FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer
GAVRETO is indicated for the treatment of adult patients with metastatic RET fusion-positive non-small cell lung cancer (NSCLC) as detected by an FDA approved test.
1.2 RET Fusion-Positive Thyroid Cancer
GAVRETO is indicated for the treatment of adult and pediatric patients 12 years of age and older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate).
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment with GAVRETO based on the presence of a RET gene fusion (NSCLC or thyroid cancer) [see Clinical Studies (14)].
Information on FDA-approved tests for RET gene fusion (NSCLC) is available at http://www.fda.gov/CompanionDiagnostics.
An FDA-approved test for the detection of RET gene fusion (thyroid cancer) is not currently available.
2.2 Recommended Dosage
The recommended dosage of GAVRETO is 400 mg orally once daily on an empty stomach (no food intake for at least 2 hours before and at least 1 hour after taking GAVRETO) [see Clinical Pharmacology (12.3)]. Continue treatment until disease progression or until unacceptable toxicity.
If a dose of GAVRETO is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for GAVRETO the next day.
Do not take an additional dose if vomiting occurs after GAVRETO but continue with the next dose as scheduled.
2.3 Dosage Modifications for Adverse Reactions
The recommended dose reductions and dosage modifications for adverse reactions are provided in Table 1 and Table 2.
| Dose Reduction | Recommended Dosage |
|---|---|
| First | 300 mg once daily |
| Second | 200 mg once daily |
| Third | 100 mg once daily |
Permanently discontinue GAVRETO in patients who are unable to tolerate 100 mg taken orally once daily.
The recommended dosage modifications for adverse reactions are provided in Table 2.
| Adverse Reaction | Severity* | Dosage Modification |
|---|---|---|
|
||
| ILD/Pneumonitis
[see Warnings and Precautions (5.1)] | Grade 1 or 2 | Withhold GAVRETO until resolution. Resume by reducing the dose as shown in Table 1. |
| Permanently discontinue GAVRETO for recurrent ILD/pneumonitis. | ||
| Grade 3 or 4 | Permanently discontinue for confirmed ILD/pneumonitis. | |
| Hypertension
[see Warnings and Precautions (5.2)] | Grade 3 | Withhold GAVRETO for Grade 3 hypertension that persists despite optimal antihypertensive therapy. Resume at a reduced dose when hypertension is controlled. |
| Grade 4 | Discontinue GAVRETO. | |
| Hepatotoxicity
[see Warnings and Precautions (5.3)] | Grade 3 or 4 | Withhold GAVRETO and monitor AST/ALT once weekly until resolution to Grade 1 or baseline. |
| Resume at reduced dose (Table 1). | ||
| For recurrent events at Grade 3 or higher, discontinue GAVRETO. | ||
| Hemorrhagic Events
[see Warnings and Precautions (5.4)] | Grade 3 or 4 | Withhold GAVRETO until recovery to baseline or Grade 0 or 1. |
| Discontinue GAVRETO for severe or life-threatening hemorrhagic events. | ||
| Other Adverse Reactions
[see Adverse Reactions (6.1)] | Grade 3 or 4 | Withhold GAVRETO until improvement to ≤ Grade 2. Resume at reduced dose (Table 1). |
| Permanently discontinue for recurrent Grade 4 adverse reactions. | ||
2.4 Dose Modification for Use withCYP3A and/or P-glycoprotein (P-gp) Inhibitors
Avoid coadministration of GAVRETO with any of the following:
- Strong CYP3A inhibitors
- Moderate CYP3A inhibitors
- P-gp inhibitors
- Combined P-gp and strong CYP3A inhibitors
- Combined P-gp and moderate CYP3A inhibitors
If coadministration with any of the above inhibitors cannot be avoided, reduce the current dose of GAVRETO as recommended in Table 3. After the inhibitor has been discontinued for 3 to 5 elimination half-lives, resume GAVRETO at the dose taken prior to initiating the inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
| Current GAVRETO Dosage | Recommended GAVRETO Dosage when Coadministered with: | |
|---|---|---|
| Combined P-gp and Strong CYP3A Inhibitors |
|
|
| 400 mg orally once daily | 200 mg orally once daily | 300 mg orally once daily |
| 300 mg orally once daily | 200 mg orally once daily | 200 mg orally once daily |
| 200 mg orally once daily | 100 mg orally once daily | 100 mg orally once daily |
2.5 Dose Modification for Use with CYP3A Inducers
Avoid coadministration of GAVRETO with any of the following:
- Strong CYP3A inducers
- Moderate CYP3A inducers
If coadministration with any of the above inducers cannot be avoided, increase the starting dose of GAVRETO as recommended in Table 4 starting on Day 7 of coadministration of GAVRETO with the inducer. After the inducer has been discontinued for at least 14 days, resume GAVRETO at the dose taken prior to initiating the inducer [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
| Current GAVRETO Dosage | Recommended GAVRETO Dosage when Coadministered with: | |
|---|---|---|
| Strong CYP3A Inducers | Moderate CYP3A Inducers | |
| 400 mg orally once daily | 800 mg orally once daily | 600 mg orally once daily |
| 300 mg orally once daily | 600 mg orally once daily | 500 mg orally once daily |
| 200 mg orally once daily | 400 mg orally once daily | 300 mg orally once daily |
3 DOSAGE FORMS AND STRENGTHS
Capsules: 100 mg, light blue, opaque, hard hydroxypropyl methylcellulose (HPMC) capsule printed with "BLU-667" on the capsule shell body and "100 mg" on the capsule shell cap.
