DESCRIPTION
Felodipine is a calcium antagonist (calcium channel blocker). Felodipine is a dihydropyridine derivative that is chemically described as ± ethyl methyl 4-(2,3-dichlorophenyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate. Its molecular formula is C18H19Cl2NO4 and its structural formula is:
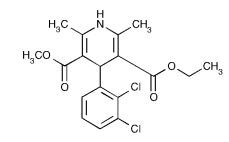
Felodipine, USP is a light yellow to yellow crystalline powder with a molecular weight of 384.26. It is insoluble in water and is freely soluble in acetone and in methanol; very slightly soluble in heptane. Felodipine is a racemic mixture.
Felodipine extended-release tablets, USP provide extended release of felodipine. They are available as tablets containing 2.5 mg, 5 mg or 10 mg of felodipine, USP for oral administration. Inactive ingredients are: calcium silicate, castor oil polyethoxylated 40, hydroxypropyl cellulose, hypromellose, lactose monohydrate, microcrystalline cellulose, propyl gallate, sodium stearyl fumarate, titanium dioxide, and triacetin. In addition, the 2.5 mg tablet strength contains FD&C Blue 2/indigo carmine aluminum lake and iron oxide yellow and the 5 mg and 10 mg strengths contain iron oxide red.
Felodipine extended-release tablets, USP complies with USP Dissolution Test I and Test II.
CLINICAL PHARMACOLOGY
Mechanism of Action
Felodipine is a member of the dihydropyridine class of calcium channel antagonists (calcium channel blockers). It reversibly competes with nitrendipine and/or other calcium channel blockers for dihydropyridine binding sites, blocks voltage-dependent Ca++ currents in vascular smooth muscle and cultured rabbit atrial cells, and blocks potassium-induced contracture of the rat portal vein.
In vitro studies show that the effects of felodipine on contractile processes are selective, with greater effects on vascular smooth muscle than cardiac muscle. Negative inotropic effects can be detected in vitro, but such effects have not been seen in intact animals.
The effect of felodipine on blood pressure is principally a consequence of a dose-related decrease of peripheral vascular resistance in man, with a modest reflex increase in heart rate (see Cardiovascular Effects). With the exception of a mild diuretic effect seen in several animal species and man, the effects of felodipine are accounted for by its effects on peripheral vascular resistance.
Pharmacokinetics and Metabolism
Following oral administration, felodipine is almost completely absorbed and undergoes extensive first-pass metabolism. The systemic bioavailability of felodipine is approximately 20%. Mean peak concentrations following the administration of felodipine extended-release tablets, USP are reached in 2.5 to 5 hours. Both peak plasma concentration and the area under the plasma concentration time curve (AUC) increase linearly with doses up to 20 mg. Felodipine is greater than 99% bound to plasma proteins.
Following intravenous administration, the plasma concentration of felodipine declined triexponentially with mean disposition half-lives of 4.8 minutes, 1.5 hours, and 9.1 hours. The mean contributions of the three individual phases to the overall AUC were 15, 40 and 45%, respectively, in the order of increasing t1/2.
Following oral administration of the immediate-release formulation, the plasma level of felodipine also declined polyexponentially with a mean terminal t1/2 of 11 to 16 hours. The mean peak and trough steady-state plasma concentrations achieved after 10 mg of the immediate-release formulation given once a day to normal volunteers, were 20 and 0.5 nmol/L, respectively. The trough plasma concentration of felodipine in most individuals was substantially below the concentration needed to effect a half-maximal decline in blood pressure (EC50) [4 to 6 nmol/L for felodipine], thus precluding once-a-day dosing with the immediate-release formulation.
