FULL PRESCRIBING INFORMATION
WARNING: CYTOPENIAS, INFUSION-RELATED REACTIONS, AND INFECTIONS
Cytopenias: Serious, including fatal, pancytopenia/marrow hypoplasia, autoimmune idiopathic thrombocytopenia, and autoimmune hemolytic anemia can occur in patients receiving CAMPATH. Single doses of CAMPATH greater than 30 mg or cumulative doses greater than 90 mg per week increase the incidence of pancytopenia [see Warnings and Precautions (5.1)].
Infusion-Related Reactions: CAMPATH administration can result in serious, including fatal, infusion-related reactions. Carefully monitor patients during infusions and withhold CAMPATH for Grade 3 or 4 infusion-related reactions. Gradually escalate CAMPATH to the recommended dose at the initiation of therapy and after interruption of therapy for 7 or more days [see Dosage and Administration (2.1) and Warnings and Precautions (5.2)].
Immunosuppression/Infections: Serious, including fatal, bacterial, viral, fungal, and protozoan infections can occur in patients receiving CAMPATH. Administer prophylaxis against Pneumocystis jirovecii pneumonia (PCP) and herpes virus infections [see Dosage and Administration (2.2) and Warnings and Precautions (5.3)].
1 INDICATIONS AND USAGE
CAMPATH is indicated as a single agent for the treatment of B-cell chronic lymphocytic leukemia (B-CLL).
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Schedule and Administration
- Administer as an intravenous infusion over 2 hours. Do not administer as intravenous push or bolus.
- Recommended Dosing Regimen
- Gradually escalate to the maximum recommended single dose of 30 mg. Escalation is required at initiation of dosing or if dosing is held ≥7 days during treatment. Escalation to 30 mg ordinarily can be accomplished in 3 to 7 days.
- Escalation Strategy:
- –
- Administer 3 mg daily until infusion-related reactions are ≤ Grade 2 [see Adverse Reactions (6.1)].
- –
- Then administer 10 mg daily until infusion-related reactions are ≤ Grade 2.
- –
- Then administer 30 mg/day three times per week on alternate days (e.g., Mon-Wed-Fri). The total duration of therapy, including dose escalation, is 12 weeks.
- Single doses of greater than 30 mg or cumulative doses greater than 90 mg per week increase the incidence of pancytopenia.
2.2 Recommended Concomitant Medications
- Premedicate with diphenhydramine (50 mg) and acetaminophen (500–1000 mg) 30 minutes prior to first infusion and each dose escalation. Institute appropriate medical management (e.g., glucocorticoids, epinephrine, meperidine) for infusion-related reactions as needed [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
- Administer trimethoprim/sulfamethoxazole double strength (DS) twice daily 3 times per week (or equivalent) as Pneumocystis jirovecii pneumonia (PCP) prophylaxis.
- Administer famciclovir 250 mg BID or equivalent as herpetic prophylaxis.
Continue PCP and herpes viral prophylaxis for a minimum of 2 months after completion of CAMPATH or until the CD4+ count is ≥200 cells/µL, whichever occurs later [see Warnings and Precautions (5.3)].
2.3 Dosage Modification
- Withhold CAMPATH during serious infection or other serious adverse reactions until resolution.
- Discontinue CAMPATH for autoimmune anemia or autoimmune thrombocytopenia.
- There are no dose modifications recommended for lymphopenia.
| Hematologic Values | Dosage Modification* |
|---|---|
|
|
| ANC <250/μL and/or platelet count ≤25,000/μL | |
| For first occurrence: | Withhold CAMPATH therapy. Resume CAMPATH at 30 mg when ANC ≥500/μL and platelet count ≥50,000/μL. |
| For second occurrence: | Withhold CAMPATH therapy. Resume CAMPATH at 10 mg when ANC ≥500/μL and platelet count ≥50,000/μL. |
| For third occurrence: | Discontinue CAMPATH therapy. |
| ≥50% decrease from baseline in patients initiating therapy with a baseline ANC ≤250/μL and/or a baseline platelet count ≤25,000/μL | |
| For first occurrence: | Withhold CAMPATH therapy. Resume CAMPATH at 30 mg upon return to baseline value(s). |
| For second occurrence: | Withhold CAMPATH therapy. Resume CAMPATH at 10 mg upon return to baseline value(s). |
| For third occurrence: | Discontinue CAMPATH therapy. |
2.4 Preparation and Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. If particulate matter is present or the solution is discolored, discard the vial. DO NOT SHAKE VIAL.
