FULL PRESCRIBING INFORMATION
WARNING: SEVERE ACUTE EXACERBATIONS OF HEPATITIS B, PATIENTS CO-INFECTED WITH HIV AND HBV, AND LACTIC ACIDOSIS AND HEPATOMEGALY
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy, including entecavir. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)] .
Limited clinical experience suggests there is a potential for the development of resistance to HIV (human immunodeficiency virus) nucleoside reverse transcriptase inhibitors if entecaviris used to treat chronic hepatitis B virus (HBV) infection in patients with HIV infection that is not being treated. Therapy with entecaviris not recommended for HIV/HBV co-infected patients who are not also receiving highly active antiretroviral therapy (HAART) [see Warnings and Precautions (5.2)] .
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogue inhibitors alone or in combination with antiretrovirals [see Warnings and Precautions (5.3)] .
1 INDICATIONS AND USAGE
Entecavir tablets are indicated for the treatment of chronic hepatitis B virus infection in adults with evidence of active viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
The following points should be considered when initiating therapy with entecavir tablets:
- In adult patients, this indication is based on clinical trial data in nucleoside-inhibitor-treatment-naïve and lamivudine-resistant subjects with HBeAg-positive and HBeAg-negative HBV infection and compensated liver disease and a more limited number of subjects with decompensated liver disease [see Clinical Studies (14.1)] .
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
2 DOSAGE AND ADMINISTRATION
2.1 Timing of Administration
Entecavir tablets should be administered on an empty stomach (at least 2 hours after a meal and 2 hours before the next meal).
2.2 Recommended Dosage in Adults
Compensated Liver Disease
The recommended dose of entecavir tablets for chronic hepatitis B virus infection in nucleoside-inhibitor-treatment-naïve adults and adolescents 16 years of age and older is 0.5 mg once daily.
The recommended dose of entecavir tablets in adults and adolescents (at least 16 years of age) with a history of hepatitis B viremia while receiving lamivudine or known lamivudine or telbivudine resistance substitutions rtM204I/V with or without rtL180M, rtL80I/V, or rtV173L is 1 mg once daily.
Decompensated Liver Disease
The recommended dose of entecavir tablets for chronic hepatitis B virus infection in adults with decompensated liver disease is 1 mg once daily.
2.3 Recommended Dosage in Pediatric Patients
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
2.4 Renal Impairment
In adult subjects with renal impairment, the apparent oral clearance of entecavir decreased as creatinine clearance decreased [see Clinical Pharmacology (12.3)] . Dosage adjustment is recommended for patients with creatinine clearance less than 50 mL/min, including patients on hemodialysis or continuous ambulatory peritoneal dialysis (CAPD), as shown in Table 2. The once-daily dosing regimens are preferred.
|
Table 2: Recommended Dosage of Entecavir Tablets in Adult Patients with Renal Impairment |
||
|
Creatinine Clearance
|
Usual Dose (0.5 mg) |
Lamivudine-Refractory or
|
|
50 or greater |
0.5 mg once daily |
1 mg once daily |
|
30 to less than 50 |
0.5 mg every 48 hours |
0.5 mg once daily
|
|
10 to less than 30 |
0.5 mg every 72 hours |
1 mg every 72 hours |
|
less than 10
|
0.5 mg every 7 days |
1 mg every 7 days |
|
* If administered on a hemodialysis day, administer entecavir tablets after the hemodialysis session. |
||
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
3 DOSAGE FORMS AND STRENGTHS
- Entecavir tablets 0.5 mg are white to off-white, oval, film-coated, biconvex bevel edged, unscored tablets, debossed with “AN” on one side and “446” on the other side.
- Entecavir tablets 1 mg are pink, oval, film-coated, biconvex bevel edged, unscored tablets, debossed with “AN” on one side and “449” on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Severe Acute Exacerbations of Hepatitis B
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy, including entecavir [see Adverse Reactions (6.1)] . Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
5.2 Patients Co-infected with HIV and HBV
Entecavir has not been evaluated in HIV/HBV co-infected patients who were not simultaneously receiving effective HIV treatment. Limited clinical experience suggests there is a potential for the development of resistance to HIV nucleoside reverse transcriptase inhibitors if entecavir is used to treat chronic hepatitis B virus infection in patients with HIV infection that is not being treated [see Microbiology (12.4)] . Therefore, therapy with entecavir is not recommended for HIV/HBV co-infected patients who are not also receiving HAART. Before initiating entecavir therapy, HIV antibody testing should be offered to all patients. Entecavir has not been studied as a treatment for HIV infection and is not recommended for this use.
5.3 Lactic Acidosis and Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogue inhibitors, including entecavir, alone or in combination with antiretrovirals. A majority of these cases have been in women. Obesity and prolonged nucleoside inhibitor exposure may be risk factors. Particular caution should be exercised when administering nucleoside analogue inhibitors to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors.
Lactic acidosis with entecavir use has been reported, often in association with hepatic decompensation, other serious medical conditions, or drug exposures. Patients with decompensated liver disease may be at higher risk for lactic acidosis. Treatment with entecavir should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Exacerbations of hepatitis after discontinuation of treatment [see Boxed Warning, Warnings and Precautions (5.1)] .
- Lactic acidosis and severe hepatomegaly with steatosis [see Boxed Warning, Warnings and Precautions (5.3)] .
6.1 Clinical Trial Experience in Adults
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Compensated Liver Disease
Assessment of adverse reactions is based on four studies (AI463014, AI463022, AI463026 and AI463027) in which 1720 subjects with chronic hepatitis B virus infection and compensated liver disease received double-blind treatment with entecavir 0.5 mg/day (n=679), entecavir 1 mg/day (n=183), or lamivudine (n=858) for up to 2 years. Median duration of therapy was 69 weeks for entecavir-treated subjects and 63 weeks for lamivudine-treated subjects in Studies AI463022 and AI463027 and 73 weeks for entecavir-treated subjects and 51 weeks for lamivudine-treated subjects in Studies AI463026 and AI463014. The safety profiles of entecavir and lamivudine were comparable in these studies.
The most common adverse reactions of any severity (≥3%) with at least a possible relation to study drug for entecavir-treated subjects were headache, fatigue, dizziness and nausea. The most common adverse reactions among lamivudine-treated subjects were headache, fatigue and dizziness. One percent of entecavir-treated subjects in these four studies compared with 4% of lamivudine-treated subjects discontinued for adverse events or abnormal laboratory test results.
Clinical adverse reactions of moderate-severe intensity and considered at least possibly related to treatment occurring during therapy in four clinical studies in which entecavir was compared with lamivudine are presented in Table 3.
|
Table 3: Clinical Adverse Reactionsa of Moderate-Severe Intensity (Grades 2 to 4) Reported in Four Entecavir Clinical Trials
Through 2 Years |
||||
| Nucleoside-Inhibitor-Naïve b | Lamivudine-Refractory c | |||
| Entecavir | Lamivudine | Entecavir | Lamivudine | |
| Body System/ | 0.5 mg | 100 mg | 1 mg | 100 mg |
| Adverse Reaction | n=679 | n=668 | n=183 | n=190 |
| Any Grade 2 to 4 adverse reaction a | 15% | 18% | 22% | 23% |
| Gastrointestinal | ||||
| Diarrhea | <1% | 0 | 1% | 0 |
| Dyspepsia | <1% | <1% | 1% | 0 |
| Nausea | <1% | <1% | <1% | 2% |
| Vomiting | <1% | <1% | <1% | 0 |
| General | ||||
| Fatigue | 1% | 1% | 3% | 3% |
| Nervous System | ||||
| Headache | 2% | 2% | 4% | 1% |
| Dizziness | <1% | <1% | 0 | 1% |
| Somnolence | <1% | <1% | 0 | 0 |
| Psychiatric | ||||
| Insomnia | <1% | <1% | 0 | <1% |
| a Includes events of possible, probable, certain, or unknown relationship to treatment regimen. | ||||
| b Studies AI463022 and AI463027. | ||||
|
c Includes Study AI463026 and the entecavir 1 mg and lamivudine treatment arms of Study AI463014, a Phase 2 multinational,
randomized, double-blind study of three doses of entecavir (0.1, 0.5 and 1 mg) once daily versus continued lamivudine 100 mg once daily for up to 52 weeks in subjects who experienced recurrent viremia on lamivudine therapy. |
||||
Laboratory Abnormalities
Frequencies of selected treatment-emergent laboratory abnormalities reported during therapy in four clinical trials of entecavir compared with lamivudine are listed in Table 4.