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, and fatal interstitial lung disease (ILD) / pneumonitis can occur in patients treated with GAVRETO. Pneumonitis occurred in 12% of patients who received GAVRETO, including 3.3% with Grade 3-4, and 0.2% with fatal reactions.
Monitor for pulmonary symptoms indicative of ILD/pneumonitis. Withhold GAVRETO and promptly investigate for ILD in any patient who presents with acute or worsening of respiratory symptoms which may be indicative of ILD (e.g., dyspnea, cough, and fever). Withhold, reduce dose or permanently discontinue GAVRETO based on severity of confirmed ILD [see Dosage and Administration (2.3)].
5.2 Hypertension
Hypertension occurred in 35% of patients, including Grade 3 hypertension in 18% of patients [see Adverse Reactions (6.1)]. Overall, 8% had their dose interrupted and 4.8% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications.
Do not initiate GAVRETO in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating GAVRETO. Monitor blood pressure after 1 week, at least monthly thereafter and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue GAVRETO based on the severity [see Dosage and Administration (2.3)].
5.3 Hepatoxicity
Serious hepatic adverse reactions occurred in 1.5% of patients treated with GAVRETO. Increased AST occurred in 49% of patients, including Grade 3 or 4 in 7% and increased ALT occurred in 37% of patients, including Grade 3 or 4 in 4.8% [see Adverse Reactions (6.1)]. The median time to first onset for increased AST was 15 days (range: 5 days to 2.5 years) and for increased ALT was 24 days (range: 7 days to 3.7 years).
Monitor AST and ALT prior to initiating GAVRETO, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose or permanently discontinue GAVRETO based on severity [see Dosage and Administration (2.3)].
5.4 Hemorrhagic Events
Serious, including fatal, hemorrhagic events can occur with GAVRETO. Grade ≥ 3 hemorrhagic events occurred in 4.1% of patients treated with GAVRETO including one patient with a fatal hemorrhagic event.
Permanently discontinue GAVRETO in patients with severe or life-threatening hemorrhage [see Dosage and Administration (2.3)].
5.5 Tumor Lysis Syndrome
Cases of tumor lysis syndrome (TLS) have been reported in patients with medullary thyroid carcinoma receiving GAVRETO [see Adverse Reactions (6.1)]. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
5.6 Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, GAVRETO has the potential to adversely affect wound healing.
Withhold GAVRETO for at least 5 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of GAVRETO after resolution of wound healing complications has not been established.
5.7 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, GAVRETO can cause fetal harm when administered to a pregnant woman. Oral administration of pralsetinib to pregnant rats during the period of organogenesis resulted in malformations and embryolethality at maternal exposures below the human exposure at the clinical dose of 400 mg once daily.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with GAVRETO and for 2 weeks after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with GAVRETO and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.1)]
- Hypertension [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Hemorrhagic Events [see Warnings and Precautions (5.4)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.5)]
- Risk of Impaired Wound Healing [see Warnings and Precautions (5.6)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population in the WARNINGS AND PRECAUTIONS reflect exposure to GAVRETO as a single agent at 400 mg orally once daily in 540 patients in ARROW [see Clinical Studies (14)]. Among 540 patients who received GAVRETO, 71% were exposed for 6 months or longer and 57% were exposed for greater than one year. The most common adverse reactions (≥ 25%) were musculoskeletal pain, constipation, hypertension, diarrhea, fatigue, edema, pyrexia, and cough. The most common Grade 3-4 laboratory abnormalities (≥ 2%) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased phosphate, decreased leukocytes, decreased sodium, increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), decreased calcium (corrected), decreased platelets, increased alkaline phosphatase, increased potassium, decreased potassium and increased bilirubin.
RET Fusion-Positive Non-Small Cell Lung Cancer
The safety of GAVRETO was evaluated as a single agent at 400 mg orally once daily in 281 patients with metastatic rearranged during transfection (RET fusion-positive) non-small cell lung cancer (NSCLC) in ARROW [see Clinical Studies (14.1)]. Among the 281 patients who received GAVRETO, 72% were exposed for 6 months or longer and 56% were exposed for ≥1 year.
The median age was 60 years (range: 26 to 87 years); 54% were female, 46% were White, 46% were Asian, and 4% were Hispanic/Latino.
Serious adverse reactions occurred in 65% of patients who received GAVRETO. The most frequent serious adverse reactions (in ≥ 2% of patients) were pneumonia, anemia, pneumonitis, pyrexia, sepsis, urinary tract infection, coronavirus infection, pleural effusion, dyspnea, musculoskeletal pain, pulmonary embolism, and seizure. Fatal adverse reactions occurred in 7% of patients; fatal adverse reactions which occurred in > 1 patient included pneumonia (n=8), sepsis (n=3) and COVID (n=3).
Permanent discontinuation due to an adverse reaction occurred in 20% of patients who received GAVRETO. Adverse reactions resulting in permanent discontinuation which occurred in ≥ 2% of patients included pneumonitis (3.2%), and pneumonia (2.8%).
Dosage interruptions due to an adverse reaction occurred in 73% of patients who received GAVRETO. Adverse reactions requiring dosage interruption in ≥ 2% of patients included anemia, pneumonia, pneumonitis, neutropenia, hypertension, increased blood creatine phosphokinase, fatigue, pyrexia, increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), coronavirus infection, diarrhea, hypophosphatemia, musculoskeletal pain, thrombocytopenia, dyspnea, hemorrhage, leukopenia, lymphopenia, edema, sepsis, and vomiting.
