USE IN PREGNANCY
When used in pregnancy during the second and third trimesters, ACE inhibitors can cause injury and even death to the developing fetus. When pregnancy is detected, enalaprilat injection should be discontinued as soon as possible. See WARNINGS, Fetal/Neonatal Morbidity and Mortality.
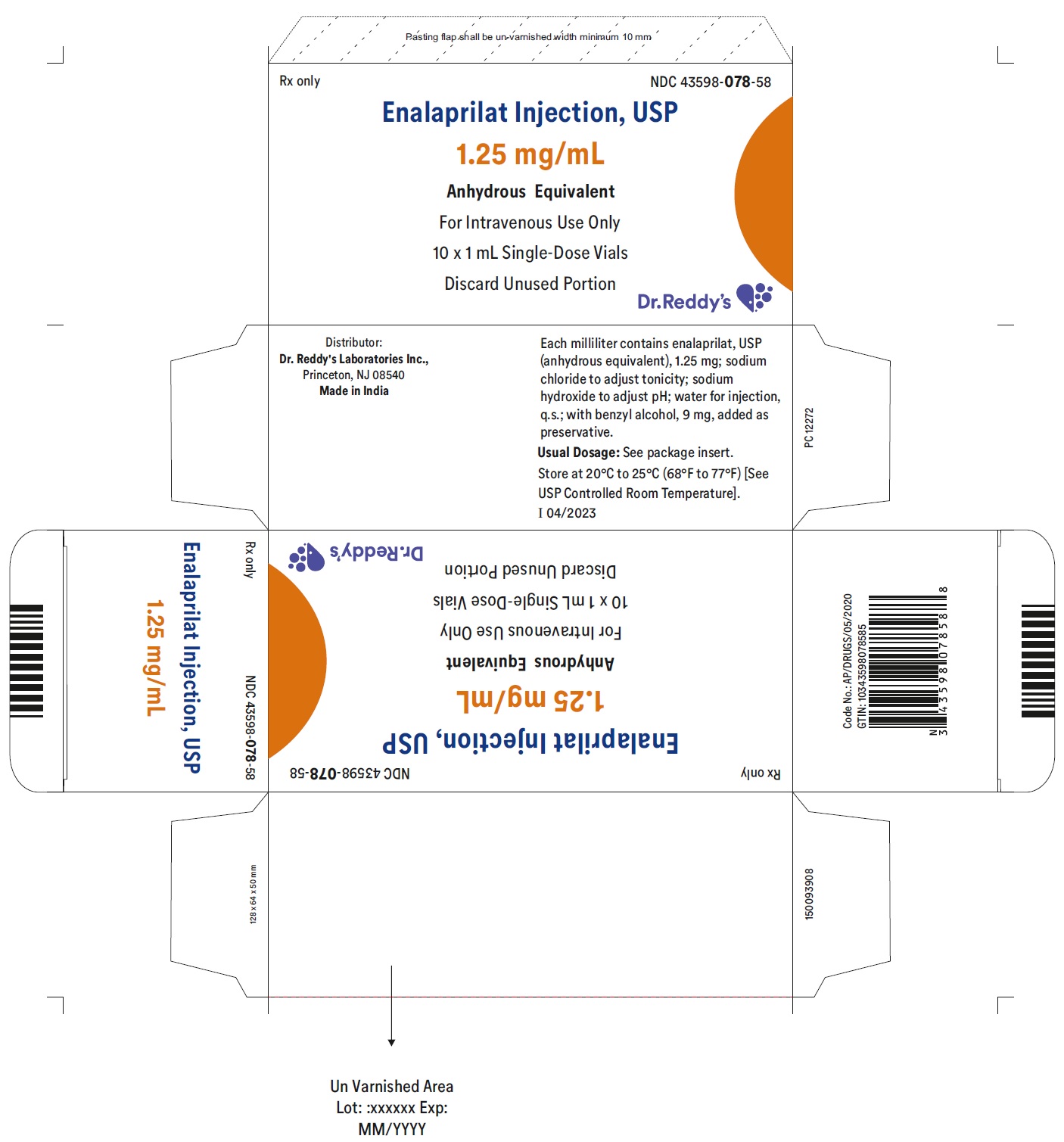
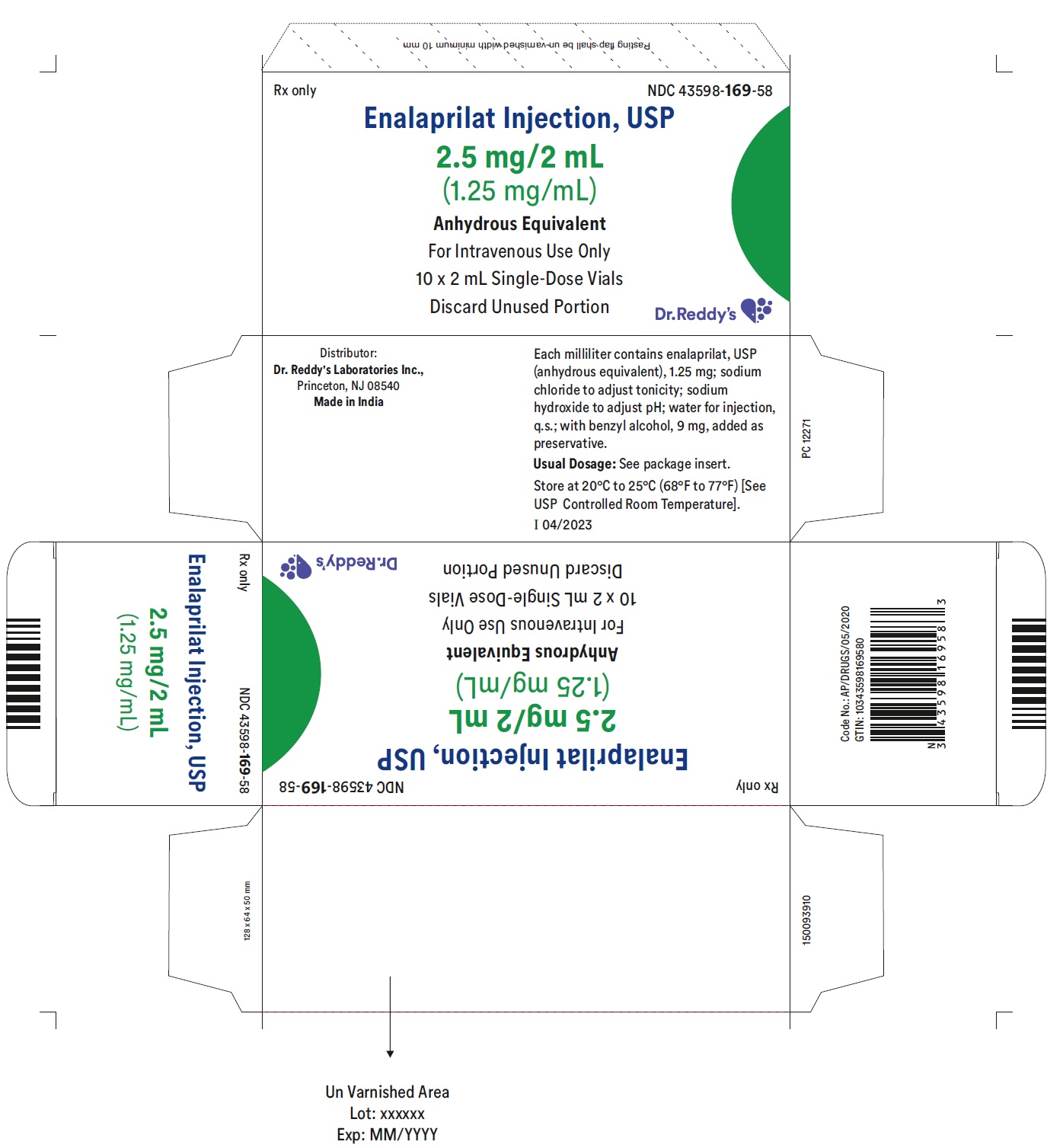
DESCRIPTION
Enalaprilat injection, USP is a sterile aqueous solution for intravenous administration. Enalaprilat is an angiotensin-converting enzyme inhibitor. It is chemically described as (S)-1-[N-(1-carboxy-3-phenylpropyl)-L-alanyl]-L-proline dihydrate. Its molecular formula is C18H24N2O5•2H2O and its structural formula is:
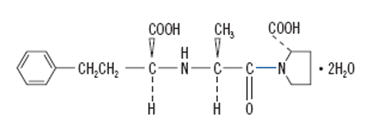
Enalaprilat, USP is a white or almost white crystalline powder with a molecular weight of 384.43. It is sparingly soluble in methanol and slightly soluble in water.