| Table 4: Selected Treatment-Emergent a Laboratory Abnormalities Reported in Four Entecavir Clinical Trials Through 2 Years | ||||
| Nucleoside-Inhibitor-Naïveb | Lamivudine-Refractoryc | |||
| Entecavir | Lamivudine | Entecavir | Lamivudine | |
| 0.5 mg | 100 mg | 1 mg | 100 mg | |
| Test | n=679 | n=668 | n=183 | n=190 |
| Any Grade 3 to 4 laboratory abnormality d | 35% | 36% | 37% | 45% |
| ALT >10 x ULN and >2 x baseline | 2% | 4% | 2% | 11% |
| ALT >5 x ULN | 11% | 16% | 12% | 24% |
| Albumin <2.5 g/dL | <1% | <1% | 0 | 2% |
| Total bilirubin >2.5 x ULN | 2% | 2% | 3% | 2% |
| Lipase ≥2.1 x ULN | 7% | 6% | 7% | 7% |
| Creatinine >3 x ULN | 0 | 0 | 0 | 0 |
| Confirmed creatinine increase ≥0.5 mg/dL | 1% | 1% | 2% | 1% |
| Hyperglycemia, fasting >250 mg/dL | 2% | 1% | 3% | 1% |
| Glycosuria e | 4% | 3% | 4% | 6% |
| Hematuria f | 9% | 10% | 9% | 6% |
| Platelets <50,000/mm 3 | <1% | <1% | <1% | <1% |
|
a On-treatment value worsened from baseline to Grade 3 or Grade 4 for all parameters except albumin (any on-treatment value <2.5 g/dL),
confirmed creatinine increase ≥0.5 mg/dL, and ALT >10 x ULN and >2 x baseline. |
||||
| b Studies AI463022 and AI463027. | ||||
|
c Includes Study AI463026 and the entecavir 1 mg and lamivudine treatment arms of Study AI463014, a Phase 2 multinational,
randomized, double-blind study of three doses of entecavir (0.1, 0.5 and 1 mg) once daily versus continued lamivudine 100 mg once daily for up to 52 weeks in subjects who experienced recurrent viremia on lamivudine therapy. |
||||
| d Includes hematology, routine chemistries, renal and liver function tests, pancreatic enzymes and urinalysis. | ||||
| e Grade 3 = 3+, large, ≥500 mg/dL; Grade 4 = 4+, marked, severe. | ||||
| f Grade 3 = 3+, large; Grade 4 = ≥4+, marked, severe, many. | ||||
| ULN=upper limit of normal. | ||||
Among entecavir-treated subjects in these studies, on-treatment ALT elevations greater than 10 times the upper limit of normal (ULN) and greater than 2 times baseline generally resolved with continued treatment. A majority of these exacerbations were associated with a ≥2 log 10/mL reduction in viral load that preceded or coincided with the ALT elevation. Periodic monitoring of hepatic function is recommended during treatment.
Exacerbations of Hepatitis after Discontinuation of Treatment
An exacerbation of hepatitis or ALT flare was defined as ALT greater than 10 times ULN and greater than 2 times the subject’s reference level (minimum of the baseline or last measurement at end of dosing). For all subjects who discontinued treatment (regardless of reason), Table 5 presents the proportion of subjects in each study who experienced post-treatment ALT flares. In these studies, a subset of subjects was allowed to discontinue treatment at or after 52 weeks if they achieved a protocol-defined response to therapy. If entecavir is discontinued without regard to treatment response, the rate of post-treatment flares could be higher [see also Warnings and Precautions (5.1)] .
| Table 5: Exacerbations of Hepatitis During Off-Treatment Follow-up, Subjects in Studies AI463022, AI463027 and AI463026 | ||
| Subjects with ALT Elevations >10 x ULN and >2 x Reference a | ||
| Entecavir | Lamivudine | |
| Nucleoside-inhibitor-naïve | ||
| HBeAg-positive | 4/174 (2%) | 13/147 (9%) |
| HBeAg-negative | 24/302 (8%) | 30/270 (11%) |
| Lamivudine-refractory | 6/52 (12%) | 0/16 |
|
a Reference is the minimum of the baseline or last measurement at end of dosing. Median time to off-treatment exacerbation was 23 weeks
for entecavir-treated subjects and 10 weeks for lamivudine-treated subjects. |
||
Decompensated Liver Disease
Study AI463048 was a randomized, open-label study of entecavir 1 mg once daily versus adefovir dipivoxil 10 mg once daily given for up to 48 weeks in adult subjects with chronic HBV infection and evidence of hepatic decompensation, defined as a Child-Turcotte-Pugh (CTP) score of 7 or higher [see Clinical Studies (14.1)] . Among the 102 subjects receiving entecavir, the most common treatment-emergent adverse events of any severity, regardless of causality, occurring through Week 48 were peripheral edema (16%), ascites (15%), pyrexia (14%), hepatic encephalopathy (10%) and upper respiratory infection (10%). Clinical adverse reactions not listed in Table 2 that were observed through Week 48 include blood bicarbonate decreased (2%) and renal failure (<1%).
Eighteen of 102 (18%) subjects treated with entecavir and 18/89 (20%) subjects treated with adefovir dipivoxil died during the first 48 weeks of therapy. The majority of deaths (11 in the entecavir group and 16 in the adefovir dipivoxil group) were due to liver-related causes such as hepatic failure, hepatic encephalopathy, hepatorenal syndrome and upper gastrointestinal hemorrhage. The rate of hepatocellular carcinoma (HCC) through Week 48 was 6% (6/102) for subjects treated with entecavir and 8% (7/89) for subjects treated with adefovir dipivoxil. Five percent of subjects in either treatment arm discontinued therapy due to an adverse event through Week 48.
No subject in either treatment arm experienced an on-treatment hepatic flare (ALT >2 x baseline and >10 x ULN) through Week 48. Eleven of 102 (11%) subjects treated with entecavir and 11/89 (13%) subjects treated with adefovir dipivoxil had a confirmed increase in serum creatinine of 0.5 mg/dL through Week 48.
HIV/HBV Co-infected
The safety profile of entecavir 1 mg (n=51) in HIV/HBV co-infected subjects enrolled in Study AI463038 was similar to that of placebo (n=17) through 24 weeks of blinded treatment and similar to that seen in non-HIV infected subjects [see Warnings and Precautions (5.2)] .
6.2 Clinical Trial Experience in Pediatric Subjects
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
6.3 Postmarketing Experience
The following adverse reactions have been reported during postmarketing use of entecavir. Because these reactions were reported voluntarily from a population of unknown size, it is not possible to reliably estimate their frequency or establish a causal relationship to entecavir exposure.
Immune system disorders: Anaphylactoid reaction.
Metabolism and nutrition disorders: Lactic acidosis.
Hepatobiliary disorders: Increased transaminases.
Skin and subcutaneous tissue disorders: Alopecia, rash.
7 DRUG INTERACTIONS
Since entecavir is primarily eliminated by the kidneys [see Clinical Pharmacology (12.3)] , co-administration of entecavir with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either entecavir or the co-administered drug. Co-administration of entecavir with lamivudine, adefovir dipivoxil, or tenofovir disoproxil fumarate did not result in significant drug interactions. The effects of co-administration of entecavir with other drugs that are renally eliminated or are known to affect renal function have not been evaluated, and patients should be monitored closely for adverse events when entecavir is co-administered with such drugs.