Dose reductions due to adverse reactions occurred in 51% of patients who received GAVRETO. Adverse reactions requiring dosage reductions in ≥ 2% of patients included anemia, neutropenia, pneumonitis, increased blood creatine phosphokinase, leukopenia, hypertension, fatigue, pneumonia, and lymphopenia.
Table 5 summarizes the adverse reactions in patients with NSCLC in ARROW.
| Adverse reaction | GAVRETO N = 281 |
|
|---|---|---|
| Grades 1 - 4 (%) | Grades 3 or 4 (%) |
|
|
||
| Gastrointestinal disorders | ||
| Constipation | 45 | 0.7 |
| Diarrhea | 30 | 2.5 |
| Nausea | 19 | 0 |
| Dry mouth | 17 | 0 |
| General Disorders and Administration Site Conditions | ||
| Edema* | 44 | 0 |
| Fatigue† | 42 | 2.5 |
| Pyrexia | 29 | 0.7 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Musculoskeletal pain‡ | 44 | 2.5 |
| Increased Blood Creatine Phosphokinase | 19 | 9 |
| Vascular | ||
| Hypertension§ | 38 | 18 |
| Respiratory, thoracic and mediastinal disorders | ||
| Cough¶ | 36 | 0.4 |
| Dyspnea | 21 | 2.1 |
| Infection and Infestations | ||
| Pneumonia# | 24 | 13 |
| Urinary tract infection | 16 | 3.6 |
| Metabolism and Nutrition Disorders | ||
| Decreased appetite | 18 | 1.1 |
| Nervous system disorders | ||
| Taste disorderÞ | 17 | 0 |
| Headacheß | 15 | 1.1 |
| Skin and subcutaneous tissue disorders | ||
| Rashà | 17 | 0 |
Clinically relevant adverse reactions occurring in < 15% of patients included pneumonitis (14%), vomiting (14%), abdominal pain (14%), and stomatitis (6%).
Table 6 summarizes the laboratory abnormalities in ARROW.
| Laboratory Abnormality | GAVRETO N=281 |
|
|---|---|---|
| Grades 1-4 (%) | Grades 3-4 (%) |
|
| Chemistry | ||
| Increased AST | 80 | 3.2 |
| Increased ALT | 58 | 3.9 |
| Decreased albumin | 52 | 0 |
| Decreased calcium (corrected) | 50 | 1.8 |
| Decreased phosphate | 50 | 17 |
| Increased creatinine | 45 | 1.4 |
| Increased alkaline phosphatase | 43 | 2.5 |
| Decreased sodium | 42 | 10 |
| Decreased Potassium | 27 | 4.6 |
| Increased Potassium | 27 | 1.8 |
| Decreased Magnesium | 25 | 0 |
| Increased Bilirubin | 20 | 1.8 |
| Hematology | ||
| Decreased leukocytes | 79 | 11 |
| Decreased hemoglobin | 78 | 18 |
| Decreased lymphocytes | 73 | 32 |
| Decreased neutrophils | 70 | 21 |
| Decreased platelets | 33 | 5 |
Clinically relevant laboratory abnormalities occurring in < 20% of patients who received GAVRETO included increased magnesium (14%).
RET-altered Thyroid Cancer
The safety of GAVRETO was evaluated as a single agent at 400 mg orally once daily in 138 patients with RET-altered Thyroid Cancer (including 19 patients with RET fusion-positive thyroid cancer) in ARROW [see Clinical Studies (14.2)]. Among the 138 patients who received GAVRETO, 68% were exposed for 6 months or longer, and 40% were exposed for greater than one year.
The median age was 59 years (range: 18 to 83 years); 36% were female, 74% were White, 17% were Asian, and 6% were Hispanic/Latino.
Serious adverse reactions occurred in 39% of patients who received GAVRETO. The most frequent serious adverse reactions (in ≥ 2% of patients) were pneumonia, pneumonitis, urinary tract infection, pyrexia, fatigue, diarrhea, dizziness, anemia, hyponatremia, and ascites. Fatal adverse reaction occurred in 2.2% of patients; fatal adverse reactions that occurred in > 1 patient included pneumonia (n=2).
Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received GAVRETO. Adverse reactions resulting in permanent discontinuation which occurred in > 1 patient included fatigue, pneumonia and anemia.
Dosage interruptions due to an adverse reaction occurred in 67% of patients who received GAVRETO. Adverse reactions requiring dosage interruption in ≥ 2% of patients included neutropenia, hypertension, diarrhea, fatigue, pneumonitis, anemia, increased blood creatine phosphokinase, pneumonia, urinary tract infection, musculoskeletal pain, vomiting, pyrexia, increased AST, dyspnea, hypocalcemia, cough, thrombocytopenia, abdominal pain, increased blood creatinine, dizziness, headache, decreased lymphocyte count, stomatitis, and syncope.
Dose reductions due to adverse reactions occurred in 44% of patients who received GAVRETO. Adverse reactions requiring dosage reductions in ≥ 2% of patients included neutropenia, anemia, hypertension, increased blood creatine phosphokinase, decreased lymphocyte count, pneumonitis, fatigue and thrombocytopenia.
