interaction Potential was not Further Evaluated Clinically
CYP Substrates: Nilotinib is a competitive inhibitor of CYP2C8, CYP2D6, and is an inducer of CYP2B6 and CYP2C8.
Substrates of Transporters: Nilotinib is an inhibitor of UGT1A1 and P-gp.
12.5 Pharmacogenomics
Tasigna can increase bilirubin levels. The (TA)7/(TA)7 genotype of UGT1A1 was associated with a statistically significant increase in the risk of hyperbilirubinemia relative to the (TA)6/(TA)6 and (TA)6/(TA)7 genotypes. However, the largest increases in bilirubin were observed in the (TA)7/(TA)7 genotype (UGT1A1*28) patients [see Warnings and Precautions (5.6)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A 2-year carcinogenicity study was conducted orally in rats at nilotinib doses of 5, 15, and 40 mg/kg/day. Exposures in animals at the highest dose tested were approximately 2- to 3-fold the human exposure (based on AUC) at the nilotinib dose of 400 mg twice daily. The study was negative for carcinogenic findings. A 26-week carcinogenicity study was conducted orally in Tg.rasH2 mice, a model genetically modified to enhance susceptibility to neoplastic transformation, at nilotinib doses of 30, 100, and 300 mg/kg/day. Nilotinib induced in the skin and subcutis statistically significant increases in the incidence of papillomas in females and of papillomas and combined papillomas and carcinomas in males at 300 mg/kg/day. The no-observed-adverse-effect-level (NOAEL) for skin neoplastic lesions was 100 mg/kg/day.
Nilotinib was not mutagenic in a bacterial mutagenesis (Ames) assay, was not clastogenic in a chromosome aberration assay in human lymphocytes, did not induce DNA damage (comet assay) in L5178Y mouse lymphoma cells, nor was it clastogenic in an in vivo rat bone marrow micronucleus assay with two oral treatments at doses up to 2000 mg/kg/dose.
There were no effects on male or female rat and female rabbit mating or fertility at doses up to 180 mg/kg in rats (approximately 4- to 7-fold for males and females, respectively, the AUC in patients at the dose of 400 mg twice daily) or 300 mg/kg in rabbits (approximately one-half the AUC in patients at the dose of 400 mg twice daily). The effect of Tasigna on human fertility is unknown. In a study where male and female rats were treated with nilotinib at oral doses of 20 to 180 mg/kg/day (approximately 1- to 6.6-fold the AUC in patients at the dose of 400 mg twice daily) during the pre-mating and mating periods and then mated, and dosing of pregnant rats continued through gestation Day 6, nilotinib increased post-implantation loss and early resorption, and decreased the number of viable fetuses and litter size at all doses tested.
14 CLINICAL STUDIES
14.1 Adult Newly Diagnosed Ph+ CML-CP
The ENESTnd (Evaluating Nilotinib Efficacy and Safety in clinical Trials-Newly Diagnosed patients) study (NCT00471497) was an open-label, multicenter, randomized trial conducted to determine the efficacy of Tasigna versus imatinib tablets in adult patients with cytogenetically confirmed newly diagnosed Ph+ CML-CP. Patients were within 6 months of diagnosis and were previously untreated for CML-CP, except for hydroxyurea and/or anagrelide. Efficacy was based on a total of 846 patients: 283 patients in the imatinib 400 mg once daily group, 282 patients in the Tasigna 300 mg twice daily group, 281 patients in the Tasigna 400 mg twice daily group.
Median age was 46 years in the imatinib group and 47 years in both Tasigna groups, with 12%, 13%, and 10% of patients greater than or equal to 65 years of age in imatinib 400 mg once daily, Tasigna 300 mg twice daily and Tasigna 400 mg twice daily treatment groups, respectively. There were slightly more male than female patients in all groups (56%, 56%, and 62% in imatinib 400 mg once daily, Tasigna 300 mg twice daily and Tasigna 400 mg twice daily treatment groups, respectively). More than 60% of all patients were Caucasian, and 25% were Asian.
The primary data analysis was performed when all 846 patients completed 12 months of treatment (or discontinued earlier). Subsequent analyses were done when patients completed 24, 36, 48, and 60 months of treatment (or discontinued earlier). The median time on treatment was approximately 61 months in all three treatment groups.