Following administration of a 10 mg dose of felodipine, the extended-release formulation, to young, healthy volunteers, mean peak and trough steady-state plasma concentrations of felodipine were 7 and 2 nmol/L, respectively. Corresponding values in hypertensive patients (mean age 64) after a 20 mg dose of felodipine extended-release were 23 and 7 nmol/L. Since the EC50 for felodipine is 4 to 6 nmol/L, a 5 mg to 10 mg dose of felodipine extended-release in some patients, and a 20 mg dose in others, would be expected to provide an antihypertensive effect that persists for 24 hours (see Cardiovascular Effects below and DOSAGE AND ADMINISTRATION).
The systemic plasma clearance of felodipine in young healthy subjects is about 0.8 L/min, and the apparent volume of distribution is about 10 L/kg.
Following an oral or intravenous dose of 14C-labeled felodipine in man, about 70% of the dose of radioactivity was recovered in urine and 10% in the feces. A negligible amount of intact felodipine is recovered in the urine and feces (< 0.5%). Six metabolites, which account for 23% of the oral dose, have been identified; none has significant vasodilating activity.
Following administration of felodipine extended-release tablets, USP to hypertensive patients, mean peak plasma concentrations at steady-state are about 20% higher than after a single dose. Blood pressure response is correlated with plasma concentrations of felodipine.
The bioavailability of felodipine extended-release is influenced by the presence of food. When administered either with a high fat or carbohydrate diet, Cmax is increased by approximately 60%; AUC is unchanged. When felodipine extended-release tablet, USP was administered after a light meal (orange juice, toast, and cereal), however, there is no effect on felodipine’s pharmacokinetics. The bioavailability of felodipine was increased approximately two-fold when taken with grapefruit juice. Orange juice does not appear to modify the kinetics of felodipine extended- release. A similar finding has been seen with other dihydropyridine calcium antagonists, but to a lesser extent than that seen with felodipine.
Geriatric Use
Plasma concentrations of felodipine, after a single dose and at steady-state, increase with age. Mean clearance of felodipine in elderly hypertensives (mean age 74) was only 45% of that of young volunteers (mean age 26). At steady-state mean AUC for young patients was 39% of that for the elderly. Data for intermediate age ranges suggest that the AUCs fall between the extremes of the young and the elderly.
Hepatic Dysfunction
In patients with hepatic disease, the clearance of felodipine was reduced to about 60% of that seen in normal young volunteers.
Renal impairment does not alter the plasma concentration profile of felodipine; although higher concentrations of the metabolites are present in the plasma due to decreased urinary excretion, these are inactive.
Animal studies have demonstrated that felodipine crosses the blood-brain barrier and the placenta.
Cardiovascular Effects
Following administration of felodipine extended-release tablets, USP a reduction in blood pressure generally occurs within 2 to 5 hours. During chronic administration, substantial blood pressure control lasts for 24 hours, with trough reductions in diastolic blood pressure approximately 40 to 50% of peak reductions. The antihypertensive effect is dose dependent and correlates with the plasma concentration of felodipine.
A reflex increase in heart rate frequently occurs during the first week of therapy; this increase attenuates over time. Heart rate increases of 5 to 10 beats per minute may be seen during chronic dosing. The increase is inhibited by beta-blocking agents.
The P-R interval of the ECG is not affected by felodipine when administered alone or in combination with a beta-blocking agent. Felodipine alone or in combination with a beta-blocking agent has been shown, in clinical and electrophysiologic studies, to have no significant effect on cardiac conduction (P-R, P-Q, and H-V intervals).
In clinical trials in hypertensive patients without clinical evidence of left ventricular dysfunction, no symptoms suggestive of a negative inotropic effect were noted; however, none would be expected in this population (see PRECAUTIONS).
Renal/Endocrine Effects
Renal vascular resistance is decreased by felodipine while glomerular filtration rate remains unchanged. Mild diuresis, natriuresis, and kaliuresis have been observed during the first week of therapy. No significant effects on serum electrolytes were observed during short- and long-term therapy.
In clinical trials in patients with hypertension, increases in plasma noradrenaline levels have been observed.