To report SUSPECTED ADVERSE REACTIONS contact AvKARE at 1-855-361-3993; email drugsafety@avkare.com; or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of entecavir in pregnant women. Because animal reproduction studies are not always predictive of human response, entecavir should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Antiretroviral Pregnancy Registry: To monitor fetal outcomes of pregnant women exposed to entecavir, an Antiretroviral Pregnancy Registry has been established. Healthcare providers are encouraged to register patients by calling 1-800-258-4263.
Animal Data
Animal reproduction studies with entecavir in rats and rabbits revealed no evidence of teratogenicity. Developmental toxicity studies were performed in rats and rabbits. There were no signs of embryofetal or maternal toxicity when pregnant animals received oral entecavir at approximately 28 (rat) and 212 (rabbit) times the human exposure achieved at the highest recommended human dose of 1 mg/day. In rats, maternal toxicity, embryofetal toxicity (resorptions), lower fetal body weights, tail and vertebral malformations, reduced ossification (vertebrae, sternebrae and phalanges) and extra lumbar vertebrae and ribs were observed at exposures 3100 times those in humans. In rabbits, embryofetal toxicity (resorptions), reduced ossification (hyoid) and an increased incidence of 13th rib were observed at exposures 883 times those in humans. In a peripostnatal study, no adverse effects on offspring occurred when rats received oral entecavir at exposures greater than 94 times those in humans.
8.2 Labor and Delivery
There are no studies in pregnant women and no data on the effect of entecavir on transmission of HBV from mother to infant. Therefore, appropriate interventions should be used to prevent neonatal acquisition of HBV.
8.3 Nursing Mothers
It is not known whether entecavir is excreted into human milk; however, entecavir is excreted into the milk of rats. Because many drugs are excreted into human milk and because of the potential for serious adverse reactions in nursing infants from entecavir, a decision should be made to discontinue nursing or to discontinue entecavir taking into consideration the importance of continued hepatitis B therapy to the mother and the known benefits of breastfeeding.
8.4 Pediatric Use
The efficacy and safety of entecavir have not been established in patients less than 2 years of age. Use of entecavir in this age group has not been evaluated because treatment of HBV in this age group is rarely required.
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
8.5 Geriatric Use
Clinical studies of entecavir did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. Entecavir is substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function [see Dosage and Administration (2.4)] .
8.7 Renal Impairment
Dosage adjustment of entecavir is recommended for patients with creatinine clearance less than 50 mL/min, including patients on hemodialysis or CAPD [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
8.8 Liver Transplant Recipients
If entecavir treatment is determined to be necessary for a liver transplant recipient who has received or is receiving an immunosuppressant that may affect renal function, such as cyclosporine or tacrolimus, renal function must be carefully monitored both before and during treatment with entecavir [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
10 OVERDOSAGE
There is limited experience of entecavir overdosage reported in patients. Healthy subjects who received single entecavir doses up to 40 mg or multiple doses up to 20 mg/day for up to 14 days had no increase in or unexpected adverse events. If overdose occurs, the patient must be monitored for evidence of toxicity, and standard supportive treatment applied as necessary.
Following a single 1 mg dose of entecavir, a 4-hour hemodialysis session removed approximately 13% of the entecavir dose.
11 DESCRIPTION
Entecavir is a guanosine nucleoside analogue with selective activity against HBV. The chemical name for entecavir is 2-amino-1,9-dihydro-9-[( 1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6 H-purin-6-one, monohydrate. Its molecular formula is C 12H 15N 5O 3•H 2O, which corresponds to a molecular weight of 295.3. Entecavir has the following structural formula:
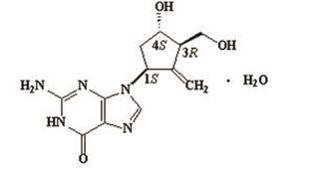
Entecavir is a white to off-white powder. It is slightly soluble in water (2.4 mg/mL), and the pH of the saturated solution in water is 7.9 at 25° C ± 0.5° C.
Entecavir film-coated tablets are available for oral administration in strengths of 0.5 mg and 1 mg of entecavir. Entecavir 0.5 mg and 1 mg film-coated tablets contain the following inactive ingredients: Crospovidone, lactose monohydrate, magnesium stearate, microcrystalline cellulose and povidone. The tablet coating contains hypromellose, iron oxide red (in 1 mg tablet only), polyethylene glycol 8000 and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
The single- and multiple-dose pharmacokinetics of entecavir were evaluated in healthy subjects and subjects with chronic hepatitis B virus infection.
Absorption
Following oral administration in healthy subjects, entecavir peak plasma concentrations occurred between 0.5 and 1.5 hours. Following multiple daily doses ranging from 0.1 to 1 mg, C max and area under the concentration-time curve (AUC) at steady-state increased in proportion to dose.
Steady-state was achieved after 6 to 10 days of once-daily administration with approximately 2-fold accumulation. For a 0.5 mg oral dose, C max at steady-state was 4.2 ng/mL and trough plasma concentration (C trough) was 0.3 ng/mL. For a 1 mg oral dose, C max was 8.2 ng/mL and C trough was 0.5 ng/mL.
Effects of food on oral absorption: Oral administration of 0.5 mg of entecavir with a standard
high-fat meal (945 kcal, 54.6 g fat) or a light meal (379 kcal, 8.2 g fat) resulted in a delay in absorption (1 to 1.5 hours fed vs. 0.75 hours fasted), a decrease in C max of 44% to 46% and a decrease in AUC of 18% to 20% [see Dosage and Administration (2)] .
Distribution
Based on the pharmacokinetic profile of entecavir after oral dosing, the estimated apparent volume of distribution is in excess of total body water, suggesting that entecavir is extensively distributed into tissues.
Binding of entecavir to human serum proteins in vitro was approximately 13%.
Metabolism and Elimination
Following administration of 14C-entecavir in humans and rats, no oxidative or acetylated metabolites were observed. Minor amounts of phase II metabolites (glucuronide and sulfate conjugates) were observed. Entecavir is not a substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. See Drug Interactions, below .
After reaching peak concentration, entecavir plasma concentrations decreased in a bi-exponential manner with a terminal elimination half-life of approximately 128 to 149 hours. The observed drug accumulation index is approximately 2-fold with once-daily dosing, suggesting an effective accumulation half-life of approximately 24 hours.
Entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at steady-state ranging from 62% to 73% of the administered dose. Renal clearance is independent of dose and ranges from 360 to 471 mL/min suggesting that entecavir undergoes both glomerular filtration and net tubular secretion [see Drug Interactions (7)] .
Special Populations
Gender: There are no significant gender differences in entecavir pharmacokinetics.
Race: There are no significant racial differences in entecavir pharmacokinetics.
Elderly: The effect of age on the pharmacokinetics of entecavir was evaluated following administration of a single 1 mg oral dose in healthy young and elderly volunteers. Entecavir AUC was 29.3% greater in elderly subjects compared to young subjects. The disparity in exposure between elderly and young subjects was most likely attributable to differences in renal function. Dosage adjustment of entecavir should be based on the renal function of the patient, rather than age [see Dosage and Administration (2.4)] .