The primary efficacy endpoint was major molecular response (MMR) at 12 months after the start of study medication. MMR was defined as less than or equal to 0.1% BCR-ABL/ABL % by international scale measured by RQ-PCR, which corresponds to a greater than or equal to 3 log reduction of BCR-ABL transcript from standardized baseline. Efficacy endpoints are summarized in Table 11.
Two patients in the Tasigna arm progressed to either accelerated phase or blast crisis (both within the first 6 months of treatment) while 12 patients on the imatinib arm progressed to either accelerated phase or blast crisis (7 patients within first 6 months, 2 patients within 6 to 12 months, 2 patients within 12 to 18 months and 1 patient within 18 to 24 months).
| Abbreviation: CI, confidence interval. aCMH test stratified by Sokal risk group. bCCyR: 0% Ph+ metaphases. Cytogenetic responses were based on the percentage of Ph+ metaphases among greater than or equal to 20 metaphase cells in each bone marrow sample. |
||
| Tasigna
300 mg twice daily | imatinib
400 mg once daily |
|
| N = 282 | N = 283 | |
| MMR at 12 months (95% CI) | 44% (38.4, 50.3) | 22% (17.6, 27.6) |
| P-Valuea | < 0.0001 | |
| CCyRb by 12 months (95% CI) | 80% (75.0, 84.6) | 65% (59.2, 70.6) |
| MMR at 24 months (95% CI) | 62% (55.8, 67.4) | 38% (31.8, 43.4) |
| CCyRb by 24 months (95% CI) | 87% (82.4, 90.6) | 77% (71.7, 81.8) |
By the 60 months, MMR was achieved by 77% of patients on Tasigna and 60% of patients on imatinib; MR4.5 was achieved by 53.5% of patients on Tasigna and 31.4% on imatinib. Median overall survival was not reached in either arm. At the time of the 60-month final analysis, the estimated survival rate was 93.7% for patients on Tasigna and 91.7% for patients on imatinib.
14.2 Adult Patients With Resistant or Intolerant Ph+ CML-CP and CML-AP
Study CAMN107A2101 (referred to as Study A2101) (NCT00109707) was a single-arm, open-label, multicenter study conducted to evaluate the efficacy and safety of Tasigna (400 mg twice daily) in patients with imatinib-resistant or -intolerant CML with separate cohorts for chronic and accelerated phase disease. The definition of imatinib resistance included failure to achieve a complete hematologic response (by 3 months), cytogenetic response (by 6 months) or major cytogenetic response (by 12 months) or progression of disease after a previous cytogenetic or hematologic response. Imatinib intolerance was defined as discontinuation of treatment due to toxicity and lack of a major cytogenetic response at time of study entry. At the time of data cut-off, 321 patients with CML-CP and 137 patients with CML-AP with a minimum follow-up of 24 months were enrolled. In this study, about 50% of CML-CP and CML-AP patients were males, over 90% (CML-CP) and 80% (CML-AP) were Caucasian, and approximately 30% were age 65 years or older.
Overall, 73% of patients were imatinib resistant while 27% were imatinib intolerant. The median time of prior imatinib treatment was approximately 32 (CML-CP) and 28 (CML-AP) months. Prior therapy included hydroxyurea in 85% of patients, interferon in 56% and stem cell or bone marrow transplant in 8%. The median highest prior imatinib dose was 600 mg per day for patients with CML-CP and CML-AP, and the highest prior imatinib dose was greater than or equal to 600 mg/day in 74% of all patients with 40% of patients receiving imatinib doses greater than or equal to 800 mg/day.
Median duration of Tasigna treatment was 18.4 months in patients with CML-CP and 8.7 months in patients with CML-AP.
The efficacy endpoint in CML-CP was unconfirmed major cytogenetic response (MCyR) which included complete and partial cytogenetic responses.
The efficacy endpoint in CML-AP was confirmed hematologic response (HR), defined as either a complete hematologic response (CHR) or no evidence of leukemia (NEL). The rates of response for CML-CP and CML-AP patients are reported in Table 12.