Clinical Studies
Felodipine produces dose-related decreases in systolic and diastolic blood pressure as demonstrated in six placebo-controlled, dose response studies using either immediate-release or extended-release dosage forms. These studies enrolled over 800 patients on active treatment, at total daily doses ranging from 2.5 to 20 mg. In those studies felodipine was administered either as monotherapy or was added to beta-blockers. The results of the two studies with felodipine extended-release given once daily as monotherapy are shown in the table below:
MEAN REDUCTIONS IN BLOOD PRESSURE (mmHg)*
| * Placebo response subtracted † Different number of patients available for peak and trough measurements |
||||
| Dose | N | Systolic/Diastolic Mean Peak Response | Mean Trough Response | Trough/Peak Ratios (%s) |
| Study 1 (8 weeks) | ||||
| 2.5 mg | 68 | 9.4/4.7 | 2.7/2.5 | 29/53 |
| 5 mg | 69 | 9.5/6.3 | 2.4/3.7 | 25/59 |
| 10 mg | 67 | 18/10.8 | 10/6 | 56/56 |
| Study 2 (4 weeks) | ||||
| 10 mg | 50 | 5.3/7.2 | 1.5/3.2 | 33/40† |
| 20 mg | 50 | 11.3/10.2 | 4.5/3.2 | 43/34† |
INDICATIONS AND USAGE
Felodipine extended-release tablets, USP are indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure lowers the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including felodipine.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than 1 drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (eg, on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
Felodipine extended-release tablets, USP may be administered with other antihypertensive agents.
CONTRAINDICATIONS
Felodipine extended-release tablets, USP are contraindicated in patients who are hypersensitive to this product.
PRECAUTIONS
General
Hypotension
Felodipine, like other calcium antagonists, may occasionally precipitate significant hypotension and, rarely, syncope. It may lead to reflex tachycardia which in susceptible individuals may precipitate angina pectoris (see ADVERSE REACTIONS).
Heart Failure
Although acute hemodynamic studies in a small number of patients with NYHA Class II or III heart failure treated with felodipine have not demonstrated negative inotropic effects, safety in patients with heart failure has not been established. Caution, therefore, should be exercised when using felodipine extended-release in patients with heart failure or compromised ventricular function, particularly in combination with a beta-blocker.
Patients with Impaired Liver Function
Patients with impaired liver function may have elevated plasma concentrations of felodipine and may respond to lower doses of felodipine extended-release; therefore, a starting dose of 2.5 mg once a day is recommended. These patients should have their blood pressure monitored closely during dosage adjustment of felodipine extended-release (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Peripheral Edema
Peripheral edema, generally mild and not associated with generalized fluid retention, was the most common adverse event in the clinical trials. The incidence of peripheral edema was both dose and age dependent. Frequency of peripheral edema ranged from about 10% in patients under 50 years of age taking 5 mg daily to about 30% in those over 60 years of age taking 20 mg daily. This adverse effect generally occurs within 2 to 3 weeks of the initiation of treatment.
Information for Patients
Patients should be instructed to take felodipine extended-release tablet, USP whole and not to crush or chew the tablets. They should be told that mild gingival hyperplasia (gum swelling) has been reported. Good dental hygiene decreases its incidence and severity.
NOTE: As with many other drugs, certain advice to patients being treated with felodipine extended-release is warranted. This information is intended to aid in the safe and effective use of this medication. It is not a disclosure of all possible adverse or intended effects.
Drug Interactions
Felodipine is metabolized by CYP3A4. Co-administration of CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, erythromycin, grapefruit juice, cimetidine) with felodipine may lead to several-fold increases in the plasma levels of felodipine, either due to an increase in bioavailability or due to a decrease in metabolism. These increases in concentration may lead to increased effects, (lower blood pressure and increased heart rate). These effects have been observed with co-administration of itraconazole (a potent CYP3A4 inhibitor). Caution should be used when CYP3A4 inhibitors are co-administered with felodipine. A conservative approach to dosing felodipine should be taken. The following specific interactions have been reported:
Itraconazole
Co-administration of another extended release formulation of felodipine with itraconazole resulted in approximately 8-fold increase in the AUC, more than 6-fold increase in the Cmax, and 2-fold prolongation in the half-life of felodipine.