Pediatrics: Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
Renal impairment: The pharmacokinetics of entecavir following a single 1 mg dose were studied in subjects (without chronic hepatitis B virus infection) with selected degrees of renal impairment, including subjects whose renal impairment was managed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD). Results are shown in Table 7 [see Dosage and Administration (2.4)] .
| Table 7: Pharmacokinetic Parameters in Subjects with Selected Degrees of Renal Function | ||||||
| Renal Function Group | ||||||
| Baseline Creatinine Clearance (mL/min) | ||||||
| Unimpaired | Mild | Moderate | Severe | Severe | Severe | |
| >80 | >50 to ≤80 | 30 to 50 | <30 |
Managed with
Hemodialysis a |
Managed
with CAPD |
|
| n=6 | n=6 | n=6 | n=6 | n=6 | n=4 | |
| C max (ng/mL) | 8.1 | 10.4 | 10.5 | 15.3 | 15.4 | 16.6 |
| (CV%) | (30.7) | (37.2) | (22.7) | (33.8) | (56.4) | (29.7) |
| AUC (0-T) (ng•h/mL) | 27.9 | 51.5 | 69.5 | 145.7 | 233.9 | 221.8 |
| (CV) | (25.6) | (22.8) | (22.7) | (31.5) | (28.4) | (11.6) |
| CLR (mL/min) | 383.2 | 197.9 | 135.6 | 40.3 | NA | NA |
| (SD) | (101.8) | (78.1) | (31.6) | (10.1) | ||
| CLT/F (mL/min) | 588.1 | 309.2 | 226.3 | 100.6 | 50.6 | 35.7 |
| (SD) | (153.7) | (62.6) | (60.1) | (29.1) | (16.5) | (19.6) |
| a Dosed immediately following hemodialysis. | ||||||
| CLR = renal clearance; CLT/F = apparent oral clearance. | ||||||
Following a single 1 mg dose of entecavir administered 2 hours before the hemodialysis session, hemodialysis removed approximately 13% of the entecavir dose over 4 hours. CAPD removed approximately 0.3% of the dose over 7 days [see Dosage and Administration (2.4)] .
Hepatic impairment: The pharmacokinetics of entecavir following a single 1 mg dose were studied in adult subjects (without chronic hepatitis B virus infection) with moderate or severe hepatic impairment (Child-Turcotte-Pugh Class B or C). The pharmacokinetics of entecavir were similar between hepatically impaired and healthy control subjects; therefore, no dosage adjustment of entecavir is recommended for patients with hepatic impairment. The pharmacokinetics of entecavir have not been studied in pediatric subjects with hepatic impairment.
Post-liver transplant: Limited data are available on the safety and efficacy of entecavir in liver transplant recipients. In a small pilot study of entecavir use in HBV-infected liver transplant recipients on a stable dose of cyclosporine A (n=5) or tacrolimus (n=4), entecavir exposure was approximately 2-fold the exposure in healthy subjects with normal renal function. Altered renal function contributed to the increase in entecavir exposure in these subjects. The potential for pharmacokinetic interactions between entecavir and cyclosporine A or tacrolimus was not formally evaluated [see Use in Specific Populations (8.8)] .
Drug Interactions
The metabolism of entecavir was evaluated in in vitro and in vivo studies. Entecavir is not a substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. At concentrations up to approximately 10,000-fold higher than those obtained in humans, entecavir inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6 and 2E1. At concentrations up to approximately 340-fold higher than those observed in humans, entecavir did not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5 and 2B6. The pharmacokinetics of entecavir are unlikely to be affected by co-administration with agents that are either metabolized by, inhibit, or induce the CYP450 system. Likewise, the pharmacokinetics of known CYP substrates are unlikely to be affected by co-administration of entecavir.
The steady-state pharmacokinetics of entecavir and co-administered drug were not altered in interaction studies of entecavir with lamivudine, adefovir dipivoxil and tenofovir disoproxil fumarate [see Drug Interactions (7)] .
12.4 Microbiology
Mechanism of Action
Entecavir, a guanosine nucleoside analogue with activity against HBV reverse transcriptase (rt), is efficiently phosphorylated to the active triphosphate form, which has an intracellular half-life of 15 hours. By competing with the natural substrate deoxyguanosine triphosphate, entecavir triphosphate functionally inhibits all three activities of the HBV reverse transcriptase: (1) base priming, (2) reverse transcription of the negative strand from the pregenomic messenger RNA, and (3) synthesis of the positive strand of HBV DNA. Entecavir triphosphate is a weak inhibitor of cellular DNA polymerases α, β and δ and mitochondrial DNA polymerase γ with K i values ranging from 18 to >160 μM.
Antiviral Activity
Entecavir inhibited HBV DNA synthesis (50% reduction, EC 50) at a concentration of 0.004 μM in human HepG2 cells transfected with wild-type HBV. The median EC 50 value for entecavir against lamivudine-resistant HBV (rtL180M, rtM204V) was 0.026 μM (range 0.010 to 0.059 μM).
The co-administration of HIV nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) with entecavir is unlikely to reduce the antiviral efficacy of entecavir against HBV or of any of these agents against HIV. In HBV combination assays in cell culture, abacavir, didanosine, lamivudine, stavudine, tenofovir, or zidovudine were not antagonistic to the anti-HBV activity of entecavir over a wide range of concentrations. In HIV antiviral assays, entecavir was not antagonistic to the cell culture anti-HIV activity of these six NRTIs or emtricitabine at concentrations greater than 100 times the C max of entecavir using the 1 mg dose.
Antiviral Activity Against HIV
A comprehensive analysis of the inhibitory activity of entecavir against a panel of laboratory and clinical HIV type 1 (HIV-1) isolates using a variety of cells and assay conditions yielded EC 50 values ranging from 0.026 to >10 μM; the lower EC 50 values were observed when decreased levels of virus were used in the assay. In cell culture, entecavir selected for an M184I substitution in HIV reverse transcriptase at micromolar concentrations, confirming inhibitory pressure at high entecavir concentrations. HIV variants containing the M184V substitution showed loss of susceptibility to entecavir.
Resistance
In Cell Culture
In cell-based assays, 8- to 30-fold reductions in entecavir phenotypic susceptibility were observed for lamivudine-resistant strains. Further reductions (>70-fold) in entecavir phenotypic susceptibility required the presence of amino acid substitutions rtM204I/V with or without rtL180M along with additional substitutions at residues rtT184, rtS202, or rtM250, or a combination of these substitutions with or without an rtI169 substitution in the HBV reverse transcriptase.
Clinical Studies
Nucleoside-inhibitor-naïve subjects: Genotypic evaluations were performed on evaluable samples (>300 copies/mL serum HBV DNA) from 562 subjects who were treated with entecavir for up to 96 weeks in nucleoside-inhibitor-naïve studies (AI463022, AI463027 and rollover study AI463901). By Week 96, evidence of emerging amino acid substitution rtS202G with rtM204V and rtL180M substitutions was detected in the HBV of 2 subjects (2/562=<1%), and 1 of them experienced virologic rebound (≥1 log 10 increase above nadir). In addition, emerging amino acid substitutions at rtM204I/V and rtL180M, rtL80I, or rtV173L, which conferred decreased phenotypic susceptibility to entecavir in the absence of rtT184, rtS202, or rtM250 changes, were detected in the HBV of 3 subjects (3/562=<1%) who experienced virologic rebound. For subjects who continued treatment beyond 48 weeks, 75% (202/269) had HBV DNA <300 copies/mL at end of dosing (up to 96 weeks).
HBeAg-positive (n=243) and -negative (n=39) treatment-naïve subjects who failed to achieve the study-defined complete response by 96 weeks were offered continued entecavir treatment in a rollover study. Complete response for HBeAg-positive was <0.7 MEq/mL (approximately 7 x 10 5 copies/mL) serum HBV DNA and HBeAg loss and, for HBeAg-negative was <0.7 MEq/mL HBV DNA and ALT normalization. Subjects received 1 mg entecavir once daily for up to an additional 144 weeks. Of these 282 subjects, 141 HBeAg-positive and 8 HBeAg-negative subjects entered the long-term follow-up rollover study and were evaluated for entecavir resistance. Of the 149 subjects entering the rollover study, 88% (131/149), 92% (137/149) and 92% (137/149) attained serum HBV DNA <300 copies/mL by Weeks 144, 192 and 240 (including end of dosing), respectively. No novel entecavir resistance-associated substitutions were identified in a comparison of the genotypes of evaluable isolates with their respective baseline isolates. The cumulative probability of developing rtT184, rtS202, or rtM250 entecavir resistance-associated substitutions (in the presence of rtM204V and rtL180M substitutions) at Weeks 48, 96, 144, 192 and 240 was 0.2%, 0.5%, 1.2%, 1.2% and 1.2%, respectively.