Each milliliter of enalaprilat injection, USP contains 1.25 mg enalaprilat, USP (anhydrous equivalent); sodium chloride to adjust tonicity; sodium hydroxide to adjust pH; water for injection, q.s.; with benzyl alcohol, 9 mg, added as a preservative.
CLINICAL PHARMACOLOGY
Enalaprilat, an angiotensin-converting enzyme (ACE) inhibitor when administered intravenously, is the active metabolite of the orally administered pro-drug, enalapril maleate. Enalaprilat is poorly absorbed orally.
Mechanism of Action
Intravenous enalaprilat, or oral enalapril, after hydrolysis to enalaprilat, inhibits ACE in human subjects and animals. ACE is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor substance, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex. Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. Although the latter decrease is small, it results in small increases of serum potassium. In hypertensive patients treated with enalapril alone for up to 48 weeks, mean increases in serum potassium of approximately 0.2 mEq/L were observed. In patients treated with enalapril plus a thiazide diuretic, there was essentially no change in serum potassium (see PRECAUTIONS). Removal of angiotensin II negative feedback on renin secretion leads to increased plasma renin activity.
ACE is identical to kininase, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effects of enalaprilat remains to be elucidated.
While the mechanism through which enalaprilat lowers blood pressure is believed to be primarily suppression of the renin-angiotensin-aldosterone system, enalaprilat has anti-hypertensive activity even in patients with low-renin hypertension. In clinical studies, black hypertensive patients (usually a low-renin hypertensive population) had a smaller average response to enalaprilat monotherapy than non-black patients.
Pharmacokinetics and Metabolism
Following intravenous administration of a single dose, the serum concentration profile of enalaprilat is polyexponential with a prolonged terminal phase, apparently representing a small fraction of the administered dose that has been bound to ACE. The amount bound does not increase with dose, indicating a saturable site of binding. The effective half-life for accumulation of enalaprilat, as determined from oral administration of multiple doses of enalapril maleate, is approximately 11 hours. Excretion of enalaprilat is primarily renal with more than 90 percent of an administered dose recovered in the urine as unchanged drug within 24 hours. Enalaprilat is poorly absorbed following oral administration.
The disposition of enalaprilat in patients with renal insufficiency is similar to that in patients with normal renal function until the glomerular filtration rate is 30 mL/min or less. With glomerular filtration rate ≤30 mL/min, peak and trough enalaprilat levels increase, time to peak concentration increases and time to steady state may be delayed. The effective half-life of enalaprilat is prolonged at this level of renal insufficiency (see DOSAGE AND ADMINISTRATION). Enalaprilat is dialyzable at the rate of 62 mL/min.
Studies in dogs indicate that enalaprilat does not enter the brain, and that enalapril crosses the blood-brain barrier poorly, if at all. Multiple doses of enalapril maleate in rats do not result in accumulation in any tissues. Milk in lactating rats contains radioactivity following administration of 14C enalapril maleate. Radioactivity was found to cross the placenta following administration of labeled drug to pregnant hamsters.
Pharmacodynamics
Enalaprilat injection results in the reduction of both supine and standing systolic and diastolic blood pressure, usually with no orthostatic component. Symptomatic postural hypotension is therefore infrequent, although it might be anticipated in volume-depleted patients (see WARNINGS). The onset of action usually occurs within fifteen minutes of administration with the maximum effect occurring within one to four hours. The abrupt withdrawal of enalaprilat has not been associated with a rapid increase in blood pressure. The duration of hemodynamic effects appears to be dose-related. However, for the recommended dose, the duration of action in most patients is approximately six hours.
Following administration of enalapril, there is an increase in renal blood flow; glomerular filtration rate is usually unchanged. The effects appear to be similar in patients with renovascular hypertension.
In a clinical pharmacology study, indomethacin or sulindac was administered to hypertensive patients receiving enalapril maleate. In this study there was no evidence of a blunting of the antihypertensive action of enalapril maleate (see PRECAUTIONS, Drug Interactions).
INDICATIONS AND USAGE
Enalaprilat injection is indicated for the treatment of hypertension when oral therapy is not practical.
Enalaprilat injection has been studied with only one other antihypertensive agent, furosemide, which showed approximately additive effects on blood pressure. Enalapril, the pro-drug of enalaprilat, has been used extensively with a variety of other antihypertensive agents, without apparent difficulty except for occasional hypotension.
In using enalaprilat injection, consideration should be given to the fact that another angiotensin-converting enzyme inhibitor, captopril, has caused agranulocytosis, particularly in patients with renal impairment or collagen vascular disease, and that available data are insufficient to show that enalaprilat injection does not have a similar risk (see WARNINGS).