Erythromycin
Co-administration of felodipine with erythromycin resulted in approximately 2.5-fold increase in the AUC and Cmax, and about 2-fold prolongation in the half-life of felodipine.
Grapefruit Juice
Co-administration of felodipine with grapefruit juice resulted in more than 2-fold increase in the AUC and Cmax, but no prolongation in the half-life of felodipine.
Cimetidine
Co-administration of felodipine with cimetidine (a non-specific CYP-450 inhibitor) resulted in an increase of approximately 50% in the AUC and the Cmax, of felodipine.
Beta-Blocking Agents
A pharmacokinetic study of felodipine in conjunction with metoprolol demonstrated no significant effects on the pharmacokinetics of felodipine. The AUC and Cmax of metoprolol, however, were increased approximately 31 and 38%, respectively. In controlled clinical trials, however, beta-blockers including metoprolol were concurrently administered with felodipine and were well tolerated.
Digoxin
When given concomitantly with felodipine extended-release the pharmacokinetics of digoxin in patients with heart failure were not significantly altered.
Anticonvulsants
In a pharmacokinetic study, maximum plasma concentrations of felodipine were considerably lower in epileptic patients on long-term anticonvulsant therapy (e.g., phenytoin, carbamazepine, or phenobarbital) than in healthy volunteers. In such patients, the mean area under the felodipine plasma concentration-time curve was also reduced to approximately 6% of that observed in healthy volunteers. Since a clinically significant interaction may be anticipated, alternative antihypertensive therapy should be considered in these patients.
Tacrolimus
Felodipine may increase the blood concentration of tacrolimus. When given concomitantly with felodipine, the tacrolimus blood concentration should be followed and the tacrolimus dose may need to be adjusted.
Other Concomitant Therapy
In healthy subjects there were no clinically significant interactions when felodipine was given concomitantly with indomethacin or spironolactone.
Interaction with Food
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2 year carcinogenicity study in rats fed felodipine at doses of 7.7, 23.1 or 69.3 mg/kg/day (up to 61 times1 the maximum recommended human dose on a mg/m2 basis), a dose-related increase in the incidence of benign interstitial cell tumors of the testes (Leydig cell tumors) was observed in treated male rats. These tumors were not observed in a similar study in mice at doses up to 138.6 mg/kg/day (61 times1 the maximum recommended human dose on a mg/m2 basis). Felodipine, at the doses employed in the 2 year rat study, has been shown to lower testicular testosterone and to produce a corresponding increase in serum luteinizing hormone in rats. The Leydig cell tumor development is possibly secondary to these hormonal effects which have not been observed in man.
In this same rat study a dose-related increase in the incidence of focal squamous cell hyperplasia compared to control was observed in the esophageal groove of male and female rats in all dose groups. No other drug-related esophageal or gastric pathology was observed in the rats or with chronic administration in mice and dogs. The latter species, like man, has no anatomical structure comparable to the esophageal groove.
Felodipine was not carcinogenic when fed to mice at doses up to 138.6 mg/kg/day (61 times1 the maximum recommended human dose on a mg/m2 basis) for periods of up to 80 weeks in males and 99 weeks in females.
Felodipine did not display any mutagenic activity in vitro in the Ames microbial mutagenicity test or in the mouse lymphoma forward mutation assay. No clastogenic potential was seen in vivo in the mouse micronucleus test at oral doses up to 2500 mg/kg (1,100 times1 the maximum recommended human dose on a mg/m2 basis) or in vitro in a human lymphocyte chromosome aberration assay.