Use aseptic technique during the preparation and administration of CAMPATH. Withdraw the necessary amount of CAMPATH from the vial into a syringe.
- To prepare the 3 mg dose, withdraw 0.1 mL into a 1 mL syringe calibrated in increments of 0.01 mL.
- To prepare the 10 mg dose, withdraw 0.33 mL into a 1 mL syringe calibrated in increments of 0.01 mL.
- To prepare the 30 mg dose, withdraw 1 mL in either a 1 mL or 3 mL syringe calibrated in 0.1 mL increments.
Inject syringe contents into 100 mL sterile 0.9% Sodium Chloride USP or 5% Dextrose in Water USP. Gently invert the bag to mix the solution. Discard syringe.
The vial contains no preservatives and is intended for single use only. DISCARD VIAL including any unused portion after withdrawal of dose.
Use within 8 hours after dilution. Store diluted CAMPATH at room temperature between 15°C to 30°C (59°F to 86°F) or refrigerated at 2°C to 8°C (36°F to 46°F). Protect from light.
3 DOSAGE FORMS AND STRENGTHS
Injection: 30 mg/1 mL as a clear, colorless solution in single-dose vial
5 WARNINGS AND PRECAUTIONS
5.1 Cytopenias
Severe, including fatal, autoimmune anemia and thrombocytopenia, and prolonged myelosuppression have been reported in patients receiving CAMPATH.
In addition, hemolytic anemia, pure red cell aplasia, bone marrow aplasia, and hypoplasia have been reported after treatment with CAMPATH at the recommended dose. Single doses of CAMPATH greater than 30 mg or cumulative doses greater than 90 mg per week increase the incidence of pancytopenia.
Withhold CAMPATH for severe cytopenias (except lymphopenia). Discontinue for autoimmune cytopenias or recurrent/persistent severe cytopenias (except lymphopenia) [see Dosage and Administration (2.3)]. No data exist on the safety of CAMPATH resumption in patients with autoimmune cytopenias or marrow aplasia [see Adverse Reactions (6.1)].
Obtain complete blood counts (CBC) at weekly intervals during CAMPATH therapy and more frequently if worsening anemia, neutropenia, or thrombocytopenia occurs. Assess CD4+ counts after treatment until recovery to ≥200 cells/µL [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.2 Infusion-Related Reactions
Adverse reactions occurring during or shortly after CAMPATH infusion include pyrexia, chills/rigors, nausea, hypotension, urticaria, dyspnea, rash, emesis, and bronchospasm [see Adverse Reactions (6.1)]. In clinical trials, the frequency of infusion-related reactions was highest in the first week of treatment. Monitor for the signs and symptoms listed above and withhold infusion for Grade 3 or 4 infusion-related reactions.
The following serious, including fatal, infusion-related reactions have been identified in postmarketing reports: syncope, pulmonary infiltrates, acute respiratory distress syndrome (ARDS), respiratory arrest, cardiac arrhythmias, myocardial infarction, acute cardiac insufficiency, cardiac arrest, angioedema, and anaphylactoid shock.
Initiate CAMPATH according to the recommended dose-escalation scheme [see Dosage and Administration (2.1)]. Premedicate patients with an antihistamine and acetaminophen prior to each dose. Institute appropriate medical management (e.g., glucocorticoids, epinephrine, meperidine) for infusion-related reactions as needed [see Dosage and Administration (2.2)]. If therapy is interrupted for 7 or more days, reinstitute CAMPATH with gradual dose escalation [see Dosage and Administration (2.1)].
5.3 Immunosuppression/Infections
CAMPATH treatment results in severe and prolonged lymphopenia with a concomitant increased incidence of opportunistic infections [see Adverse Reactions (6.1)]. Administer PCP and herpes viral prophylaxis during treatment with CAMPATH and for a minimum of 2 months after completion of CAMPATH or until the CD4+ count is ≥200 cells/µL, whichever occurs later [see Dosage and Administration (2.2)]. Prophylaxis does not eliminate these infections.
Routinely monitor patients for CMV infection during treatment with CAMPATH and for at least 2 months following completion of CAMPATH. Withhold CAMPATH for serious infections and during antiviral treatment for CMV infection or confirmed CMV viremia (defined as polymerase chain reaction [PCR] positive CMV in ≥2 consecutive samples obtained 1 week apart). Initiate therapeutic ganciclovir (or equivalent) for CMV infection or confirmed CMV viremia.