Table 7 summarizes the adverse reactions occurring in RET-altered Thyroid Cancer Patients in ARROW.
| Adverse Reactions | GAVRETO N=138 |
|
|---|---|---|
| Grades 1-4 (%) | Grades 3-4 (%) |
|
|
||
| Musculoskeletal | ||
| Musculoskeletal Pain* | 42 | 0.7† |
| Gastrointestinal | ||
| Constipation | 41 | 0.7† |
| Diarrhea‡ | 34 | 5† |
| Abdominal Pain§ | 17 | 0.7† |
| Dry mouth | 17 | 0 |
| Stomatitis¶ | 17 | 0.7† |
| Nausea | 17 | 0.7† |
| Vascular | ||
| Hypertension | 40 | 21† |
| General | ||
| Fatigue# | 38 | 6† |
| EdemaÞ | 29 | 0 |
| Pyrexia | 22 | 2.2† |
| Respiratory | ||
| Coughß | 27 | 1.4† |
| Dyspneaà | 22 | 2.2† |
| Nervous System | ||
| Headacheè | 24 | 0 |
| Peripheral Neuropathyð | 20 | 0 |
| Dizzinessø | 19 | 0.7† |
| Dysgeusiaý | 17 | 0 |
| Skin and Subcutaneous | ||
| Rash£ | 24 | 0 |
| Metabolism and Nutrition | ||
| Decreased Appetite | 15 | 0 |
Clinically relevant adverse reactions in < 15% of patients who received GAVRETO included tumor lysis syndrome and increased creatine phosphokinase.
Table 8 summarizes the laboratory abnormalities occurring in RET-altered Thyroid Cancer Patients in ARROW.
| Laboratory Abnormality | GAVRETO N=138 |
|
|---|---|---|
| Grades 1-4 (%) | Grades 3-4 (%) |
|
| Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available, which ranged from 135 to 138 patients. | ||
| Chemistry | ||
| Decreased calcium (corrected) | 70 | 9 |
| Increased AST | 69 | 4.3 |
| Increased ALT | 43 | 3.6 |
| Increased creatinine | 41 | 0 |
| Decreased albumin | 41 | 1.5 |
| Decreased sodium | 28 | 2.2 |
| Decreased phosphate | 28 | 8 |
| Decreased magnesium | 27 | 0.7 |
| Increased potassium | 26 | 1.4 |
| Increased bilirubin | 24 | 1.4 |
| Increased alkaline phosphatase | 22 | 1.4 |
| Hematology | ||
| Decreased lymphocytes | 67 | 27 |
| Decreased hemoglobin | 63 | 13 |
| Decreased neutrophils | 59 | 16 |
| Decreased platelets | 31 | 2.9 |
Clinically relevant laboratory abnormalities in patients who received GAVRETO included increased phosphate (40%).
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on GAVRETO
Strong or Moderate CYP3A and/or P-gp Inhibitors
Concomitant use with a strong or moderate CYP3A inhibitor and/or a P-gp inhibitor increases pralsetinib exposure [Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to GAVRETO. Avoid coadministration of GAVRETO with a strong or moderate CYP3A and/or P-gp inhibitor. If coadministration with any of the above inhibitors cannot be avoided, reduce the GAVRETO dose [see Dosage and Administration (2.4)].
Strong or Moderate CYP3A Inducers
Concomitant use with a strong CYP3A inducer decreases pralsetinib exposure [see Clinical Pharmacology (12.3)], which may decrease efficacy of GAVRETO. Avoid concomitant use of GAVRETO with strong or moderate CYP3A inducers. If coadministration of GAVRETO with strong or moderate CYP3A inducers cannot be avoided, increase the GAVRETO dose [see Dosage and Administration (2.5)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, GAVRETO can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on GAVRETO use in pregnant women to inform drug-associated risk. Oral administration of pralsetinib to pregnant rats during the period of organogenesis resulted in malformations and embryolethality at maternal exposures below the human exposure at the clinical dose of 400 mg once daily (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study, once daily oral administration of pralsetinib to pregnant rats during the period of organogenesis resulted in 100% post-implantation loss at dose levels ≥ 20 mg/kg (approximately 1.8 times the human exposure based on area under the curve [AUC] at the clinical dose of 400 mg). Post-implantation loss also occurred at the 10 mg/kg dose level (approximately 0.6 times the human exposure based on AUC at the clinical dose of 400 mg). Once daily oral administration of pralsetinib at dose levels ≥ 5 mg/kg (approximately 0.2 times the human AUC at the clinical dose of 400 mg) resulted in an increase in visceral malformations and variations (absent or small kidney and ureter, absent uterine horn, malpositioned kidney or testis, retroesophageal aortic arch) and skeletal malformations and variations (vertebral and rib anomalies and reduced ossification).
8.2 Lactation
Risk Summary
There are no data on the presence of pralsetinib or its metabolites in human milk or their effects on either the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with GAVRETO and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, GAVRETO can cause embryolethality and malformations at doses resulting in exposures below the human exposure at the clinical dose of 400 mg daily [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating GAVRETO [see Use in Specific Populations (8.1)].
Contraception
GAVRETO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Infertility
Based on histopathological findings in the reproductive tissues of male and female rats and a dedicated fertility study in which animals of both sexes were treated and mated to each other, GAVRETO may impair fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of GAVRETO have been established in pediatric patients aged 12 years and older for RET fusion-positive thyroid cancer. Use of GAVRETO in this age group is supported by evidence from an adequate and well-controlled study of GAVRETO in adults with additional population pharmacokinetic data demonstrating that age and body weight had no clinically meaningful effect on the pharmacokinetics of pralsetinib, that the exposure of pralsetinib is expected to be similar between adults and pediatric patients age 12 years and older, and that the course of RET fusion-positive thyroid cancer is sufficiently similar in adults and pediatric patients to allow extrapolation of data in adults to pediatric patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.2)].