Lamivudine-refractory subjects: Genotypic evaluations were performed on evaluable samples from 190 subjects treated with entecavir for up to 96 weeks in studies of lamivudine-refractory HBV (AI463026, AI463014, AI463015 and rollover study AI463901). By Week 96, resistance-associated amino acid substitutions at rtS202, rtT184, or rtM250, with or without rtI169 changes, in the presence of amino acid substitutions rtM204I/V with or without rtL180M, rtL80V, or rtV173L/M emerged in the HBV from 22 subjects (22/190=12%), 16 of whom experienced virologic rebound (≥1 log 10 increase above nadir) and 4 of whom were never suppressed <300 copies/mL. The HBV from 4 of these subjects had entecavir resistance substitutions at baseline and acquired further changes on entecavir treatment. In addition to the 22 subjects, 3 subjects experienced virologic rebound with the emergence of rtM204I/V and rtL180M, rtL80V, or rtV173L/M. For isolates from subjects who experienced virologic rebound with the emergence of resistance substitutions (n=19), the median fold-change in entecavir EC 50 values from reference was 19-fold at baseline and 106-fold at the time of virologic rebound. For subjects who continued treatment beyond 48 weeks, 40% (31/77) had HBV DNA <300 copies/mL at end of dosing (up to 96 weeks).
Lamivudine-refractory subjects (n=157) who failed to achieve the study-defined complete response by Week 96 were offered continued entecavir treatment. Subjects received 1 mg entecavir once daily for up to an additional 144 weeks. Of these subjects, 80 subjects entered the long-term follow-up study and were evaluated for entecavir resistance. By Weeks 144, 192 and 240 (including end of dosing), 34% (27/80), 35% (28/80) and 36% (29/80), respectively, attained HBV DNA <300 copies/mL. The cumulative probability of developing rtT184, rtS202, or rtM250 entecavir resistance-associated substitutions (in the presence of rtM204I/V with or without rtL180M substitutions) at Weeks 48, 96, 144, 192 and 240 was 6.2%, 15%, 36.3%, 46.6% and 51.5%, respectively. The HBV of 6 subjects developed rtA181C/G/S/T amino acid substitutions while receiving entecavir, and of these, 4 developed entecavir resistance-associated substitutions at rtT184, rtS202, or rtM250 and 1 had an rtT184S substitution at baseline. Of 7 subjects whose HBV had an rtA181 substitution at baseline, 2 also had substitutions at rtT184, rtS202, or rtM250 at baseline and another 2 developed them while on treatment with entecavir.
Cross-resistance
Cross-resistance has been observed among HBV nucleoside analogue inhibitors. In cell-based assays, entecavir had 8- to 30-fold less inhibition of HBV DNA synthesis for HBV containing lamivudine and telbivudine resistance substitutions rtM204I/V with or without rtL180M than for wild-type HBV. Substitutions rtM204I/V with or without rtL180M, rtL80I/V, or rtV173L, which are associated with lamivudine and telbivudine resistance, also confer decreased phenotypic susceptibility to entecavir. The efficacy of entecavir against HBV harboring adefovir resistance- associated substitutions has not been established in clinical trials. HBV isolates from lamivudine-refractory subjects failing entecavir therapy were susceptible in cell culture to adefovir but remained resistant to lamivudine. Recombinant HBV genomes encoding adefovir resistance-associated substitutions at either rtN236T or rtA181V had 0.3- and 1.1-fold shifts in susceptibility to entecavir in cell culture, respectively.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term oral carcinogenicity studies of entecavir in mice and rats were carried out at exposures up to approximately 42 times (mice) and 35 times (rats) those observed in humans at the highest recommended dose of 1 mg/day. In mouse and rat studies, entecavir was positive for carcinogenic findings.
In mice, lung adenomas were increased in males and females at exposures 3 and 40 times those in humans. Lung carcinomas in both male and female mice were increased at exposures 40 times those in humans. Combined lung adenomas and carcinomas were increased in male mice at exposures 3 times and in female mice at exposures 40 times those in humans. Tumor development was preceded by pneumocyte proliferation in the lung, which was not observed in rats, dogs, or monkeys administered entecavir, supporting the conclusion that lung tumors in mice may be a species-specific event. Hepatocellular carcinomas were increased in males and combined liver adenomas and carcinomas were also increased at exposures 42 times those in humans. Vascular tumors in female mice (hemangiomas of ovaries and uterus and hemangiosarcomas of spleen) were increased at exposures 40 times those in humans. In rats, hepatocellular adenomas were increased in females at exposures 24 times those in humans; combined adenomas and carcinomas were also increased in females at exposures 24 times those in humans. Brain gliomas were induced in both males and females at exposures 35 and 24 times those in humans. Skin fibromas were induced in females at exposures 4 times those in humans.
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
Mutagenesis
Entecavir was clastogenic to human lymphocyte cultures. Entecavir was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains in the presence or absence of metabolic activation, a mammalian-cell gene mutation assay, and a transformation assay with Syrian hamster embryo cells. Entecavir was also negative in an oral micronucleus study and an oral DNA repair study in rats.
Impairment of Fertility
In reproductive toxicology studies, in which animals were administered entecavir at up to 30 mg/kg for up to 4 weeks, no evidence of impaired fertility was seen in male or female rats at systemic exposures greater than 90 times those achieved in humans at the highest recommended dose of 1 mg/day. In rodent and dog toxicology studies, seminiferous tubular degeneration was observed at exposures 35 times or greater than those achieved in humans. No testicular changes were evident in monkeys.
14 CLINICAL STUDIES
The safety and efficacy of entecavir were evaluated in three Phase 3 active-controlled trials [see Clinical Studies (14.1, 14.2)] . These studies included 1633 subjects 16 years of age or older with chronic hepatitis B virus infection (serum HBsAg-positive for at least 6 months) accompanied by evidence of viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR assay). Subjects had persistently elevated ALT levels at least 1.3 times ULN and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral hepatitis. The safety and efficacy of entecavir were also evaluated in a study of 191 HBV-infected subjects with decompensated liver disease and in a study of 68 subjects co-infected with HBV and HIV [see Clinical Studies (14.1)] .
14.1 Outcomes in Adults
At 48 Weeks
The safety and efficacy of entecavir in adults were evaluated in three Phase 3 active-controlled trials. These studies included 1633 subjects 16 years of age or older with chronic hepatitis B virus infection (serum HBsAg-positive for at least 6 months) accompanied by evidence of viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR assay). Subjects had persistently elevated ALT levels at least 1.3 times ULN and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral hepatitis. The safety and efficacy of entecavir were also evaluated in a study of 191 HBV-infected subjects with decompensated liver disease and in a study of 68 subjects co-infected with HBV and HIV.
Nucleoside-inhibitor-naïve Subjects with Compensated Liver Disease
HBeAg-positive: Study AI463022 was a multinational, randomized, double-blind study of entecavir 0.5 mg once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 709 (of 715 randomized) nucleoside-inhibitor-naïve subjects with chronic hepatitis B virus infection, compensated liver disease, and detectable HBeAg. The mean age of subjects was 35 years, 75% were male, 57% were Asian, 40% were Caucasian and 13% had previously received interferon-α. At baseline, subjects had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor ® PCR assay was 9.66 log 10 copies/mL, and mean serum ALT level was 143 U/L. Paired, adequate liver biopsy samples were available for 89% of subjects.