Median durations of response had not been reached at the time of data analysis.
| aCytogenetic response criteria: Complete (0% Ph+ metaphases) or partial (1% to 35%). Cytogenetic responses were based on the percentage of Ph-positive metaphases among greater than or equal to 20 metaphase cells in each bone marrow sample. bHematologic response = CHR + NEL (all responses confirmed after 4 weeks). |
|
| Cytogenetic response rate (unconfirmed) (%)a | |
| Chronic phase
(n = 321) |
|
| Major (95% CI) | 51% (46% – 57%) |
| Complete (95% CI) | 37% (32% – 42%) |
| Partial (95% CI) | 15% (11% – 19%) |
| Accelerated phase
(n = 137) |
|
| Hematologic response rate (confirmed) (95% CI)b | 39% (31% – 48%) |
| Complete hematologic response rate (95% CI) | 30% (22% – 38%) |
| No evidence of leukemia (95% CI) | 9% (5% – 16%) |
CHR (CML-CP): WBC less than 10 x 109/L, platelets less than 450,000/mm3, no blasts or promyelocytes in peripheral blood, less than 5% myelocytes + metamyelocytes in bone marrow, less than 20% basophils in peripheral blood, and no extramedullary involvement.
CHR (CML-AP): neutrophils greater than or equal to 1.5 x 109/L, platelets greater than or equal to 100 x 109/L, no myeloblasts in peripheral blood, myeloblasts less than 5% in bone marrow, and no extramedullary involvement.
NEL: same criteria as for CHR but neutrophils greater than or equal to 1.0 x 109/L and platelets greater than or equal to 20 x 109/L without transfusions or bleeding.
Adult Patients With Chronic Phase
The MCyR rate in 321 CML-CP patients was 51%. The median time to MCyR among responders was 2.8 months (range, 1 to 28 months). The median duration of MCyR cannot be estimated. The median duration of exposure on this single arm-trial was 18.4 months. Among the CML-CP patients who achieved MCyR, 62% of them had MCyR lasting more than 18 months. The CCyR rate was 37%.
Adult Patients With Accelerated Phase
The overall confirmed hematologic response rate in 137 patients with CML-AP was 39%. The median time to first hematologic response among responders was 1 month (range, 1 to 14 months). Among the CML-AP patients who achieved HR, 44% of them had a response lasting for more than 18 months.
After imatinib failure, 24 different BCR-ABL mutations were noted in 42% of chronic phase and 54% of accelerated phase CML patients who were evaluated for mutations.
14.3 Treatment Discontinuation in Newly Diagnosed Ph+ CML-CP Patients Who Have Achieved a Sustained Molecular Response (MR4.5)
The ENESTfreedom (Evaluating Nilotinib Efficacy and Safety in clinical Trials-freedom) study (NCT01784068) is an open-label, multicenter, single-arm study, where 215 adult patients with Ph+ CML-CP treated with Tasigna in first-line for ≥ 2 years who achieved MR4.5 as measured with the MolecularMD MRDx® BCR-ABL Test were enrolled to continue Tasigna treatment for an additional 52 weeks (Tasigna consolidation phase).
Of the 215 patients, 190 patients (88.4%) entered the “Treatment-Free Remission” (TFR) phase after achieving a sustained molecular response (MR4.5) during the consolidation phase, defined by the following criteria:
- The 4 last quarterly assessments (taken every 12 weeks) were at least MR4 (BCR-ABL/ABL ≤ 0.01% IS), and maintained for 1 year
- The last assessment being MR4.5 (BCR-ABL/ABL ≤ 0.0032% IS)
- No more than two assessments falling between MR4 and MR4.5 (0.0032% IS < BCR-ABL/ABL ≤ 0.01% IS).
The median age of patients who entered the TFR phase was 55 years, 49.5% were females, and 21.1% of the patients were ≥ 65 years of age. BCR-ABL levels were monitored every 4 weeks during the first 48 weeks of the TFR phase. Monitoring frequency was intensified to every 2 weeks upon the loss of MR4.0. Biweekly monitoring ended at one of the following time points:
- Loss of MMR requiring patient to reinitiate Tasigna treatment
- When the BCR-ABL levels returned to a range between MR4.0 and MR4.5
- When the BCR-ABL levels remained lower than MMR for 4 consecutive measurements (8 weeks from initial loss of MR4.0).
Any patient with loss of MMR during the TFR phase reinitiated Tasigna treatment at 300 mg twice daily or at a reduced dose level of 400 mg once daily if required from the perspective of tolerance, within 5 weeks after the collection date of the blood sample demonstrating loss of MMR. Patients who required reinitiation of Tasigna treatment were monitored for BCR-ABL levels every 4 weeks for the first 24 weeks and then every 12 weeks thereafter in patients who regained MMR.