The safety and effectiveness of GAVRETO have not been established in pediatric patients with RET fusion-positive NSCLC or in pediatric patients younger than 12 years old with RET fusion-positive thyroid cancer.
Animal Toxicity Data
In a 4-week repeat-dose toxicology study in non-human primates, physeal dysplasia in the femur occurred at doses resulting in exposures similar to the human exposure (AUC) at the clinical dose of 400 mg. In rats there were findings of increased physeal thickness in the femur and sternum as well as tooth (incisor) abnormalities (fractures, dentin matrix alteration, ameloblast/odontoblast degeneration, necrosis) in both 4- and 13-week studies at doses resulting in exposures similar to the human exposure (AUC) at the clinical dose of 400 mg. Recovery was not assessed in the 13-week toxicology study, but increased physeal thickness in the femur and incisor degeneration did not show evidence of complete recovery in the 28-day rat study.
Monitor growth plates in adolescent patients with open growth plates. Consider interrupting or discontinuing therapy based on the severity of any growth plate abnormalities and based on an individual risk-benefit assessment.
8.5 Geriatric Use
Of the 540 patients in ARROW who received the recommended dose of GAVRETO at 400 mg once daily, 31% were 65 years or and over, while 7% were 75 years and over.
No overall differences in pharmacokinetics (PK), safety or effectiveness were observed between patients aged 65 years or older and younger patients.
11 DESCRIPTION
Pralsetinib is an oral receptor tyrosine kinase inhibitor. The chemical name for pralsetinib is (cis)-N-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-(5-methyl-1H-pyrazol-3-ylamino)pyrimidin-2-yl)cyclohexanecarboxamide. The molecular formula for pralsetinib is C27H32FN9O2, and the molecular weight is 533.61 g/mol. Pralsetinib has the following structure:
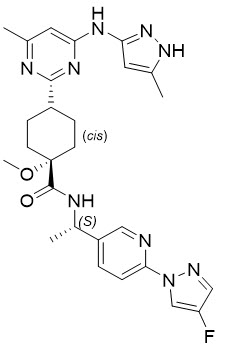
The solubility of pralsetinib in aqueous media decreases over the range pH 1.99 to pH 7.64 from 0.880 mg/mL to < 0.001 mg/mL, indicating a decrease in solubility with increasing pH.
GAVRETO (pralsetinib) is supplied for oral use as immediate release hydroxypropyl methylcellulose (HPMC) hard capsules containing 100 mg pralsetinib. The capsules also contain inactive ingredients:
citric acid, hydroxypropyl methylcellulose (HPMC), magnesium stearate, microcrystalline cellulose (MCC), pregelatinized starch and sodium bicarbonate. The capsule shell consists of FD&C Blue #1 (Brilliant Blue FCF), hypromellose and titanium dioxide. The white printing ink contains butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, shellac, strong ammonia solution and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pralsetinib is a kinase inhibitor of wild-type RET and oncogenic RET fusions (CCDC6-RET) and mutations (RET V804L, RET V804M and RET M918T) with half maximal inhibitory concentrations (IC50s) less than 0.5 nM. In purified enzyme assays, pralsetinib inhibited DDR1, TRKC, FLT3, JAK1-2, TRKA, VEGFR2, PDGFRB, and FGFR1 at higher concentrations that were still clinically achievable at Cmax. In cellular assays, pralsetinib inhibited RET at approximately 14-, 40-, and 12-fold lower concentrations than VEGFR2, FGFR2, and JAK2, respectively.
Certain RET fusion proteins and activating point mutations can drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to uncontrolled cell proliferation. Pralsetinib exhibited anti-tumor activity in cultured cells and animal tumor implantation models harboring oncogenic RET fusions or mutations including KIF5B-RET, CCDC6-RET, RET M918T, RET C634W, RET V804E, RET V804L and RET V804M. In addition, pralsetinib prolonged survival in mice implanted intracranially with tumor models expressing KIF5B-RET or CCDC6-RET.
12.2 Pharmacodynamics
Pralsetinib exposure-response relationships and the time course of pharmacodynamics response have not been fully characterized.
12.3 Pharmacokinetics
At 400 mg GAVRETO once daily under fasting conditions, the steady state mean [% coefficient of variation (CV%)] of maximum observed plasma concentration (Cmax) and area under the concentration-time curve (AUC0-24h) of pralsetinib was 2,470 (55%) ng/mL and 36,700 (66%) h•ng/mL, respectively. Pralsetinib Cmax and AUC increased inconsistently over the dose range of 60 mg to 600 mg once daily (0.15 to 1.5 times the recommended dose). Pralsetinib plasma concentrations reached steady state by 3 to 5 days. The mean accumulation ratio was approximately 2-fold after once-daily oral administration.
Absorption
The median time to peak concentration (Tmax) ranged from 2 to 4 hours following single doses of pralsetinib 60 mg to 600 mg.
Food Effect
Following administration of a single dose of 200 mg with a high-fat meal, (approximately 800 to 1000 calories with 50 to 60% of calories from fat), the mean Cmax of pralsetinib was increased by 2.0-fold, the mean AUC0-INF was increased by 2.2-fold, and the median Tmax was delayed from 4 to 8.5 hours, compared to the fasted state.