In considering use of enalaprilat, it should be noted that in controlled clinical trials ACE inhibitors have an effect on blood pressure that is less in black patients than in non-blacks. In addition, it should be noted that black patients receiving ACE inhibitors have been reported to have a higher incidence of angioedema compared to non-blacks (see WARNINGS, Angioedema).
CONTRAINDICATIONS
Enalaprilat injection is contraindicated in patients who are hypersensitive to any component of this product and in patients with a history of angioedema related to previous treatment with an angiotensin-converting enzyme inhibitor and in patients with hereditary or idiopathic angioedema.
WARNINGS
Hypotension
Excessive hypotension is rare in uncomplicated hypertensive patients but is a possible consequence of the use of enalaprilat especially in severely salt/volume depleted persons such as those treated vigorously with diuretics or patients on dialysis. Patients at risk for excessive hypotension, sometimes associated with oliguria and/or progressive azotemia, and rarely with acute renal failure and/or death, include those with the following conditions or characteristics: heart failure, hyponatremia, high dose diuretic therapy, recent intensive diuresis or increase in diuretic dose, renal dialysis, or severe volume and/or salt depletion of any etiology. It may be advisable to eliminate the diuretic, reduce the diuretic dose or increase salt intake cautiously before initiating therapy with enalaprilat injection in patients at risk for excessive hypotension who are able to tolerate such adjustments (see PRECAUTIONS, Drug Interactions;ADVERSE REACTIONS, and DOSAGE AND ADMINISTRATION). In patients with heart failure, with or without associated renal insufficiency, excessive hypotension has been observed and may be associated with oliguria and/or progressive azotemia, and rarely with acute renal failure and/or death. Because of the potential for an excessive fall in blood pressure especially in these patients, therapy should be followed closely whenever the dose of enalaprilat is adjusted and/or diuretic is increased. Similar considerations may apply to patients with ischemic heart or cerebrovascular disease, in whom an excessive fall in blood pressure could result in a myocardial infarction or cerebrovascular accident.
If hypotension occurs, the patient should be placed in the supine position and, if necessary, receive an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further doses, which usually can be given without difficulty once the blood pressure has increased after volume expansion.
Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors (including enalaprilat injection) may be subject to a variety of adverse reactions, some of them serious.
Angioedema: Angioedema of the face, extremities, lips, tongue, glottis and/or larynx has been reported in patients treated with angiotensin-converting enzyme inhibitors, including enalaprilat. This may occur at any time during treatment. In such cases, enalaprilat injection should be promptly discontinued and appropriate therapy and monitoring should be provided until complete and sustained resolution of signs and symptoms has occurred. In instances where swelling has been confined to the face and lips, the condition has generally resolved without treatment, although antihistamines have been useful in relieving symptoms. Angioedema associated with laryngeal edema may be fatal. Where there is involvement of the tongue, glottis or larynx, likely to cause airway obstruction, appropriate therapy, e.g., subcutaneous epinephrine solution 1:1000 (0.3 mL to 0.5 mL) and/or measures necessary to ensure a patent airway, should be promptly provided (see ADVERSE REACTIONS).
Patients with a history of angioedema unrelated to ACE inhibitor therapy may be at increased risk of angioedema while receiving an ACE inhibitor (see also INDICATIONS AND USAGE and CONTRAINDICATIONS).
Anaphylactoid reactions during desensitization: Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions were avoided when ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Anaphylactoid reactions during membrane exposure: Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
Neutropenia/Agranulocytosis
Another angiotensin-converting enzyme inhibitor, captopril, has been shown to cause agranulocytosis and bone marrow depression, rarely in uncomplicated patients but more frequently in patients with renal impairment especially if they also have a collagen vascular disease. Available data from clinical trials of enalapril are insufficient to show that enalapril does not cause agranulocytosis in similar rates. Marketing experience has revealed cases of neutropenia, or agranulocytosis in which a causal relationship to enalapril cannot be excluded. Periodic monitoring of white blood cell counts in patients with collagen vascular disease and renal disease should be considered.
Hepatic Failure
Rarely, ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis, and (sometimes) death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice or marked elevations of hepatic enzymes should discontinue the ACE inhibitor and receive appropriate medical follow-up.
Fetal/Neonatal Morbidity and Mortality
ACE inhibitors can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature. When pregnancy is detected, ACE inhibitors should be discontinued as soon as possible.
The use of ACE inhibitors during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to the ACE-inhibitor exposure.