Efficacy was based on the 96-week analysis data cut-off date, by which time, 91 patients (47.9%) discontinued from the TFR phase due to loss of MMR, and 1 (0.5%), 1 (0.5%), 1 (0.5%) and 3 patients (1.6%) due to death from unknown cause, physician decision, lost to follow-up and subject decision, respectively. Among the 91 patients who discontinued the TFR phase due to loss of MMR, 88 patients restarted Tasigna treatment and 3 patients permanently discontinued from the study.
By the 96-week data cut-off, of the 88 patients who restarted treatment due to loss of MMR in the TFR phase, 87 patients (98.9%) patients regained MMR (one patient discontinued study permanently due to subject decision after 7.1 weeks of retreatment without regaining MMR) and 81 patients (92.0%) regained MR4.5 by the time of the cut-off date. The cumulative rate of MMR and MR4.5 regained at 24 weeks since treatment reinitiation was 97.7% (86/88 patients) and 86.4% (76/88 patients), respectively.
| Abbreviation: CI, confidence interval. 1Patients in MMR at the specified time point in the TFR phase. 2Based on the time to event (loss of MMR) data during the TFR phase. |
|||
| Patients who entered the treatment free remission (TFR) phase (full analysis Set, N = 190) | |||
| Patients in TFR phase1
at the specified time point | Loss of MMR2 by the specified time point
|
||
| % | 95% CI | % | |
| 24 weeks | 62.1 | (54.8, 69.0) | 35.8 |
| 48 weeks | 51.6 | (44.2, 58.9) | 45.8 |
| 96 weeks | 48.9 | (41.6, 56.3) | 47.9 |
Among the 190 patients in the TFR phase, 98 patients had a treatment-free survival (TFS) event (defined as discontinuation from TFR phase due to any reason, loss of MMR, death due to any cause, progression to AP/BC up to the end of TFR phase, or reinitiation of treatment due to any cause in the study) by the 96-week cut-off date.
Figure 1: Kaplan-Meier Estimate of Treatment-Free Survival After Start of TFR (Full Analysis Set ENESTfreedom)
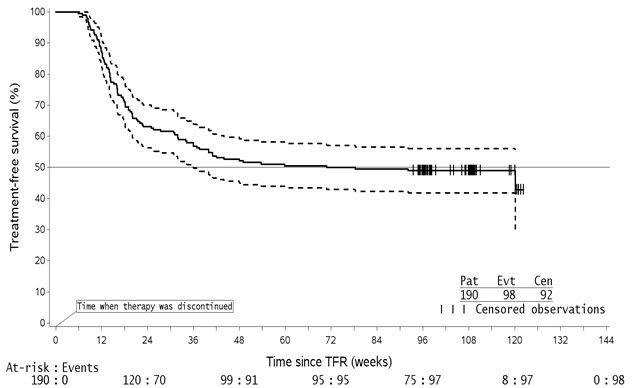
- For a given time point, the points on the dashed curves represent the 95% confidence limits for the associated KM estimate on the solid curve.
- By the time of the 96-week data cut-off date, one single patient lost MMR at Week 120, at the time when only 8 patients were considered at risk. This explains the artificial drop at the end of the curve.
14.4 Treatment Discontinuation in Ph+ CML-CP Patients Who Have Achieved a Sustained Molecular Response (MR4.5) on Tasigna Following Prior Imatinib Therapy
The ENESTop (Evaluating Nilotinib Efficacy and Safety in clinical Trials-STop) study (NCT01698905) is an open-label, multicenter, single-arm study, where 163 adult patients with Ph+ CML-CP taking tyrosine kinase inhibitors (TKIs) for ≥ 3 years (imatinib as initial TKI therapy for more than 4 weeks without documented MR4.5 on imatinib at the time of switch to Tasigna, then switched to Tasigna for at least 2 years), and who achieved MR4.5 on Tasigna treatment as measured with the MolecularMD MRDx® BCR-ABL Test were enrolled to continue Tasigna treatment for an additional 52 weeks (Tasigna consolidation phase). Of the 163 patients, 126 patients (77.3%) entered the TFR phase after achieving a sustained molecular response (MR4.5) during the consolidation phase, defined by the following criterion:
- The 4 last quarterly assessments (taken every 12 weeks) showed no confirmed loss of MR4.5 (BCR-ABL/ABL ≤ 0.0032% IS) during 1 year.