Distribution
The mean (CV%) apparent volume of distribution (Vd/F) of pralsetinib is 303 L (68%). Protein binding of pralsetinib is 97% and is independent of concentration. The blood-to-plasma ratio is 0.6 to 0.7.
Elimination
The mean (±standard deviation) plasma elimination half-life (T½) of pralsetinib is 16 hours (10) following single doses and 20 hours (12) following multiple doses of pralsetinib. The mean (CV%) apparent oral clearance (CL/F) of pralsetinib is 11 L/h (66%) at steady state.
Specific Populations
No clinically significant differences in the PK of pralsetinib were observed based on age (19 to 87 years), sex, race (White, Black, or Asian), and body weight (32 to 128 kg). Mild and moderate renal impairment (CLcr 30 - 89 mL/min) had no effect on the exposure of pralsetinib. Pralsetinib has not been studied in patients with severe renal impairment (CLcr < 15 mL/min).
Drug Interaction Studies
Clinical Studies and Model-Informed Approach
CYP3A Inhibitors: Coadministration of multiple doses of CYP3A inhibitors increases pralsetinib Cmax and AUC.
| Type of inhibitor | Coadministered CYP3A Inhibitor | Increase in pralsetinib Cmax | Increase in pralsetinib AUC |
|---|---|---|---|
| Observed | |||
| P-gp and Strong CYP3A Inhibitor | Itraconazole (200 mg twice daily on Day 1, followed by 200 mg once daily) | 1.8-fold | 3.5-fold |
| Predicted | |||
| Strong CYP3A Inhibitors | Voriconazole (400 mg twice daily on Day 1, followed by 200 mg twice daily) | 1.2-fold | 2.2-fold |
| Combined P-gp and Moderate CYP3A Inhibitors | Verapamil (80 mg three times daily) | 1.6-fold | 2.1-fold |
| Moderate CYP3A Inhibitors | Fluconazole (400 mg once daily) | 1.2-fold | 1.7-fold |
P-gp Inhibitors: Coadministration of cyclosporine (single 600 mg dose) with a single 200 mg dose of pralsetinib in healthy subjects increased pralsetinib AUC0-inf by 1.8-fold and Cmax by 1.5-fold, relative to a 200 mg dose of pralsetinib administered alone.
Strong CYP3A Inducers: Coadministration of rifampin 600 mg once daily with a single GAVRETO 400 mg dose decreased pralsetinib AUC0-INF by 68% and Cmax by 30%.
Moderate CYP3A Inducers: Coadministration of multiple doses of efavirenz (600 mg once daily) is predicted to decrease the pralsetinib AUC by 45% and Cmax by 18%.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Pralsetinib is a time-dependent inhibitor of CYP3A and an inhibitor of CYP2C8, CYP2C9, and CYP3A, but not an inhibitor of CYP1A2, CYP2B6, CYP2C19 or CYP2D6 at clinically relevant concentrations.
Pralsetinib is an inducer of CYP2C8, CYP2C9, and CYP3A, but not an inducer of CYP1A2, CYP2B6, or CYP2C19 at clinically relevant concentrations.
Transporter Systems: Pralsetinib is a substrate of P-gp and BCRP, but not a substrate of BSEP, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, OAT1, or OAT3.
Pralsetinib is an inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, MATE1, MATE2-K, and BSEP, but not an inhibitor of OCT1, OCT2, and OAT1A3 at clinically relevant concentrations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with pralsetinib have not been conducted. Pralsetinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay with or without metabolic activation and was not clastogenic in either an in vitro micronucleus assay in TK6 cells or an in vivo bone marrow micronucleus assay in rats.
In a dedicated fertility and early embryonic development study conducted in treated male rats mated to treated female rats, although pralsetinib did not have clear effects on male or female mating performance or ability to become pregnant, at the 20 mg/kg dose level (approximately 2.9 times the human exposure (AUC) at the clinical dose of 400 mg based on toxicokinetic data from the 13-week rat toxicology study) 82% of female rats had totally resorbed litters, with 92% post-implantation loss (early resorptions); post-implantation loss occurred at doses as low as 5 mg/kg (approximately 0.35 times the human exposure (AUC) at the clinical dose of 400 mg based on toxicokinetic data from the 13-week rat toxicology study). In a separate fertility and early embryonic development study in which male rats administered 20 mg/kg pralsetinib were mated to untreated female rats, there were no clear pralsetinib-related effects on intrauterine survival of embryos or on male reproductive performance at a dose approximately 1.7 times the human exposure (AUC) at the clinical dose of 400 mg. In a 13-week repeat-dose toxicology study, male rats exhibited histopathological evidence of tubular degeneration/atrophy in the testis with secondary cellular debris and reduced sperm in the lumen of the epididymis, which correlated with lower mean testis and epididymis weights and gross observations of soft and small testis. Female rats exhibited degeneration of the corpus luteum in the ovary. For both sexes, these effects were observed at pralsetinib doses ≥ 10 mg/kg/day, approximately 1 times the human exposure based on AUC at the clinical dose of 400 mg.