These adverse effects do not appear to have resulted from intrauterine ACE-inhibitor exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to ACE inhibitors only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should make every effort to discontinue the use of enalaprilat injection as soon as possible.
Rarely (probably less often than once in every thousand pregnancies), no alternative to ACE inhibitors will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses, and serial ultrasound examinations should be performed to assess the intraamniotic environment.
If oligohydramnios is observed, enalaprilat injection should be discontinued unless it is considered lifesaving for the mother. Contraction stress testing (CST), a non-stress test (NST), or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Infants with histories of in utero exposure to ACE inhibitors should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as means of reversing hypotension and/or substituting for disordered renal function. Enalapril, which crosses the placenta, has been removed from neonatal circulation by peritoneal dialysis with some clinical benefit, and theoretically may be removed by exchange transfusion, although there is no experience with the latter procedure.
No teratogenic effects of oral enalapril were seen in studies of pregnant rats and rabbits. On a body surface area basis, the doses used were 57 times and 12 times, respectively, the maximum recommended human daily dose (MRHDD).
PRECAUTIONS
General
Aortic Stenosis/Hypertrophic Cardiomyopathy: As with all vasodilators, enalapril should be given with caution to patients with obstruction in the outflow tract of the left ventricle.
Impaired Renal Function: As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. In patients with severe heart failure whose renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with angiotensin-converting enzyme inhibitors, including enalapril or enalaprilat, may be associated with oliguria and/or progressive azotemia and rarely with acute renal failure and/or death.
In clinical studies in hypertensive patients with unilateral or bilateral renal artery stenosis, increases in blood urea nitrogen and serum creatinine were observed in 20 percent of patients receiving enalapril. These increases were almost always reversible upon discontinuation of enalapril or enalaprilat and/or diuretic therapy. In such patients renal function should be monitored during the first few weeks of therapy.
Some hypertensive patients with no apparent pre-existing renal vascular disease have developed increases in blood urea and serum creatinine, usually minor and transient, especially when enalaprilat has been given concomitantly with a diuretic. This is more likely to occur in patients with pre-existing renal impairment. Dosage reduction of enalaprilat and/or discontinuation of the diuretic may be required.
Evaluation of the hypertensive patient should always include assessment of renal function (see DOSAGE AND ADMINISTRATION).
Hyperkalemia: Elevated serum potassium (greater than 5.7 mEq/L) was observed in approximately one percent of hypertensive patients in clinical trials receiving enalapril. In most cases these were isolated values which resolved despite continued therapy. Hyperkalemia was a cause of discontinuation of therapy in 0.28 percent of hypertensive patients. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus, and the concomitant use of potassium-sparing agents or potassium supplements, which should be used cautiously, if at all, with enalaprilat injection (see Drug Interactions).
Cough: Presumably due to the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. ACE inhibitor-induced cough should be considered in the differential diagnosis of cough.
Surgery/Anesthesia: In patients undergoing major surgery or during anesthesia with agents that produce hypotension, enalapril may block angiotensin II formation secondary to compensatory renin release. If hypotension occurs and is considered to be due to this mechanism, it can be corrected by volume expansion.
Drug Interactions
Hypotension-Patients on Diuretic Therapy: Patients on diuretics and especially those in whom diuretic therapy was recently instituted, may occasionally experience an excessive reduction of blood pressure after initiation of therapy with enalaprilat. The possibility of hypotensive effects with enalaprilat can be minimized by administration of an intravenous infusion of normal saline, discontinuing the diuretic or increasing the salt intake prior to initiation of treatment with enalaprilat. If it is necessary to continue the diuretic, provide close medical supervision for at least one hour after the initial dose of enalaprilat (see WARNINGS).
Agents Causing Renin Release: The antihypertensive effect of enalaprilat injection appears to be augmented by antihypertensive agents that cause renin release (e.g., diuretics).
Non-Steroidal Anti-Inflammatory Agents: In some patients with compromised renal function who are being treated with non-steroidal anti-inflammatory drugs, the coadministration of enalapril may result in a further deterioration of renal function. These effects are usually reversible.
In a clinical pharmacology study, indomethacin or sulindac was administered to hypertensive patients receiving enalapril maleate. In this study there was no evidence of a blunting of the antihypertensive action of enalapril maleate. However, reports suggest that NSAIDs may diminish the antihypertensive effect of ACE inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE inhibitors.