The median age of patients who entered the TFR phase was 56 years, 55.6% were females, and 27.8% of the patients were ≥ 65 years of age. The median actual dose intensity during the 52-week Tasigna consolidation phase was 771.8 mg/day with 52.4%, 29.4%, 0.8%, 16.7%, and 0.8% of patients receiving a daily Tasigna dose of 800 mg, 600 mg, 450 mg, 400 mg and 300 mg just before entry into the TFR phase, respectively.
Patients who entered the TFR phase but experienced two consecutive measurements of BCR-ABL/ABL > 0.01% IS were considered having a confirmed loss of MR4.0, triggering reinitiation of Tasigna treatment. Patients with loss of MMR in the TFR phase immediately restarted Tasigna treatment without confirmation. All patients who restarted Tasigna therapy had BCR-ABL transcript levels monitored every 4 weeks for the first 24 weeks, then once every 12 weeks.
Efficacy was based on the 96-week analysis data cut-off date, by which time, 61 patients (48.4%) had discontinued from the TFR phase: 58 patients (46.0%) due to loss of MMR or confirmed loss of MR4.0, 2 patients (1.6%) due to subject/guardian decision and one patient (0.8%) due to pregnancy. Among the 58 patients who discontinued from the TFR phase due to confirmed loss of MR4.0 or loss of MMR, 56 patients restarted Tasigna therapy and 2 patients permanently discontinued from the study.
By the 96-week data cut-off, of the 56 patients who restarted Tasigna treatment due to confirmed loss of MR4.0 or loss of MMR in the TFR phase, 52 patients (92.9%) regained MR4.0 and MR4.5; 4 patients (7.1%) did not regain MR4.0 by the time of the cut-off date. The cumulative rate of MR4 and MR4.5 regained by 48-weeks since treatment reinitiation, was 92.9% (52/56 patients) and 91.1% (51/56 patients), respectively.
| Abbreviation: CI, confidence interval. 1Patients without loss of MMR or confirmed loss of MR4 by specified time point of TFR phase. 2Based on the time to event (loss of MMR or confirmed loss of MR4) data during the TFR phase. |
|||
| Patients who entered the treatment free remission (TFR) phase (full analysis set, N = 126) | |||
| Patients in TFR phase1
at the specified time point | Loss of MMR or confirmed loss of MR42 by the specified time point
|
||
| % | 95% CI | % | |
| 24 weeks | 60.3 | (51.2, 68.9) | 38.9 |
| 48 weeks | 57.9 | (48.8, 66.7) | 41.3 |
| 96 weeks | 53.2 | (44.1, 62.1) | 43.7 |
Among the 126 patients in the TFR phase, 61 patients (48.4%) had a treatment-free survival (TFS) event (defined as discontinuation from TFR phase due to any reason, loss of MMR, confirmed loss of MR4, death due to any cause, progression to AP/BC up to the end of TFR phase, or reinitiation of treatment due to any cause in the study) on or before the 96-month cut-off date.
Figure 2: Kaplan-Meier Estimate of Treatment-Free Survival after Start of TFR (Full Analysis Set ENESTop)
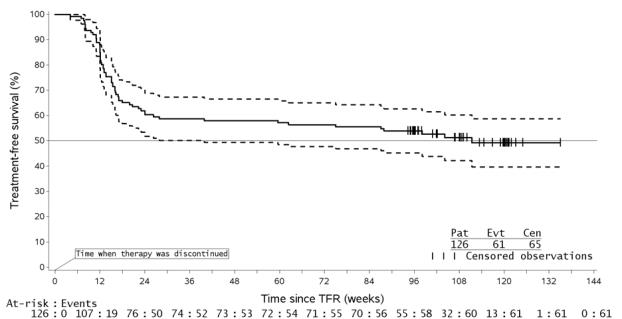
- For a given time point, the points on the dashed curves represent the 95% confidence limits for the associated KM estimate on the solid curve.