Epstein-Barr virus (EBV) infection, including severe and fatal EBV-associated hepatitis, has been reported in patients who received CAMPATH.
Monitor for sign and symptoms of EBV infections. Withhold CAMPATH for EBV reactivation or severe infection.
Administer only irradiated blood products to avoid transfusion associated Graft versus Host Disease (TAGVHD), unless emergent circumstances dictate immediate transfusion.
In patients who received CAMPATH as initial therapy, recovery of CD4+ counts to ≥200 cells/µL occurred by 6 months following completion of CAMPATH; however, at 2 months post treatment, the median was 183 cells/µL. In previously treated patients who received CAMPATH, the median time to recovery of CD4+ counts to ≥200 cells/µL was 2 months; however, full recovery (to baseline) of CD4+ and CD8+ counts may take more than 12 months [see Adverse Reactions (6)].
5.4 Immunization
The safety of immunization with live viral vaccines following CAMPATH therapy has not been studied. Do not administer live viral vaccines to patients or infants born to patients receiving CAMPATH. The ability to generate an immune response to any vaccine following CAMPATH therapy has not been studied.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label:
- Cytopenias [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- Immunosuppression/Infections [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data below reflect exposure to CAMPATH in 296 patients with CLL of whom 147 were previously untreated and 149 received at least 2 prior chemotherapy regimens. The median duration of exposure was 11.7 weeks for previously untreated patients and 8 weeks for previously treated patients.
The most common adverse reactions with CAMPATH are: infusion-related reactions (pyrexia, chills, hypotension, urticaria, nausea, rash, tachycardia, dyspnea), cytopenias (neutropenia, lymphopenia, thrombocytopenia, anemia), infections (CMV viremia, CMV infection, other infections), gastrointestinal symptoms (nausea, emesis, abdominal pain), and neurological symptoms (insomnia, anxiety). The most common serious adverse reactions are cytopenias, infusion-related reactions, and immunosuppression/infections.
Lymphopenia
Severe lymphopenia and a rapid and sustained decrease in lymphocyte subsets occurred in previously untreated and previously treated patients following administration of CAMPATH. In previously untreated patients, the median CD4+ was 0 cells/μL at one month after treatment and 238 cells/μL [25%–75% interquartile range 115 to 418 cells/μL at 6 months post treatment [see Warnings and Precautions (5.3)].
Neutropenia
In previously untreated patients, the incidence of Grade 3 or 4 neutropenia was 42% with a median time to onset of 31 days and a median duration of 37 days. In previously treated patients, the incidence of Grade 3 or 4 neutropenia was 64% with a median duration of 28 days. Ten percent of previously untreated patients and 17% of previously treated patients received granulocyte colony stimulating factors.
Anemia
In previously untreated patients, the incidence of Grade 3 or 4 anemia was 12% with a median time to onset of 31 days and a median duration of 8 days. In previously treated patients, the incidence of Grade 3 or 4 anemia was 38%. Seventeen percent of previously untreated patients and 66% of previously treated patients received either erythropoiesis stimulating agents, transfusions or both.
Thrombocytopenia
In previously untreated patients, the incidence of Grade 3 or 4 thrombocytopenia was 14% with a median time to onset of 9 days and a median duration of 14 days. In previously treated patients, the incidence of Grade 3 or 4 thrombocytopenia was 52% with a median duration of 21 days. Autoimmune thrombocytopenia was reported in 2% of previously treated patients with one fatality.
Infusion-Related Reactions
Infusion-related reactions, which included pyrexia, chills, hypotension, urticaria, and dyspnea, were common. Grade 3 and 4 pyrexia and/or chills occurred in approximately 10% of previously untreated patients and in approximately 35% of previously treated patients. The occurrence of infusion-related reactions was greatest during the initial week of treatment and decreased with subsequent doses of CAMPATH. All patients were pretreated with antipyretics and antihistamines; additionally, 43% of previously untreated patients received glucocorticoid pre-treatment.
Infections
In the study of previously untreated patients, patients were tested weekly for CMV using a PCR assay from initiation through completion of therapy, and every 2 weeks for the first 2 months following therapy. CMV infection occurred in 16% (23/147) of previously untreated patients; approximately one-third of these infections were serious or life threatening. In studies of previously treated patients in which routine CMV surveillance was not required, CMV infection was documented in 6% (9/149) of patients; nearly all of these infections were serious or life threatening.