13.2 Animal Toxicology and/or Pharmacology
In 28-day rat and monkey toxicology studies, once daily oral administration of pralsetinib resulted in histologic necrosis and hemorrhage in the heart of preterm decedents at exposures ≥ 1.3 times and ≥ 3.1 times, respectively, the human exposure based on AUC at the clinical dose of 400 mg. Pralsetinib induced hyperphosphatemia (rats) and multi-organ mineralization (rats and monkeys) in 13-week toxicology studies at exposures approximately 2.8 times and ≥ 0.13 times, respectively, the human exposure based on AUC at the clinical dose of 400 mg.
14 CLINICAL STUDIES
14.1 Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer
The efficacy of GAVRETO was evaluated in patients with RET fusion-positive metastatic NSCLC in a multicenter, non-randomized, open-label, multi-cohort clinical trial (ARROW, NCT03037385). The study enrolled, in separate cohorts, patients with metastatic RET fusion-positive NSCLC who had progressed on platinum-based chemotherapy and treatment-naïve patients with metastatic NSCLC. Identification of a RET gene fusion was determined by local laboratories using next generation sequencing (NGS), fluorescence in situ hybridization (FISH), and other tests. Among the 237 patients in the efficacy population(s) described in this section, samples from 40% of patients were retrospectively tested with the LIFE Technologies Corporation Oncomine Dx Target Test (ODxTT). Patients with asymptomatic central nervous system (CNS) metastases, including patients with stable or decreasing steroid use within 2 weeks prior to study entry, were enrolled. Patients received GAVRETO 400 mg orally once daily until disease progression or unacceptable toxicity.
The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR), as assessed by a blinded independent central review (BICR) according to RECIST v1.1.
Metastatic RET Fusion-Positive NSCLC Previously Treated with Platinum Chemotherapy
Efficacy was evaluated in 130 patients with RET fusion-positive NSCLC with measurable disease who were previously treated with platinum chemotherapy enrolled into a cohort of ARROW.
The median age was 59 years (range: 26 to 85); 51% were female, 40% were White, 50% were Asian, 4.6% were Hispanic/Latino. ECOG performance status was 0-1 (95%) or 2 (3.8%), 99% of patients had metastatic disease, and 41% had either a history of or current CNS metastasis. Patients received a median of 2 prior systemic therapies (range 1–6); 42% had prior anti-PD-1/PD-L1 therapy and 27% had prior kinase inhibitors. A total of 48% of the patients received prior radiation therapy. RET fusions were detected in 80% of patients using NGS (37% tumor samples; 15% blood or plasma samples, 28% unknown), 13% using FISH, and 2% using other methods. The most common RET fusion partners were KIF5B (70%) and CCDC6 (19%).
Efficacy results for RET fusion-positive NSCLC patients who received prior platinum-based chemotherapy are summarized in Table 10.
| Efficacy Parameter | GAVRETO N=130 |
|---|---|
| NE = not estimable | |
| Overall Response Rate (ORR)* (95% CI) | 63 (54, 71) |
| Complete Response, % | 6 |
| Partial Response, % | 57 |
| Duration of Response (DOR) | N=82 |
| Median, months (95% CI) | 38.8 (14.8, NE) |
| Patients with DOR ≥ 12-months†, % | 66 |
For the 54 patients who received an anti-PD-1 or anti-PD-L1 therapy, either sequentially or concurrently with platinum-based chemotherapy, an exploratory subgroup analysis of ORR was 59% (95% CI: 45, 72) and the median DOR was 22.3 months (95% CI: 8.0, NE).
Among the 130 patients with RET-fusion positive NSCLC, 10 had measurable CNS metastases at baseline as assessed by BICR. No patients received radiation therapy (RT) to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 7 of these 10 patients including 2 patients with a CNS complete response; 71% of responders had a DOR of ≥ 6 months.
Treatment-naïve RET Fusion-Positive NSCLC
Efficacy was evaluated in 107 patients with treatment-naïve RET fusion-positive NSCLC with measurable disease enrolled into ARROW.
The median age was 62 years (range 30 to 87); 53% were female, 49% were White, 45% were Asian, and 2.8% were Hispanic or Latino. ECOG performance status was 0-1 for 99% of the patients and 98% of patients had metastatic disease; 28% had either history of or current CNS metastasis. RET fusions were detected in 68% of patients using NGS (30% tumor samples; 17% blood or plasma; 22% unknown) and 19% using FISH. The most common RET fusion partners were KIF5B (71%) and CCDC6 (18%).
Efficacy results for treatment-naïve RET fusion-positive NSCLC are summarized in Table 11.
| Efficacy Parameter | GAVRETO N=107 |
|---|---|
| Overall Response Rate (ORR)* (95% CI) | 78 (68, 85) |
| Complete Response, % | 7 |
| Partial Response, % | 71 |
| Duration of Response (DOR) | N=83 |
| Median, months (95% CI) | 13.4 (9.4, 23.1) |
| Patients with DOR ≥ 12-months†, % | 45 |
14.2 RET Fusion-Positive Thyroid Cancer
The efficacy of GAVRETO was evaluated in RET fusion-positive metastatic thyroid cancer patients in a multicenter, open-label, multi-cohort clinical trial (ARROW, NCT03037385). All patients with RET fusion-positive thyroid cancer were required to have disease progression following standard therapy, measurable disease by RECIST version 1.1, and have RET fusion status as detected by local testing (89% NGS tumor samples and 11% using FISH).