HBeAg-negative (anti-HBe-positive/HBV DNA-positive): Study AI463027 was a multinational, randomized, double-blind study of entecavir 0.5 mg once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 638 (of 648 randomized) nucleoside-inhibitor-naïve subjects with HBeAg-negative (HBeAb-positive) chronic hepatitis B virus infection and compensated liver disease. The mean age of subjects was 44 years, 76% were male, 39% were Asian, 58% were Caucasian and 13% had previously received interferon-α. At baseline, subjects had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 7.58 log 10 copies/mL, and mean serum ALT level was 142 U/L. Paired, adequate liver biopsy samples were available for 88% of subjects.
In Studies AI463022 and AI463027, entecavir was superior to lamivudine on the primary efficacy endpoint of Histologic Improvement, defined as a 2-point or greater reduction in Knodell Necroinflammatory Score with no worsening in Knodell Fibrosis Score at Week 48, and on the secondary efficacy measures of reduction in viral load and ALT normalization. Histologic Improvement and change in Ishak Fibrosis Score are shown in Table 8. Selected virologic, biochemical and serologic outcome measures are shown in Table 9.
|
Table 8: Histologic Improvement and Change in Ishak Fibrosis Score at Week 48, Nucleoside-Inhibitor-Naïve Subjects in Studies AI463022
and AI463027 |
||||
| Study AI463022 (HBeAg-Positive) | Study AI463027 (HBeAg-Negative) | |||
| Entecavir | Lamivudine | Entecavir | Lamivudine | |
| 0.5 mg | 100 mg | 0.5 mg | 100 mg | |
| n=314a | n=314a | n=296a | n=287a | |
|
Histologic Improvement
(Knodell Scores) | ||||
| Improvement b | 72% | 62% | 70% | 61% |
| No improvement | 21% | 24% | 19% | 26% |
| Ishak Fibrosis Score | ||||
| Improvement c | 39% | 35% | 36% | 38% |
| No change | 46% | 40% | 41% | 34% |
| Worsening c | 8% | 10% | 12% | 15% |
| Missing Week 48 biopsy | 7% | 14% | 10% | 13% |
| a Subjects with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥2). | ||||
| b ≥2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the Knodell Fibrosis Score. | ||||
| c For Ishak Fibrosis Score, improvement = ≥1-point decrease from baseline and worsening = ≥1-point increase from baseline. | ||||
|
Table 9: Selected Virologic, Biochemical and Serologic Endpoints at Week 48, Nucleoside-Inhibitor-Naïve Subjects in Studies AI463022
and AI463027 |
||||
|
Study AI463022
(HBeAg-Positive) |
Study AI463027
(HBeAg-Negative) |
|||
| Entecavir | Lamivudine | Entecavir | Lamivudine | |
| 0.5 mg | 100 mg | 0.5 mg | 100 mg | |
| n=354 | n=355 | n=325 | n=313 | |
| HBV DNA a | ||||
| Proportion undetectable (<300 copies/mL) | 67% | 36% | 90% | 72% |
| Mean change from baseline (log 10 copies/mL) | −6.86 | −5.39 | −5.04 | −4.53 |
| ALT normalization (≤1 x ULN) | 68% | 60% | 78% | 71% |
| HBeAg seroconversion | 21% | 18% | NA | NA |
| a Roche COBAS Amplicor PCR assay [lower limit of quantification (LLOQ) = 300 copies/mL]. | ||||
Histologic Improvement was independent of baseline levels of HBV DNA or ALT.
Lamivudine-refractory Subjects with Compensated Liver Disease
Study AI463026 was a multinational, randomized, double-blind study of entecavir in 286 (of 293 randomized) subjects with lamivudine-refractory chronic hepatitis B virus infection and compensated liver disease. Subjects receiving lamivudine at study entry either switched to entecavir 1 mg once daily (with neither a washout nor an overlap period) or continued on lamivudine 100 mg for a minimum of 52 weeks. The mean age of subjects was 39 years, 76% were male, 37% were Asian, 62% were Caucasian and 52% had previously received interferon-α. The mean duration of prior lamivudine therapy was 2.7 years, and 85% had lamivudine resistance substitutions at baseline by an investigational line probe assay. At baseline, subjects had a mean Knodell Necroinflammatory Score of 6.5, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 9.36 log 10 copies/mL, and mean serum ALT level was 128 U/L. Paired, adequate liver biopsy samples were available for 87% of subjects.
Entecavir was superior to lamivudine on a primary endpoint of Histologic Improvement (using the Knodell Score at Week 48). These results and change in Ishak Fibrosis Score are shown in Table 10. Table 11 shows selected virologic, biochemical and serologic endpoints.
|
Table 10: Histologic Improvement and Change in Ishak Fibrosis Score at Week 48, Lamivudine-Refractory Subjects in Study
AI463026 |
||
|
Entecavir
1 mg n=124 a |
Lamivudine
100 mg n=116 a |
|
| Histologic Improvement (Knodell Scores) | ||
| Improvement b | 55% | 28% |
| No improvement | 34% | 57% |
| Ishak Fibrosis Score | ||
| Improvement c | 34% | 16% |
| No change | 44% | 42% |
| Worsening c | 11% | 26% |
| Missing Week 48 biopsy | 11% | 16% |
| a Subjects with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥2). | ||
| b ≥2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the Knodell Fibrosis Score. | ||
| c For Ishak Fibrosis Score, improvement = ≥1-point decrease from baseline and worsening = ≥1-point increase from baseline. | ||
|
Table 11: Selected Virologic, Biochemical and Serologic Endpoints at Week 48, Lamivudine-Refractory Subjects in Study
AI463026 |
||
|
Entecavir
1 mg n=141 |
Lamivudine
100 mg n=145 |
|
| HBV DNA a | ||
| Proportion undetectable (<300 copies/mL) | 19% | 1% |
| Mean change from baseline (log 10 copies/mL) | −5.11 | −0.48 |
| ALT normalization (≤1 x ULN) | 61% | 15% |
| HBeAg seroconversion | 8% | 3% |
| a Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL). | ||
Histologic Improvement was independent of baseline levels of HBV DNA or ALT.
Subjects with Decompensated Liver Disease
Study AI463048 was a randomized, open-label study of entecavir 1 mg once daily versus adefovir dipivoxil 10 mg once daily in 191 (of 195 randomized) adult subjects with HBeAg-positive or -negative chronic HBV infection and evidence of hepatic decompensation, defined as a Child-Turcotte-Pugh (CTP) score of 7 or higher. Subjects were either HBV-treatment-naïve or previously treated, predominantly with lamivudine or interferon-α.
In Study AI463048, 100 subjects were randomized to treatment with entecavir and 91 subjects to treatment with adefovir dipivoxil. Two subjects randomized to treatment with adefovir dipivoxil actually received treatment with entecavir for the duration of the study. The mean age of subjects was 52 years, 74% were male, 54% were Asian, 33% were Caucasian and 5% were Black/African American. At baseline, subjects had a mean serum HBV DNA by PCR of 7.83 log 10 copies/mL and mean ALT level of 100 U/L; 54% of subjects were HBeAg-positive; 35% had genotypic evidence of lamivudine resistance. The baseline mean CTP score was 8.6. Results for selected study endpoints at Week 48 are shown in Table 10.