14.5 Pediatric Patients With Newly Diagnosed Ph+ CML-CP or Resistant or Intolerant Ph+ CML-CP
The safety and efficacy of Tasigna in pediatric patients with Ph+ CML-CP have been investigated in two studies: Study CAMN107A2120 (NCT01077544), an open-label, single-arm, multi-center study that evaluated the pharmacokinetics, safety, and preliminary efficacy of Tasigna in pediatric patients with Ph+ CML resistant or intolerant to imatinib or dasatinib (n = 11), and Study CAMN107A2203 (NCT01844765), an open-label, single-arm, multi-center study evaluating the efficacy and safety of Tasigna in pediatric patients (from 2 to less than 18 years of age) with Ph+ CML-CP resistant or intolerant to imatinib or dasatinib (n = 33) and newly diagnosed Ph+ CML-CP (n = 25). In both studies, patients received Tasigna treatment at a dose of 230 mg/m2 twice daily, rounded to the nearest 50 mg dose (to a maximum single dose of 400 mg). Data up to 12 cycles was pooled from a total of 69 pediatric patients (from 2 to less than 18 years of age) with either newly diagnosed Ph+ CML-CP (n = 25; 6 children from 2 to less than 12 years and 19 adolescents from 12 to less than 18 years) or imatinib/dasatinib resistant or intolerant Ph+ CML-CP (n = 44; 18 children from 2 to less than 12 years and 26 adolescents from 12 to less than 18 years).
In patients with resistant or intolerant CML, the major molecular response [(MMR); BCR-ABL/ABL ≤ 0.1% IS] rate was 40.9% (18/44, 95% CI: 26.3%, 56.8%) at 12 cycles (28 days per cycle). In patients with newly diagnosed CML, the MMR rate was 60.0% (15/25, 95% CI: 38.7%, 78.9%) at 12 cycles. In patients with resistant or intolerant CML, the cumulative MMR rate was 47.7% (21/44) by Cycle 12. In patients with newly diagnosed CML, the cumulative MMR rate was 64.0% (16/25) by Cycle 12.
Among the 21 patients with resistant or intolerant CML who were in MMR at any time on treatment, the median time to first MMR was 2.8 months (range, 0.0 to 11.3). For the 17 patients with newly diagnosed CML who achieved MMR, the median time to first MMR was 5.6 months (range, 2.7 to 16.6).
Study CAMN107A2203 provided long term data with follow up of approximately 5 years.
By the time of final analysis, the median time on treatment with Tasigna was 51.9 months (range, 1.4 to 61.2 months) for patients with newly diagnosed CML and 60.5 months (range: 0.7 to 63.5 months) for patients with resistant or intolerant CML.
In the patients with resistant or intolerant CML, the major molecular response (MMR; BCR-ABL/ABL ≤0.1% IS) rates were 57.6%, 57.6% by Cycles 24, and 36, respectively. The MMR rate increased to 60.6% by Cycle 48 and was the same until end of study (Cycle 66). In the patients with newly diagnosed CML, the MMR rates were 68.0% by Cycle 24. The MMR rate increased to 76.0% by Cycle 36 and was the same until end of study (Cycle 66).
Among patients with resistant or intolerant CML, 12.1% of patients achieved BCR-ABL/ABL ≤ 0.0032% IS (MR4.5) by Cycle 66. Among patients with newly diagnosed CML, the percentage of patients who achieved MR4.5 was 44%.
None of the 20 patients with resistant or intolerant CML who achieved MMR at any time on treatment by Cycle 66 had confirmed loss of MMR by the end of Cycle 66 or at the time of early discontinuation. Among the 19 patients with newly diagnosed CML who achieved MMR at any time on treatment by the end of Cycle 66, three patients had confirmed loss of MMR. The median durations of MMR could not be estimated in either population as more than half responders did not have a confirmed loss of response by the study end. Range of duration of response was 0.03 to 61 months for resistant or intolerant CML patients and 2.8 to 57.9 months for newly diagnosed CML patients. One patient with resistant or intolerant CML progressed to AP/BC after 10.1 months on treatment.
16 HOW SUPPLIED/STORAGE AND HANDLING
Tasigna (nilotinib) 50 mg capsules are red opaque cap and light yellow opaque body hard gelatin capsules, size 4 with black radial imprint “NVR/ABL.” Tasigna (nilotinib) 150 mg capsules are red opaque hard gelatin capsules, size 1 with black axial imprint “NVR/BCR.” Tasigna (nilotinib) 200 mg capsules are light yellow opaque hard gelatin capsules, size 0 with the red axial imprint “NVR/TKI.” Tasigna 50 mg capsules are supplied in bottles and Tasigna 150 mg and 200 mg capsules are supplied in blister packs.