Other infections were reported in approximately 50% of patients across all studies. Grade 3 to 5 sepsis ranged from 3% to 10% across studies and was higher in previously treated patients. Grade 3 to 4 febrile neutropenia ranged from 5% to 10% across studies and was higher in previously treated patients. Infection-related fatalities occurred in 2% of previously untreated patients and 16% of previously treated patients. There were 198 episodes of other infection in 109 previously untreated patients; 16% were bacterial, 7% were fungal, 4% were other viral, and in 73% the organism was not identified.
Cardiac
Cardiac dysrhythmias occurred in approximately 14% of previously untreated patients. The majority were tachycardias and were temporally associated with infusion; dysrhythmias were Grade 3 or 4 in 1% of patients.
Previously Untreated Patients
Table 1 contains selected adverse reactions observed in 294 patients randomized (1:1) to receive CAMPATH or chlorambucil as first line therapy for B-CLL. CAMPATH was administered at a dose of 30 mg intravenously three times weekly for up to 12 weeks. The median duration of therapy was 11.7 weeks with a median weekly dose of 82 mg (25%–75% interquartile range: 69–90 mg).
| CAMPATH (n=147) | Chlorambucil (n=147) | ||||
|---|---|---|---|---|---|
| All Grades†
% | Grades 3–4 % | All Grades % | Grades 3–4 % |
||
|
|||||
| Blood and Lymphatic System Disorders | Lymphopenia | 97 | 97 | 9 | 1 |
| Neutropenia | 77 | 42 | 51 | 26 | |
| Anemia | 76 | 13 | 54 | 18 | |
| Thrombocytopenia | 71 | 13 | 70 | 14 | |
| General Disorders and Administration Site Conditions | Pyrexia | 69 | 10 | 11 | 1 |
| Chills | 53 | 3 | 1 | 0 | |
| Infections and Infestations | CMV viremia‡ | 55 | 4 | 8 | 0 |
| CMV infection | 16 | 5 | 0 | 0 | |
| Other infections | 74 | 21 | 65 | 10 | |
| Skin and Subcutaneous Tissue Disorders | Urticaria | 16 | 2 | 1 | 0 |
| Rash | 13 | 1 | 4 | 0 | |
| Erythema | 4 | 0 | 1 | 0 | |
| Vascular Disorders | Hypotension | 16 | 1 | 0 | 0 |
| Hypertension | 14 | 5 | 2 | 1 | |
| Nervous System Disorders | Headache | 14 | 1 | 8 | 0 |
| Tremor | 3 | 0 | 1 | 0 | |
| Respiratory, Thoracic and Mediastinal Disorders | Dyspnea | 14 | 4 | 7 | 3 |
| Gastrointestinal Disorders | Diarrhea | 10 | 1 | 4 | 0 |
| Psychiatric Disorders | Insomnia | 10 | 0 | 3 | 0 |
| Anxiety | 8 | 0 | 1 | 0 | |
| Cardiac Disorders | Tachycardia | 10 | 0 | 1 | 0 |
Previously Treated Patients
Additional safety information was obtained from 3 single arm studies of 149 previously treated patients with CLL administered 30 mg CAMPATH intravenously three times weekly for 4 to 12 weeks (median cumulative dose 673 mg [range 2–1106 mg]; median duration of therapy 8.0 weeks). Adverse reactions in these studies not listed in Table 1 that occurred at an incidence rate of >5% were fatigue, nausea, emesis, musculoskeletal pain, anorexia, dysesthesia, mucositis, and bronchospasm.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies with the incidence of antibodies to other alemtuzumab products may be misleading.
Using an ELISA assay, anti-human antibodies (HAHA) were detected in 11 of 133 (8.3%) previously untreated patients. In addition, two patients were weakly positive for neutralizing activity. Limited data suggest that the anti-CAMPATH antibodies did not adversely affect tumor response. Four of 211 (1.9%) previously treated patients were found to have antibodies to CAMPATH following treatment.
6.3 Postmarketing Experience
CAMPATH
The following adverse reactions have been identified during postapproval use of CAMPATH. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
General Disorders and Administration Site Conditions: Fatal infusion-related reactions.
Cardiovascular Disorders: Congestive heart failure, cardiomyopathy, decreased ejection fraction (some patients had been previously treated with cardiotoxic agents).