A fertility study in which male and female rats were administered doses of 3.8, 9.6 or 26.9 mg/kg/day (up to 24 times1 the maximum recommended human dose on a mg/m2 basis) showed no significant effect of felodipine on reproductive performance.
1 Based on patient weight of 50 kg
Pregnancy
Pregnancy Category C
Teratogenic Effects
Studies in pregnant rabbits administered doses of 0.46, 1.2, 2.3 and 4.6 mg/kg/day (from 0.8 to 8 times1 the maximum recommended human dose on a mg/m2 basis) showed digital anomalies consisting of reduction in size and degree of ossification of the terminal phalanges in the fetuses. The frequency and severity of the changes appeared dose related and were noted even at the lowest dose. These changes have been shown to occur with other members of the dihydropyridine class and are possibly a result of compromised uterine blood flow. Similar fetal anomalies were not observed in rats given felodipine.
In a teratology study in cynomolgus monkeys, no reduction in the size of the terminal phalanges was observed, but an abnormal position of the distal phalanges was noted in about 40% of the fetuses.
Nonteratogenic Effects
A prolongation of parturition with difficult labor and an increased frequency of fetal and early postnatal deaths were observed in rats administered doses of 9.6 mg/kg/day (8 times1 the maximum human dose on a mg/m2 basis) and above.
Significant enlargement of the mammary glands, in excess of the normal enlargement for pregnant rabbits, was found with doses greater than or equal to 1.2 mg/kg/day (2.1 times the maximum human dose on a mg/m2 basis). This effect occurred only in pregnant rabbits and regressed during lactation. Similar changes in the mammary glands were not observed in rats or monkeys.
There are no adequate and well-controlled studies in pregnant women. If felodipine is used during pregnancy, or if the patient becomes pregnant while taking this drug, she should be apprised of the potential hazard to the fetus, possible digital anomalies of the infant, and the potential effects of felodipine on labor and delivery and on the mammary glands of pregnant females.
1 Based on patient weight of 50 kg
Nursing Mothers
It is not known whether this drug is secreted in human milk and because of the potential for serious adverse reactions from felodipine in the infant, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of felodipine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Pharmacokinetics, however, indicate that the availability of felodipine is increased in older patients (see CLINICAL PHARMACOLOGY: Geriatric Use). In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
In controlled studies in the United States and overseas, approximately 3000 patients were treated with felodipine as either the extended-release or the immediate-release formulation.
The most common clinical adverse events reported with felodipine extended-release administered as monotherapy at the recommended dosage range of 2.5 mg to 10 mg once a day were peripheral edema and headache. Peripheral edema was generally mild, but it was age and dose related and resulted in discontinuation of therapy in about 3% of the enrolled patients. Discontinuation of therapy due to any clinical adverse event occurred in about 6% of the patients receiving felodipine extended-release, principally for peripheral edema, headache, or flushing.
Adverse events that occurred with an incidence of 1.5% or greater at any of the recommended doses of 2.5 mg to 10 mg once a day (felodipine extended-release, N = 861; Placebo, N = 334), without regard to causality, are compared to placebo and are listed by dose in the table below. These events are reported from controlled clinical trials with patients who were randomized to a fixed dose of felodipine extended-release tablets, USP or titrated from an initial dose of 2.5 mg or 5 mg once a day. A dose of 20 mg once a day has been evaluated in some clinical studies. Although the antihypertensive effect of felodipine extended-release tablets, USP is increased at 20 mg once a day, there is a disproportionate increase in adverse events, especially those associated with vasodilatory effects (see DOSAGE AND ADMINISTRATION).