| Table 12: Selected Endpoints at Week 48, Subjects with Decompensated Liver Disease, Study AI463048 | ||
|
Entecavir
1 mg n=100 a |
Adefovir Dipivoxil
10 mg n=91 a |
|
| HBV DNA b | ||
| Proportion undetectable (<300 copies/mL) | 57% | 20% |
| Stable or improved CTP score c | 61% | 67% |
| HBsAg loss | 5% | 0 |
| Normalization of ALT (≤1 x ULN) d | 49/78 (63%) | 33/71 (46%) |
| a Endpoints were analyzed using intention-to-treat (ITT) method, treated subjects as randomized. | ||
| b Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL). | ||
| c Defined as decrease or no change from baseline in CTP score. | ||
| d Denominator is subjects with abnormal values at baseline. | ||
| ULN=upper limit of normal. | ||
Subjects Co-infected with HIV and HBV
Study AI463038 was a randomized, double-blind, placebo-controlled study of entecavir versus placebo in 68 subjects co-infected with HIV and HBV who experienced recurrence of HBV viremia while receiving a lamivudine-containing highly active antiretroviral (HAART) regimen. Subjects continued their lamivudine-containing HAART regimen (lamivudine dose 300 mg/day) and were assigned to add either entecavir 1 mg once daily (51 subjects) or placebo (17 subjects) for 24 weeks followed by an open-label phase for an additional 24 weeks where all subjects received entecavir. At baseline, subjects had a mean serum HBV DNA level by PCR of 9.13 log 10 copies/mL. Ninety-nine percent of subjects were HBeAg-positive at baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable at approximately 2 log 10 copies/mL through 24 weeks of blinded therapy. Virologic and biochemical endpoints at Week 24 are shown in Table 13. There are no data in patients with HIV/HBV co-infection who have not received prior lamivudine therapy. Entecavir has not been evaluated in HIV/HBV co-infected patients who were not simultaneously receiving effective HIV treatment [see Warnings and Precautions (5.2)] .
| Table 13: Virologic and Biochemical Endpoints at Week 24, Study AI463038 | ||
|
Entecavir1 mga
n=51 |
Placeboa
n=17 |
|
| HBV DNA b | ||
| Proportion undetectable (<300 copies/mL) | 6% | 0 |
| Mean change from baseline (log 10 copies/mL) | −3.65 | +0.11 |
| ALT normalization (≤1 x ULN) | 34% c | 8% c |
| a All subjects also received a lamivudine-containing HAART regimen. | ||
| b Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL). | ||
|
c Percentage of subjects with abnormal ALT (>1 x ULN) at baseline who achieved ALT normalization (n=35 for entecavir and n=12 for
placebo). |
||
For subjects originally assigned to entecavir, at the end of the open-label phase (Week 48), 8% of subjects had HBV DNA <300 copies/mL by PCR, the mean change from baseline HBV DNA by PCR was −4.20 log 10 copies/mL, and 37% of subjects with abnormal ALT at baseline had ALT normalization (≤1 x ULN).
Beyond 48 Weeks
The optimal duration of therapy with entecavir is unknown. According to protocol-mandated criteria in the Phase 3 clinical trials, subjects discontinued entecavir or lamivudine treatment after 52 weeks according to a definition of response based on HBV virologic suppression (<0.7 MEq/mL by bDNA assay) and loss of HBeAg (in HBeAg-positive subjects) or ALT <1.25 x ULN (in HBeAg-negative subjects) at Week 48. Subjects who achieved virologic suppression but did not have serologic response (HBeAg-positive) or did not achieve ALT <1.25 x ULN (HBeAg-negative) continued blinded dosing through 96 weeks or until the response criteria were met. These protocol-specified subject management guidelines are not intended as guidance for clinical practice.
Nucleoside-inhibitor-naïve Subjects
Among nucleoside-inhibitor-naïve, HBeAg-positive subjects (Study AI463022), 243 (69%) entecavir-treated subjects and 164 (46%) lamivudine-treated subjects continued blinded treatment for up to 96 weeks. Of those continuing blinded treatment in Year 2, 180 (74%) entecavir subjects and 60 (37%) lamivudine subjects achieved HBV DNA <300 copies/mL by PCR at the end of dosing (up to 96 weeks). 193 (79%) entecavir subjects achieved ALT ≤1 x ULN compared to 112 (68%) lamivudine subjects, and HBeAg seroconversion occurred in 26 (11%) entecavir subjects and 20 (12%) lamivudine subjects.
Among nucleoside-inhibitor-naïve, HBeAg-positive subjects, 74 (21%) entecavir subjects and 67 (19%) lamivudine subjects met the definition of response at Week 48, discontinued study drugs, and were followed off treatment for 24 weeks. Among entecavir responders, 26 (35%) subjects had HBV DNA <300 copies/mL, 55 (74%) subjects had ALT ≤1 x ULN, and 56 (76%) subjects sustained HBeAg seroconversion at the end of follow-up. Among lamivudine responders, 20 (30%) subjects had HBV DNA <300 copies/mL, 41 (61%) subjects had ALT ≤1 x ULN, and 47 (70%) subjects sustained HBeAg seroconversion at the end of follow-up.
Among nucleoside-inhibitor-naïve, HBeAg-negative subjects (Study AI463027), 26 (8%) entecavir-treated subjects and 28 (9%) lamivudine-treated subjects continued blinded treatment for up to 96 weeks. In this small cohort continuing treatment in Year 2, 22 entecavir and 16 lamivudine subjects had HBV DNA <300 copies/mL by PCR, and 7 and 6 subjects, respectively, had ALT ≤1 x ULN at the end of dosing (up to 96 weeks).
Among nucleoside-inhibitor-naïve, HBeAg-negative subjects, 275 (85%) entecavir subjects and 245 (78%) lamivudine subjects met the definition of response at Week 48, discontinued study drugs, and were followed off treatment for 24 weeks. In this cohort, very few subjects in each treatment arm had HBV DNA <300 copies/mL by PCR at the end of follow-up. At the end of follow-up, 126 (46%) entecavir subjects and 84 (34%) lamivudine subjects had ALT ≤1 x ULN.
Lamivudine-refractory Subjects
Among lamivudine-refractory subjects (Study AI463026), 77 (55%) entecavir-treated subjects and 3 (2%) lamivudine subjects continued blinded treatment for up to 96 weeks. In this cohort of entecavir subjects, 31 (40%) subjects achieved HBV DNA <300 copies/mL, 62 (81%) subjects had ALT ≤1 x ULN, and 8 (10%) subjects demonstrated HBeAg seroconversion at the end of dosing.
16 HOW SUPPLIED
Entecavir tablets
0.5 mg, are supplied as white to off-white, oval, film-coated, biconvex bevel edged, unscored tablet, debossed with “AN” on one side and “446” on the other side.
They are available as follows:
Bottles of 30: NDC 42291-261-30
Entecavir tablets
1 mg, are supplied as pink, oval, film-coated, biconvex bevel edged, unscored tablet, debossed with “AN” on one side and “449” on the other side.
They are available as follows:
Bottles of 30: NDC 42291-262-30
Storage
Entecavir tablets should be stored in a tightly closed container at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30° C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from light.
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information).
Information about Treatment
Physicians should inform their patients of the following important points when initiating entecavir treatment:
- Patients should remain under the care of a physician while taking entecavir. They should discuss any new symptoms or concurrent medications with their physician.
- Patients should be advised that treatment with entecavir has not been shown to reduce the risk of transmission of HBV to others through sexual contact or blood contamination.
- Patients should be advised to take entecavir on an empty stomach (at least 2 hours after a meal and 2 hours before the next meal).
- Patients should be advised to take a missed dose as soon as remembered unless it is almost time for the next dose. Patients should not take two doses at the same time.
- Patients should be advised that treatment with entecavir will not cure HBV.
- Patients should be informed that entecavir may lower the amount of HBV in the body, may lower the ability of HBV to multiply and infect new liver cells, and may improve the condition of the liver.
- Patients should be informed that it is not known whether entecavir will reduce their chances of getting liver cancer or cirrhosis.
Post-treatment Exacerbation of Hepatitis
Patients should be informed that deterioration of liver disease may occur in some cases if treatment is discontinued, and that they should discuss any change in regimen with their physician.
HIV/HBV Co-infection
Patients should be offered HIV antibody testing before starting entecavir therapy. They should be informed that if they have HIV infection and are not receiving effective HIV treatment, entecavir may increase the chance of HIV resistance to HIV medication.