Cerebrovascular Disorders: Cervicocephalic arterial dissection, stroke, including hemorrhagic and ischemic stroke.
Gastrointestinal Disorders: Acute acalculous cholecystitis.
Immune System Disorders: Goodpasture's syndrome, Graves' disease, aplastic anemia, Guillain-Barré syndrome, chronic inflammatory demyelinating polyradiculoneuropathy, serum sickness, fatal transfusion associated graft versus host disease, hemophagocytic lymphohistiocytosis (HLH).
Infections: Epstein-Barr virus (EBV) infection, progressive multifocal leukoencephalopathy (PML), reactivation of latent viruses.
Metabolism and Nutrition Disorders: Tumor lysis syndrome.
Neoplasms: EBV-associated lymphoproliferative disorder.
Nervous System Disorders: Optic neuropathy.
Renal and Urinary Disorders: Glomerular nephropathies.
Other Alemtuzumab Products
The following adverse reactions have been identified during postapproval use of another alemtuzumab product. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Endocrine Disorders: Hypothyroidism, hyperthyroidism, and thyroiditis.
Nervous System Disorders: Autoimmune encephalitis.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, CAMPATH may cause fetal harm when administered to a pregnant woman. Available data from published cohort studies in pregnant women are insufficient to establish a CAMPATH-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Alemtuzumab was embryolethal in pregnant huCD52 transgenic mice when administered during organogenesis (see Data). Human IgG antibodies are known to cross the placental barrier; therefore, CAMPATH may be transmitted from the mother to the developing fetus. Advise women of the potential risk to the fetus. Infants born to pregnant women treated with CAMPATH may be at increased risk of infection (see Clinical Considerations).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal adverse reactions
Monoclonal antibodies are transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Consider the risks and benefits of administering live or live-attenuated vaccines to infants exposed to CAMPATH in utero [see Warnings and Precautions (5.3, 5.4)].
Data
Animal data
When alemtuzumab was administered to pregnant huCD52 transgenic mice during organogenesis (gestation days [GD] 6–10 or GD 11–15) at intravenous doses of 3 or 10 mg/kg, no teratogenic effects were observed. However, there was an increase in embryolethality (increased postimplantation loss and the number of dams with all fetuses dead or resorbed) in pregnant animals dosed during GD 11–15. In a separate study in pregnant huCD52 transgenic mice, administration of alemtuzumab during organogenesis (GD 6–10 or GD 11–15) at intravenous doses of 3 or 10 mg/kg, decreases in B-lymphocyte and T-lymphocyte populations were observed in the offspring at both doses tested.
In pregnant huCD52 transgenic mice administered alemtuzumab at intravenous doses of 3 or 10 mg/kg/day throughout gestation and lactation, there was an increase in pup deaths during the lactation period at 10 mg/kg. Decreases in T-lymphocyte and B-lymphocyte populations and in antibody response were observed in offspring at both doses tested.
8.2 Lactation
Risk Summary
There are no data on the presence of alemtuzumab in human milk, effects on milk production, or the breastfed child. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to alemtuzumab are unknown. Alemtuzumab was detected in the milk of lactating huCD52 transgenic mice administered alemtuzumab (see Data). Maternal IgG is known to be present in human milk and when a drug is present in animal milk, it is likely that the drug will be present in human milk.
Because of the potential for serious adverse reactions from CAMPATH in a breastfed child, including reduced lymphocyte counts, advise lactating women not to breastfeed during treatment with CAMPATH and for at least 3 months following the last dose.
Data
Alemtuzumab was detected in the milk of lactating huCD52 transgenic mice following intravenous administration of alemtuzumab at a dose of 10 mg/kg on postpartum days 8–12. Serum levels of alemtuzumab were similar in lactating mice and offspring on postpartum day 13 and were associated with evidence of pharmacological activity (decrease in lymphocyte counts) in the offspring.
8.3 Females and Males of Reproductive Potential
CAMPATH may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating CAMPATH therapy.
Infertility
Based on findings from animal studies, alemtuzumab may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)]. The reversibility of the effect on fertility is unknown.
8.4 Pediatric Use
Safety and effectiveness of CAMPATH have not been established in pediatric patients.