| Body System Adverse Events | Placebo N = 334 | 2.5 mg N = 255 | 5 mg N = 581 | 10 mg N = 408 |
| Body as a Whole | ||||
| Peripheral Edema | 3.3 (0) | 2.0 (0) | 8.8 (2.2) | 17.4 (2.5) |
| Asthenia | 3.3 (0) | 3.9 (0) | 3.3 (0) | 2.2 (0) |
| Warm Sensation | 0 (0) | 0 (0) | 0.9 (0.2) | 1.5 (0) |
| Cardiovascular | ||||
| Palpitation | 2.4 (0) | 0.4 (0) | 1.4 (0.3) | 2.5 (0.5) |
| Digestive | ||||
| Nausea | 1.5 (0.9) | 1.2 (0) | 1.7 (0.3) | 1 (0.7) |
| Dyspepsia | 1.2 (0) | 3.9 (0) | 0.7 (0) | 0.5 (0) |
| Constipation | 0.9 (0) | 1.2 (0) | 0.3 (0) | 1.5 (0.2) |
| Nervous | ||||
| Headache | 10.2 (0.9) | 10.6 (0.4) | 11.0 (1.7) | 14.7 (2.0) |
| Dizziness | 2.7 (0.3) | 2.7 (0) | 3.6 (0.5) | 3.7 (0.5) |
| Paresthesia | 1.5 (0.3) | 1.6 (0) | 1.2 (0) | 1.2 (0.2) |
| Respiratory | ||||
| Upper Respiratory Infection | 1.8 (0) | 3.9 (0) | 1.9 (0) | 0.7 (0) |
| Cough | 0.3 (0) | 0.8 (0) | 1.2 (0) | 1.7 (0) |
| Rhinorrhea | 0 (0) | 1.6 (0) | 0.2 (0) | 0.2 (0) |
| Sneezing | 0 (0) | 1.6 (0) | 0 (0) | 0 (0) |
| Skin | ||||
| Rash | 0.9 (0) | 2 (0) | 0.2 (0) | 0.2 (0) |
| Flushing | 0.9 (0) | 3.9 (0) | 5.3 (0.7) | 6.9 (1.2) |
| *Patients in titration studies may have been exposed to more than one dose level of felodipine extended-release tablets. | ||||
Adverse events that occurred in 0.5% up to 1.5% of patients who received felodipine extended-release in all controlled clinical trials at the recommended dosage range of 2.5 mg to 10 mg once a day, and serious adverse events that occurred at a lower rate, or events reported during marketing experience (those lower rate events are in italics) are listed below. These events are listed in order of decreasing severity within each category, and the relationship of these events to administration of felodipine extended-release is uncertain:
Body as a Whole: Chest pain, facial edema, flu-like illness
Cardiovascular: Myocardial infarction, hypotension, syncope, angina pectoris, arrhythmia, tachycardia, premature beats
Digestive: Abdominal pain, diarrhea, vomiting, dry mouth, flatulence, acid regurgitation
Endocrine: Gynecomastia
Hematologic: Anemia
Metabolic: ALT (SGPT) increased
Musculoskeletal: Arthralgia, back pain, leg pain, foot pain, muscle cramps, myalgia, arm pain, knee pain, hip pain
Nervous/Psychiatric: Insomnia, depression, anxiety disorders, irritability, nervousness, somnolence, decreased libido
Respiratory: Dyspnea, pharyngitis, bronchitis, influenza, sinusitis, epistaxis, respiratory infection
Skin: Angioedema, contusion, erythema, urticaria, leukocytoclastic vasculitis
Special Senses: Visual disturbances
Urogenital: Impotence, urinary frequency, urinary urgency, dysuria, polyuria.
Gingival Hyperplasia: Gingival hyperplasia, usually mild, occurred in < 0.5% of patients in controlled studies. This condition may be avoided or may regress with improved dental hygiene. (see PRECAUTIONS: Information for Patients).
Clinical Laboratory Test Findings
Serum Electrolytes
No significant effects on serum electrolytes were observed during short- and long-term therapy (see CLINICAL PHARMACOLOGY: Renal/Endocrine Effects).
Serum Glucose
No significant effects on fasting serum glucose were observed in patients treated with felodipine extended-release in the U.S. controlled study.