50 mg
Bottle of 120 capsules………………………………………………………………NDC 0078-0951-66
150 mg
Carton of 4 blister packs of (4x28)…………………………………………………NDC 0078-0592-87
Blisters of 28 capsules………………………………………………………………NDC 0078-0592-51
200 mg
Carton of 4 blister packs of (4x28)…………………………………………………NDC 0078-0526-87
Blisters of 28 capsules………………………………………………………………NDC 0078-0526-51
Tasigna (nilotinib) capsules should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
A Medication Guide is required for distribution with Tasigna. The complete text of the Medication Guide is reprinted at the end of this document.
Myelosuppression
Advise patients that treatment with Tasigna can cause serious thrombocytopenia, neutropenia, and anemia. Advise patients to seek immediate medical attention if symptoms suggestive of low blood counts occur, such as fever, chills or other signs of infection, unexplained bleeding or bruising, or unexplained weakness or shortness of breath [see Warnings and Precautions (5.1)].
QT Prolongation
Advise patients that Tasigna can cause possibly life-threatening, abnormal heart beat. Advise patients to seek immediate medical attention if symptoms of abnormal heart beat occur, such as feeling light-headed, faint or experiencing an irregular heartbeat [see Warnings and Precautions (5.2)].
Cardiac and Arterial Vascular Occlusive Events
Advise patients that cardiovascular events (including ischemic heart disease, peripheral arterial occlusive disease, and ischemic cerebrovascular events) have been reported. Advise patients to seek immediate medical attention if any symptoms suggestive of a cardiovascular event occur, such as chest or leg pain, numbness or weakness, or problems walking or speaking occur suddenly [see Warnings and Precautions (5.4)].
Pancreatitis and Elevated Serum Lipase
Advise patients that Tasigna can increase the risk of pancreatitis and that patients with a previous history of pancreatitis may be at greater risk. Advise patients to seek immediate medical attention if symptoms suggestive of pancreatitis occur, such as sudden stomach area pain with accompanying nausea and vomiting [see Warnings and Precautions (5.5)].
Hepatotoxicity
Advise patients that Tasigna can increase the risk of hepatotoxicity and that patients with previous history of liver diseases may be at risk. Advise patients to seek immediate medical attention if any symptoms suggestive of hepatotoxicity occur, such as stomach pain, yellow skin and eyes, and dark-colored urine [see Warnings and Precautions (5.6)].
Tumor Lysis Syndrome
Advise patients that Tasigna can cause TLS and to seek immediate medical attention if any symptoms suggestive of TLS occur, such as an abnormal heartbeat or less urine production [see Warnings and Precautions (5.8)].
Hemorrhage
Advise patients that serious hemorrhagic events, including fatal events, have occurred in patients with CML treated with Tasigna. Advise patients to seek immediate medical attention if symptoms suggestive of hemorrhage occur, such as uncontrolled bleeding, changes in eyesight, unconsciousness, or sudden headache or sudden confusion in surroundings [see Warnings and Precautions (5.9)].
Fluid Retention
Advise patients that Tasigna can cause fluid retention and to seek immediate medical attention if any symptoms suggestive of fluid retention, such as shortness of breath, rapid weight gain, or swelling occur [see Warnings and Precautions (5.13)].
Effects on Growth and Development in Pediatric Patients
Inform pediatric patients and their caregivers of the possibility of developing growth abnormalities. Growth retardation has been reported in pediatric patients treated with Tasigna. Therefore, monitor growth and development in pediatric patients [see Warnings and Precautions (5.14)].
Treatment-Free Remission (TFR)
Advise patients that frequent monitoring is required to detect possible loss of remission if TFR is attempted. Advise patients that musculoskeletal symptoms, such as muscle pain, pain in extremity, joint pain, bone pain, or spinal pain, may occur more frequently than before treatment discontinuation [see Warnings and Precautions (5.16)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.15), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment and for 14 days after receiving the last dose of Tasigna [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with Tasigna and for 14 days after the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
Advise patients that Tasigna and certain other medicines, including over the counter medications or herbal supplements (such as St. John’s Wort), can interact with each other [see Drug Interactions (7)].