Other Cardiovascular Agents: Enalaprilat injection has been used concomitantly with digitalis, beta adrenergic-blocking agents, methyldopa, nitrates, calcium-blocking agents, hydralazine and prazosin without evidence of clinically significant adverse interactions.
Agents Increasing Serum Potassium: Enalaprilat injection attenuates potassium loss caused by thiazide-type diuretics. Potassium-sparing diuretics (e.g., spironolactone, triamterene, or amiloride), potassium supplements, or potassium-containing salt substitutes may lead to significant increases in serum potassium. Therefore, if concomitant use of these agents is indicated because of demonstrated hypokalemia, they should be used with caution and with frequent monitoring of serum potassium.
Lithium: Lithium toxicity has been reported in patients receiving lithium concomitantly with drugs which cause elimination of sodium, including ACE inhibitors. A few cases of lithium toxicity have been reported in patients receiving concomitant enalapril and lithium and were reversible upon discontinuation of both drugs. It is recommended that serum lithium levels be monitored frequently if enalapril is administered concomitantly with lithium.
Gold: Nitritoid reactions (symptoms include facial flushing, nausea, vomiting and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE inhibitor therapy.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been done with enalaprilat injection.
Enalaprilat injection is the bioactive form of its ethyl ester, enalapril maleate. There was no evidence of a tumorigenic effect when enalapril was administered for 106 weeks to male and female rats at doses up to 90 mg/kg/day or for 94 weeks to male and female mice at doses up to 90 and 180 mg/kg/day, respectively. These doses are 26 times (in rats and female mice) and 13 times (in male mice) the maximum recommended human daily dose (MRHDD) when compared on a body surface area basis.
Enalaprilat injection was not mutagenic in the Ames microbial mutagen test with or without metabolic activation. Enalapril showed no drug-related changes in the following genotoxicity studies: rec-assay, reverse mutation assay with E. coli, sister chromatid exchange with cultured mammalian cells, the micronucleus test with mice, and in an in vivo cytogenic study using mouse bone marrow. There were no adverse effects on reproductive performance of male and female rats treated with up to 90 mg/kg/day of enalapril (26 times the MRHDD when compared on a body surface area basis).
Pregnancy
Pregnancy Categories C (first trimester) and D (second and third trimesters) See WARNINGS, Fetal/Neonatal Morbidity and Mortality.
Nursing Mothers
Enalapril and enalaprilat have been detected in human breast milk. Because of the potential for serious adverse reactions in nursing infants from enalapril, a decision should be made whether to discontinue nursing or to discontinue enalaprilat injection, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of enalaprilat did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection. Evaluation of the hypertensive patient should always include assessment of renal function (see DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
Enalaprilat has been found to be generally well tolerated in controlled clinical trials involving 349 patients (168 with hypertension, 153 with congestive heart failure and 28 with coronary artery disease). The most frequent clinically significant adverse experience was hypotension (3.4 percent), occurring in eight patients (5.2 percent) with congestive heart failure, three (1.8 percent) with hypertension and one with coronary artery disease. Other adverse experiences occurring in greater than one percent of patients were: headache (2.9 percent) and nausea (1.1 percent).
Adverse experiences occurring in 0.5 to 1 percent of patients in controlled clinical trials included: myocardial infarction, fatigue, dizziness, fever, rash and constipation.
Angioedema: Angioedema has been reported in patients receiving enalaprilat, with an incidence higher in black than in non-black patients. Angioedema associated with laryngeal edema may be fatal. If angioedema of the face, extremities, lips, tongue, glottis and/or larynx occurs, treatment with enalaprilat should be discontinued and appropriate therapy instituted immediately (see WARNINGS).
Cough: See PRECAUTIONS, Cough.
Enalapril Maleate
Since enalapril is converted to enalaprilat, those adverse experiences associated with enalapril might also be expected to occur with enalaprilat injection.
The following adverse experiences have been reported with enalapril and, within each category, are listed in order of decreasing severity.
Body as a Whole: Syncope, orthostatic effects, anaphylactoid reactions (see WARNINGS, Anaphylactoid reactions during membrane exposure), chest pain, abdominal pain, asthenia.
Cardiovascular: Cardiac arrest; myocardial infarction or cerebrovascular accident, possibly secondary to excessive hypotension in high risk patients (see WARNINGS,Hypotension); pulmonary embolism and infarction; pulmonary edema; rhythm disturbances including atrial tachycardia and bradycardia; atrial fibrillation; orthostatic hypotension; angina pectoris; palpitation, Raynaud's phenomenon.