The median age was 61 years (range: 46 to 74); 67% were male, 78% were White, 22% were Asian, 11% were Hispanic/Latino. All patients (100%) had papillary thyroid cancer. ECOG performance status was 0-1 (100%), all patients (100%) had metastatic disease, and 56% had a history of CNS metastases. Patients had received a median of 2 prior therapies (range 1-8). Prior systemic treatments included prior radioactive iodine (100%) and prior sorafenib and/or lenvatinib (56%).
Efficacy results are summarized in Table 12.
| Efficacy Parameters | GAVRETO N=9 |
|---|---|
| NR = Not Reached; NE = Not Estimable | |
| Overall Response Rate (ORR)* (95% CI) | 89 (52, 100) |
| Complete Response, % | 0 |
| Partial Response, % | 89 |
| Duration of Response (DOR) | N=8 |
| Median in months (95% CI) | NR (NE, NE) |
| Patients with DOR ≥ 6 months†, % | 100 |
16 HOW SUPPLIED/STORAGE AND HANDLING
GAVRETO (pralsetinib) 100 mg, light blue, opaque, immediate release, hydroxypropyl methylcellulose (HPMC) hard capsule printed with "BLU-667" on the capsule shell body and "100 mg" on the capsule shell cap are supplied as follows:
- Bottles of 60 capsules (NDC 50242-210-60).
- Bottles of 90 capsules (NDC 50242-210-90).
- Bottles of 120 capsules (NDC 50242-210-12).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
ILD/Pneumonitis
Advise patients to contact their healthcare provider if they experience new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Hypertension
Advise patients that they will require regular blood pressure monitoring and to contact their healthcare provider if they experience symptoms of increased blood pressure or elevated readings [see Warnings and Precautions (5.2)].
Hepatotoxicity
Advise patients that hepatotoxicity can occur and to immediately contact their healthcare provider for signs or symptoms of hepatotoxicity [see Warnings and Precautions (5.3)].
Hemorrhagic Events
Advise patients that GAVRETO may increase the risk for bleeding and to contact their healthcare provider if they experience any signs or symptoms of bleeding [see Warnings and Precautions (5.4)].
Tumor Lysis Syndrome
Advise patients to contact their healthcare provider promptly to report any signs and symptoms of TLS [see Warnings and Precautions (5.5)].
Risk of Impaired Wound Healing
Advise patients that GAVRETO may impair wound healing. Advise patients that temporary interruption of GAVRETO is recommended prior to any elective surgery [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.7), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective non-hormonal contraception during the treatment with GAVRETO and for 2 weeks after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with GAVRETO and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with GAVRETO and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential that GAVRETO may impair fertility [See Use in Specific Populations (8.3)].
Drug Interactions
Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7.1)].
Administration
Advise patients to take GAVRETO on an empty stomach (no food intake for at least 2 hours before and at least 1 hour after taking GAVRETO) [see Dosage and Administration (2.2)].
GAVRETO® [pralsetinib]
Manufactured for:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
Jointly marketed by: Genentech USA, Inc. and Blueprint Medicines Corporation
GAVRETO® is a registered trademark of Blueprint Medicines Corporation.
© 2024 Genentech, Inc.
© 2024 Blueprint Medicines Corporation
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 08/2023 | ||||
| PATIENT INFORMATION
GAVRETO® (gav-REH-toh) (pralsetinib) capsules |
|||||
| What is GAVRETO? | |||||
GAVRETO is a prescription medicine used to treat certain cancers caused by abnormal rearranged during transfection (RET) genes in:
|
|||||
| Your healthcare provider will perform a test to make sure that GAVRETO is right for you. | |||||
It is not known if GAVRETO is safe and effective when used to treat cancers caused by abnormal RET genes:
|
|||||
Before taking GAVRETO, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking GAVRETO with other medicines may affect how GAVRETO works. | |||||
How should I take GAVRETO?
|
|||||
| What are the possible side effects of GAVRETO? | |||||
GAVRETO may cause serious side effects, including:
|
|||||
|
|
|
|||
|
|||||
|
|
||||
|
|||||
|
|
||||
|
|||||
|
|
||||
|
|||||
|
|
||||
|
|||||
| The most common side effects of GAVRETO include: | |||||
|
|
||||
The most common severe abnormal blood test results with GAVRETO include:
|
|||||
| These are not all the possible side effects of GAVRETO. | |||||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||||
How should I store GAVRETO?
|
|||||
| Keep GAVRETO and all medicines out of the reach of children. | |||||
| General information about the safe and effective use of GAVRETO. | |||||
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use GAVRETO for a condition for which it was not prescribed. Do not give GAVRETO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about GAVRETO that is written for health professionals. | |||||
| What are the ingredients in GAVRETO? | |||||
| Active ingredient: pralsetinib | |||||
| Inactive ingredients: citric acid, hydroxypropyl methylcellulose (HPMC), magnesium stearate, microcrystalline cellulose (MCC), pregelatinized starch and sodium bicarbonate. | |||||
| Capsule shell: FD&C Blue #1 (Brilliant Blue FCF), hypromellose and titanium dioxide. | |||||
| White printing ink: butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, shellac, strong ammonia solution and titanium dioxide. | |||||
| Manufactured for: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 | |||||
| Jointly marketed by: Genentech USA, Inc. and Blueprint Medicines Corporation | |||||
| GAVRETO® is a registered trademark of Blueprint Medicines Corporation. | |||||
| © 2023 Genentech, Inc. | |||||
| © 2023 Blueprint Medicines Corporation. | |||||
| For more information, go to www.GAVRETO.com or call 1-888-835-2555. | |||||