Taking Tasigna
Advise patients to take Tasigna doses twice daily approximately 12 hours apart. The capsules should be swallowed whole with water.
Advise patients to take Tasigna on an empty stomach. No food should be consumed for at least 2 hours before the dose is taken and for at least 1 hour after the dose is taken. Patients should not consume grapefruit products and other foods that are known to inhibit CYP3A4 at any time during Tasigna treatment [see Dosage and Administration (2.1), Drug Interactions (7.1, 7.2)].
If the patient missed a dose of Tasigna, the patient should take the next scheduled dose at its regular time. The patient should not take two doses at the same time.
Should patients be unable to swallow capsules, the contents of each capsule may be dispersed in one teaspoon of applesauce and the mixture swallowed immediately (within 15 minutes).
Compliance
Advise patients of the following:
- Continue taking Tasigna every day for as long as their doctor tells them.
- This is a long-term treatment.
- Do not change dose or stop taking Tasigna without first consulting their doctor.
- If a dose is missed, take the next dose as scheduled. Do not take a double dose to make up for the missed capsules.
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2024-08
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: September 2021 | |
| Medication Guide
TASIGNA® (ta-sig-na) (nilotinib) capsules |
||
| What is the most important information I should know about Tasigna? Tasigna can cause a possible life-threatening heart problem called QTc prolongation. QTc prolongation causes an irregular heartbeat, which may lead to sudden death. Your healthcare provider should check the electrical activity of your heart with a test called an electrocardiogram (ECG): |
||
| • before starting Tasigna • 7 days after starting Tasigna | • with any dose changes • regularly during Tasigna treatment |
|
You may lower your chances for having QTc prolongation with Tasigna if you:
Call your healthcare provider right away if you feel lightheaded, faint, or have an irregular heartbeat during treatment with Tasigna. These can be symptoms of QTc prolongation. |
||
| What is Tasigna?
Tasigna is a prescription medicine used to treat:
It is not known if Tasigna is safe and effective in children younger than 1 year of age with newly diagnosed, resistant, or intolerant Ph+ CML in chronic phase. The long-term effects of treating children with Tasigna for a long period of time are not known. |
||
| Who should not take Tasigna?
Do not take if you have:
|
||
Before taking Tasigna, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. If you need to take antacids (medicines to treat heartburn) do not take them at the same time that you take Tasigna. If you take:
Tasigna can interact with many medicines and supplements and increase your chance for serious and life-threatening side effects. See “What is the most important information I should know about Tasigna?” |
||
How should I take Tasigna?
Your healthcare provider may change your dose. Your healthcare provider may have you stop Tasigna for some time or lower your dose if you have side effects with it.
|
||
| What are the possible side effects of Tasigna?
Tasigna may cause serious side effects, including:
|
||
|
|
|
|
||
| The most common side effects of Tasigna in adults and children include: | ||
| • nausea • rash • headache • tiredness • itching • vomiting | • diarrhea • cough • constipation • muscle and joint pain • runny or stuffy nose, sneezing, sore throat • fever • night sweats | |
|
Side effects in adult patients attempting treatment free remission:
|
||
| • muscle pain • arm and leg pain • joint pain | • bone pain • spine pain | |
|
Tell your healthcare provider if you or your child have any side effect that bothers you or does not go away. These are not all of the possible side effects of Tasigna. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store Tasigna?
Keep Tasigna and all medicines out of the reach of children. |
||
| General information about the safe and effective use of Tasigna.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Tasigna for a condition for which it was not prescribed. Do not give Tasigna to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about Tasigna that is written for health professionals. |
||
| What are the ingredients in Tasigna?
Active ingredient: nilotinib Inactive ingredients: colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate and poloxamer 188. The capsules contain gelatin, iron oxide (red), iron oxide (yellow), iron oxide (black), and titanium dioxide. Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936 © Novartis For more information, go to www.Tasigna.com or call 1-866-411-8274. |
||
T2021-128
PRINCIPAL DISPLAY PANEL
NDC 0078-0951-66
120 capsules
Rx Only
Tasigna®
(nilotinib) capsules
50 mg
Dispense with Medication Guide
NOVARTIS