Digestive: Ileus, pancreatitis, hepatic failure, hepatitis (hepatocellular [proven on rechallenge] or cholestatic jaundice) (see WARNINGS, Hepatic Failure), melena, diarrhea, vomiting, dyspepsia, anorexia, glossitis, stomatitis, dry mouth.
Hematologic: Rare cases of neutropenia, thrombocytopenia and bone marrow depression.
Musculoskeletal: Muscle cramps.
Nervous/Psychiatric: Depression, vertigo, confusion, ataxia, somnolence, insomnia, nervousness, peripheral neuropathy (e.g., paresthesia, dysesthesia), dream abnormality.
Respiratory: Bronchospasm, dyspnea, pneumonia, bronchitis, cough, rhinorrhea, sore throat and hoarseness, asthma, upper respiratory infection, pulmonary infiltrates, eosinophilic pneumonitis.
Skin: Exfoliative dermatitis, toxic epidermal necrolysis, Stevens-Johnson syndrome, pemphigus, herpes zoster, erythema multiforme, urticaria, pruritus, alopecia, flushing, diaphoresis, photosensitivity.
Special Senses: Blurred vision, taste alteration, anosmia, tinnitus, conjunctivitis, dry eyes, tearing.
Urogenital: Renal failure, oliguria, renal dysfunction (see PRECAUTIONS and DOSAGE AND ADMINISTRATION), urinary tract infection, flank pain, gynecomastia, impotence.
Miscellaneous: A symptom complex has been reported which may include some or all of the following: a positive ANA, an elevated erythrocyte sedimentation rate, arthralgia/arthritis, myalgia/myositis, fever, serositis, vasculitis, leukocytosis, eosinophilia, photosensitivity, rash and other dermatologic manifestations.
Hypotension: Combining the results of clinical trials in patients with hypertension or congestive heart failure, hypotension (including postural hypotension, and other orthostatic effects) was reported in 2.3 percent of patients following the initial dose of enalapril or during extended therapy. In the hypertensive patients, hypotension occurred in 0.9 percent and syncope occurred in 0.5 percent of patients. Hypotension or syncope was a cause for discontinuation of therapy in 0.1 percent of hypertensive patients (see WARNINGS).
Fetal/Neonatal Morbidity and Mortality: See WARNINGS, Fetal/Neonatal Morbidity and Mortality.
Clinical Laboratory Test Findings
Serum Electrolytes: Hyperkalemia (see PRECAUTIONS), hyponatremia.
Creatinine, Blood Urea Nitrogen: In controlled clinical trials minor increases in blood urea nitrogen and serum creatinine, reversible upon discontinuation of therapy, were observed in about 0.2 percent of patients with essential hypertension treated with enalapril alone. Increases are more likely to occur in patients receiving concomitant diuretics or in patients with renal artery stenosis (see PRECAUTIONS).
Hematology: Small decreases in hemoglobin and hematocrit (mean decreases of approximately 0.3 g percent and 1 vol percent, respectively) occur frequently in hypertensive patients treated with enalapril but are rarely of clinical importance unless another cause of anemia coexists. In clinical trials, less than 0.1 percent of patients discontinued therapy due to anemia. Hemolytic anemia, including cases of hemolysis in patients with G-6-PD deficiency, has been reported; a causal relationship to enalapril cannot be excluded.
Liver Function Tests: Elevations of liver enzymes and/or serum bilirubin have occurred (see WARNINGS, Hepatic Failure).
OVERDOSAGE
In clinical studies, some hypertensive patients received a maximum dose of 80 mg of enalaprilat intravenously over a fifteen minute period. At this high dose, no adverse effects beyond those as associated with the recommended dosages were observed.
A single intravenous dose of ≤4,167 mg/kg of enalaprilat was associated with lethality in female mice. No lethality occurred after an intravenous dose of 3,472 mg/kg. The most likely manifestation of overdosage would be hypotension, for which the usual treatment would be intravenous infusion of normal saline solution.
Enalaprilat may be removed from general circulation by hemodialysis and has been removed from neonatal circulation by peritoneal dialysis (see WARNINGS, Anaphylactoid reactions during membrane exposure).
DOSAGE AND ADMINISTRATION
FOR INTRAVENOUS ADMINISTRATION ONLY
The dose in hypertension is 1.25 mg every six hours administered intravenously over a five minute period. A clinical response is usually seen within 15 minutes. Peak effects after the first dose may not occur for up to four hours after dosing. The peak effects of the second and subsequent doses may exceed those of the first.