Manufactured for:
AvKARE
Pulaski, TN 38478
Mfg. Rev. 11-2019-06
AV 11/20 (P)
Manufactured by:
Amneal Pharmaceuticals of New York, LLC
Brookhaven, NY 11719
Mfg. Rev. 11-2019-06
Patient Information
Entecavir (en-TEK-a-vir) Tablets
Read this Patient Information before you start taking entecavir tablets and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about Entecavir Tablets?
1. Your hepatitis B virus (HBV) infection may get worse if you stop taking entecavir tablets. This usually happens within 6 months after stopping entecavir tablets.
- Take entecavir tablets exactly as prescribed.
- Do not run out of entecavir tablets.
- Do not stop entecavir tablets without talking to your healthcare provider.
- Your healthcare provider should monitor your health and do regular blood tests to check your liver if you stop taking entecavir tablets.
2. If you have or get HIV that is not being treated with medicines while taking entecavir tablets, the HIV virus may develop resistance to certain HIV medicines and become harder to treat. You should get an HIV test before you start taking entecavir tablets and anytime after that when there is a chance you were exposed to HIV.
Entecavir tablets can cause serious side effects including:
3. Lactic acidosis (buildup of acid in the blood). Some people who have taken entecavir tablets or medicines like entecavir(a nucleoside analogue) have developed a serious condition called lactic acidosis. Lactic acidosis is a serious medical emergency that can cause death. Lactic acidosis must be treated in the hospital. Reports of lactic acidosis with entecavir tablets generally involved patients who were seriously ill due to their liver disease or other medical condition.
Call your healthcare provider right away if you get any of the following signs or symptoms of lactic acidosis:
- You feel very weak or tired.
- You have unusual (not normal) muscle pain.
- You have trouble breathing.
- You have stomach pain with nausea and vomiting.
- You feel cold, especially in your arms and legs.
- You feel dizzy or light-headed.
- You have a fast or irregular heartbeat.
4. Serious liver problems. Some people who have taken medicines like entecavir tablets have developed serious liver problems called hepatotoxicity, with liver enlargement (hepatomegaly) and fat in the liver (steatosis). Hepatomegaly with steatosis is a serious medical emergency that can cause death.
Call your healthcare provider right away if you get any of the following signs or symptoms of liver problems:
- Your skin or the white part of your eyes turns yellow (jaundice).
- Your urine turns dark.
- Your bowel movements (stools) turn light in color.
- You don’t feel like eating food for several days or longer.
- You feel sick to your stomach (nausea).
- You have lower stomach pain.
You may be more likely to get lactic acidosis or serious liver problems if you are female, very overweight, or have been taking nucleoside analogue medicines, like entecavir tablets, for a long time.
What are Entecavir Tablets?
Entecavir tablets are a prescription medicine used to treat chronic hepatitis B virus (HBV) in adults who have active liver disease.
- Entecavir tablets will not cure HBV.
- Entecavir tablets may lower the amount of HBV in the body.
- Entecavir tablets may lower the ability of HBV to multiply and infect new liver cells.
- Entecavir tablets may improve the condition of your liver.
- It is not known whether entecavir tablets will reduce your chances of getting liver cancer or liver damage (cirrhosis), which may be caused by chronic HBV infection.
- It is not known if entecavir tablets are safe and effective for use in children less than 2 years of age.
What should I tell my healthcare provider before taking Entecavir Tablets?
Before you take entecavir tablets, tell your healthcare provider if you:
- have kidney problems. Your entecavir tablets dose or schedule may need to be changed.
- have received medicine for HBV before. Some people, especially those who have already been treated with certain other medicines for HBV infection, may develop resistance to entecavir tablets. These people may have less benefit from treatment with entecavir tablets and may have worsening of hepatitis after resistant virus appears. Your healthcare provider will test the level of the hepatitis B virus in your blood regularly.
- have any other medical conditions.
- are pregnant or plan to become pregnant. It is not known if entecavir tablets will harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
Antiretroviral Pregnancy Registry. If you take entecavir tablets while you are pregnant, talk to your healthcare provider about how you can take part in the entecavir tablets Antiretroviral Pregnancy Registry. The purpose of the pregnancy registry is to collect information about the health of you and your baby.
- are breastfeeding or plan to breastfeed. It is not known if entecavir can pass into your breast milk. You and your healthcare provider should decide if you will take entecavir tablets or breastfeed.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Especially tell your healthcare provider if you have taken a medicine to treat HBV in the past.
Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider and pharmacist when you get a new medicine.
How should I take Entecavir Tablets?
- Take entecavir tablets exactly as your healthcare provider tells you to.
- Your healthcare provider will tell you how much entecavir tablets to take.
- Your healthcare provider will tell you when and how often to take entecavir tablets.
- Take entecavir tablets on an empty stomach, at least 2 hours after a meal and at least 2 hours before the next meal.
- Do not change your dose or stop taking entecavir tablets without talking to your healthcare provider.
- If you miss a dose of entecavir tablets, take it as soon as you remember and then take your next dose at its regular time. If it is almost time for your next dose, skip the missed dose. Do not take two doses at the same time. Call your healthcare provider or pharmacist if you are not sure what to do.
- When your supply of entecavir tablets starts to run low, call your healthcare provider or pharmacy for a refill. Do not run out of entecavir tablets.
- If you take too much entecavir tablets, call your healthcare provider or go to the nearest emergency room right away.
What are the possible side effects of Entecavir Tablets?
Entecavir tablets may cause serious side effects. See “What is the most important information I should know about Entecavir Tablets?”
The most common side effects of entecavir tablets include:
- headache
- tiredness
- dizziness
- nausea
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of entecavir. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
How should I store Entecavir Tablets?
- Store entecavir tablets at room temperature, between 68° F to 77° F (20° C to 25° C).
- Keep entecavir tablets in a tightly closed container. Protect from light.
- Do not store entecavir tablets in a damp place such as a bathroom medicine cabinet or near the kitchen sink.
- Safely throw away entecavir tablets that are out of date or no longer needed. Dispose of unused medicines through community take-back disposal programs when available or place entecavir tablets in an unrecognizable closed container in the household trash.
Keep entecavir tablets and all medicines out of the reach of children.
General information about the safe and effective use of entecavir tablets
Entecavir tablets do not stop you from spreading the hepatitis B virus (HBV) to others by sex, sharing needles, or being exposed to your blood. Talk with your healthcare provider about safe sexual practices that protect your partner. Never share needles. Do not share personal items that can have blood or body fluids on them, like toothbrushes or razor blades. A shot (vaccine) is available to protect people at risk from becoming infected with HBV.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use entecavir tablets for a condition for which it was not prescribed. Do not give entecavir tablets to other people, even if they have the same symptoms you have. It may harm them.
This Patient Information leaflet summarizes the most important information about entecavir tablets. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about entecavir tablets that is written for health professionals.
For more information, go to www.avkare.com or call 1-855-361-3993.
What are the ingredients in Entecavir Tablets?
Active ingredient: entecavir
Inactive ingredients in entecavir tablets: crospovidone, lactose monohydrate, magnesium stearate, microcrystalline cellulose and povidone.
Tablet film-coat: hypromellose, iron oxide red (in 1 mg tablet only), polyethylene glycol 8000, and titanium dioxide.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Pediatric use information is approved for Bristol-Myers Squibb Company’s Baraclude ® (entecavir) tablets. However, due to Bristol-Myers Squibb Company’s marketing exclusivity rights, this drug product is not labeled with that information.
Manufactured for:
AvKARE
Pulaski, TN 38478
Mfg. Rev. 11-2019-06
AV 11/20 (P)
Manufactured by:
Amneal Pharmaceuticals of New York, LLC
Brookhaven, NY 11719
Mfg. Rev. 11-2019-06