Liver Enzymes
One of two episodes of elevated serum transaminases decreased once drug was discontinued in clinical studies; no follow-up was available for the other patient.
OVERDOSAGE
Oral doses of 240 mg/kg and 264 mg/kg in male and female mice, respectively, and 2390 mg/kg and 2250 mg/kg in male and female rats, respectively, caused significant lethality.
In a suicide attempt, one patient took 150 mg felodipine together with 15 tablets each of atenolol and spironolactone and 20 tablets of nitrazepam. The patient's blood pressure and heart rate were normal on admission to hospital; he subsequently recovered without significant sequelae.
Overdosage might be expected to cause excessive peripheral vasodilation with marked hypotension and possibly bradycardia.
If severe hypotension occurs, symptomatic treatment should be instituted. The patient should be placed supine with the legs elevated. The administration of intravenous fluids may be useful to treat hypotension due to overdosage with calcium antagonists. In case of accompanying bradycardia, atropine (0.5 to 1 mg) should be administered intravenously. Sympathomimetic drugs may also be given if the physician feels they are warranted.
It has not been established whether felodipine can be removed from the circulation by hemodialysis.
To obtain up-to-date information about the treatment of overdose, consult your Regional Poison-Control Center. Telephone numbers of certified poison-control centers are listed in the Physicians’ Desk Reference (PDR). In managing overdose, consider the possibilities of multiple-drug overdoses, drug-drug interactions, and unusual drug kinetics in your patient.
DOSAGE AND ADMINISTRATION
The recommended starting dose is 5 mg once a day. Depending on the patient's response, the dosage can be decreased to 2.5 mg or increased to 10 mg once a day. These adjustments should occur generally at intervals of not less than 2 weeks. The recommended dosage range is 2.5 to 10 mg once daily. In clinical trials, doses above 10 mg daily showed an increased blood pressure response but a large increase in the rate of peripheral edema and other vasodilatory adverse events (see ADVERSE REACTIONS). Modification of the recommended dosage is usually not required in patients with renal impairment.
Felodipine extended-release tablets, USP should regularly be taken either without food or with a light meal (see CLINICAL PHARMACOLOGY, Pharmacokinetics and Metabolism). Felodipine extended-release tablets, USP should be swallowed whole and not crushed or chewed.
Geriatric Use
Patients over 65 years of age are likely to develop higher plasma concentrations of felodipine (see CLINICAL PHARMACOLOGY). In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range (2.5 mg daily). Elderly patients should have their blood pressure closely monitored during any dosage adjustment.
Patients with Impaired Liver Function
Patients with impaired liver function may have elevated plasma concentrations of felodipine and may respond to lower doses of felodipine extended-release tablets, USP; therefore, patients should have their blood pressure monitored closely during dosage adjustment of felodipine extended-release tablets, USP (see CLINICAL PHARMACOLOGY).
HOW SUPPLIED
Felodipine Extended-release Tablets, USP are available containing 2.5 mg, 5 mg or 10 mg of Felodipine, USP.
The 2.5 mg tablet is a round, green tablet, debossed with “E 136” on one side and plain on the other side. They are available as follows:
Bottles of 100 tablets NDC 0603-3581-21
Bottles of 500 tablets NDC 0603-3581-28
The 5 mg tablet is a round, pink tablet, debossed with “E 137” on one side and plain on the other side. They are available as follows:
Bottles of 100 tablets NDC 0603-3582-21
Bottles of 500 tablets NDC 0603-3582-28
The 10 mg tablet is a red film-coated, round tablet with “E 138” debossed on one side and a plain face on the other side. They are available as follows:
Bottles of 100 tablets NDC 0603-3583-21
Bottles of 500 tablets NDC 0603-3583-28
Store at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.]
Protect from light.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Manufactured by:
Patheon Inc.
Whitby, Ontario, Canada L1N 5Z5
Distributed by:
Par Pharmaceutical
Chestnut Ridge, NY 10977
Revised: 01/2018