No dosage regimen for enalaprilat injection has been clearly demonstrated to be more effective in treating hypertension than 1.25 mg every six hours. However, in controlled clinical studies in hypertension, doses as high as 5 mg every six hours were well tolerated for up to 36 hours. There has been inadequate experience with doses greater than 20 mg per day.
In studies of patients with hypertension, enalaprilat has not been administered for periods longer than 48 hours. In other studies, patients have received enalaprilat for as long as seven days.
The dose for patients being converted to enalaprilat injection from oral therapy for hypertension with enalapril maleate is 1.25 mg every six hours. For conversion from intravenous to oral therapy, the recommended initial dose of oral enalapril maleate, is 5 mg once a day with subsequent dosage adjustments as necessary.
Patients On Diuretic Therapy
For patients on diuretic therapy the recommended starting dose for hypertension is 0.625 mg administered intravenously over a five minute period; also see below, Patients at Risk of Excessive Hypotension. A clinical response is usually seen within 15 minutes. Peak effects after the first dose may not occur for up to four hours after dosing, although most of the effect is usually apparent within the first hour. If after one hour there is an inadequate clinical response, the 0.625 mg dose may be repeated. Additional doses of 1.25 mg may be administered at six hour intervals.
For conversion from intravenous to oral therapy, the recommended initial dose of oral enalapril maleate for patients who have responded to 0.625 mg of enalaprilat every six hours is 2.5 mg once a day with subsequent dosage adjustment as necessary.
Dosage Adjustment in Renal Impairment
The usual dose of 1.25 mg of enalaprilat injection every six hours is recommended for patients with a creatinine clearance > 30 mL/min (serum creatinine of up to approximately 3 mg/dL). For patients with creatinine clearance ≤ 30 mL/min (serum creatinine ≥ 3 mg/dL), the initial dose is 0.625 mg (see WARNINGS).
If after one hour there is an inadequate clinical response, the 0.625 mg dose may be repeated. Additional doses of 1.25 mg may be administered at six hour intervals.
For dialysis patients, see below, Patients at Risk of Excessive Hypotension.
For conversion from intravenous to oral therapy, the recommended initial dose of oral enalapril maleate is 5 mg once a day for patients with creatinine clearance > 30 mL/min and 2.5 mg once daily for patients with creatinine clearance ≤ 30 mL/min. Dosage should then be adjusted according to blood pressure response.
Patients at Risk of Excessive Hypotension
Hypertensive patients at risk of excessive hypotension include those with the following concurrent conditions or characteristics: heart failure, hyponatremia, high dose diuretic therapy, recent intensive diuresis or increase in diuretic dose, renal dialysis, or severe volume and/or salt depletion of any etiology (see WARNINGS). Single doses of enalaprilat injection as low as 0.2 mg have produced excessive hypotension in normotensive patients with these diagnoses. Because of the potential for an extreme hypotensive response in these patients, therapy should be started under very close medical supervision. The starting dose should be no greater than 0.625 mg administered intravenously over a period of no less than five minutes and preferably longer (up to one hour).
Patients should be followed closely whenever the dose of enalaprilat injection is adjusted and/or diuretic is increased.
Administration
Enalaprilat injection should be administered as a slow intravenous infusion, as indicated above. It may be administered as provided or diluted with up to 50 mL of a compatible diluent.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to use whenever solution and container permit.
Compatibility and Stability
Enalaprilat injection as supplied and mixed with the following intravenous diluents has been found to maintain full activity for 24 hours at room temperature:
5 percent Dextrose Injection
0.9 percent Sodium Chloride Injection
0.9 percent Sodium Chloride Injection in 5 percent Dextrose
5 percent Dextrose in Lactated Ringer's Injection
ISOLYTE***E (*** Registered trademark of B. Braun)
HOW SUPPLIED
Enalaprilat Injection USP, 1.25 mg/mL is a clear, colorless solution free from visible particulate matter and is available as follows:
| Enalaprilat Injection USP, 1.25 mg/mL | NDC 43598-078-58 | 10 vials per carton |
| Enalaprilat Injection USP, 2.5 mg/2 mL | NDC 43598-169-58 | 10 vials per carton |
Storage
Store at 20°C to 25°C (68°F to 77°F) [See USP Controlled Room Temperature].
To report SUSPECTED ADVERSE REACTIONS, contact Dr. Reddy’s Laboratories Inc., at 1-888-375-3784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Distributor:
Dr. Reddy’s Laboratories Inc.,
Princeton, NJ 08540
Made in India
Revised: 04/2023