8.5 Geriatric Use
Of 147 previously untreated B-CLL patients treated with CAMPATH, 35% were ≥ age 65 and 4% were ≥ age 75. Of 149 previously treated patients with B-CLL, 44% were ≥65 years of age and 10% were ≥75 years of age. Clinical studies of CAMPATH did not include sufficient number of subjects age 65 and over to determine whether they respond differently than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
10 OVERDOSAGE
Across all clinical experience, the reported maximum single dose received was 90 mg. Bone marrow aplasia, infections, or severe infusion-related reactions occurred in patients who received a dose higher than recommended.
One patient who received an 80 mg dose intravenously experienced acute bronchospasm, cough, and dyspnea, followed by anuria and death. Another patient received two 90 mg doses intravenously one day apart during the second week of treatment and experienced a rapid onset of bone marrow aplasia.
There is no known specific antidote for CAMPATH overdosage. Discontinue CAMPATH and provide supportive therapy.
11 DESCRIPTION
Alemtuzumab, a CD52-directed cytolytic antibody, is a recombinant DNA-derived humanized monoclonal antibody (CAMPATH-1H). CAMPATH-1H is an IgG1 kappa antibody with human variable framework and constant regions, and complementarity-determining regions from a murine (rat) monoclonal antibody (CAMPATH-1G). The CAMPATH-1H antibody has an approximate molecular weight of 150 kD. CAMPATH is produced in mammalian cell (Chinese hamster ovary) suspension culture in a medium containing neomycin. Neomycin is not detectable in the final product.
CAMPATH (alemtuzumab) injection is a sterile, clear, colorless, isotonic solution (pH 6.8–7.4) in a single-dose vial for intravenous use. Each single-dose vial of CAMPATH contains 30 mg alemtuzumab, 8.0 mg sodium chloride, 1.44 mg dibasic sodium phosphate, 0.2 mg potassium chloride, 0.2 mg monobasic potassium phosphate, 0.1 mg polysorbate 80, and 0.0187 mg disodium edetate dihydrate. No preservatives are added.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CAMPATH binds to CD52, an antigen present on the surface of B and T lymphocytes, a majority of monocytes, macrophages, NK cells, and a subpopulation of granulocytes. A proportion of bone marrow cells, including some CD34+ cells, express variable levels of CD52. The proposed mechanism of action is antibody-dependent cellular-mediated lysis following cell surface binding of CAMPATH to the leukemic cells.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of multiple doses of alemtuzumab (12 mg/day for 5 days) on the QTc interval was evaluated in a single-arm study in 53 patients without malignancy. No large changes in the mean QTc interval (i.e., >20 ms) were detected in the study. A mean increase in heart rate of 22 to 26 beats/min was observed for at least 2 hours following the initial infusion of alemtuzumab. This increase in heart rate was not observed with subsequent doses.
12.3 Pharmacokinetics
CAMPATH pharmacokinetics were characterized in a study of 30 previously treated B-CLL patients in whom CAMPATH was administered at the recommended dose and schedule. After 12 weeks of dosing, patients exhibited a 7-fold increase in mean AUC.
Distribution
After the last 30 mg dose, the mean volume of distribution at steady-state was 0.18 L/kg (range 0.1 to 0.4 L/kg).
Elimination
CAMPATH pharmacokinetics displayed nonlinear elimination kinetics. Systemic clearance decreased with repeated administration due to decreased receptor-mediated clearance (i.e., loss of CD52 receptors in the periphery). Mean half-life was 11 hours (range 2 to 32 hours) after the first 30 mg dose and was 6 days (range 1 to 14 days) after the last 30 mg dose.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies to assess the carcinogenic or genotoxic potential of CAMPATH have not been conducted.
In fertility studies, alemtuzumab (3 or 10 mg/kg intravenously) was administered to huCD52 transgenic male mice on 5 consecutive days prior to cohabitation with untreated wild-type females. No effect on fertility or reproductive performance was observed. However, adverse effects on sperm parameters (including abnormal morphology [detached/no head] and reduced total count and motility) were observed at both doses tested.
When alemtuzumab (3 or 10 mg/kg intravenously) was administered to huCD52 transgenic female mice for 5 consecutive days prior to cohabitation with untreated wild-type males, there was a decrease in the average number of corpora lutea and implantation sites and an increase in postimplantation loss, resulting in fewer viable embryos at the higher dose tested.
14 CLINICAL STUDIES
14.1 Previously Untreated B-CLL Patients
CAMPATH was evaluated in an open-label, randomized (1:1) active-controlled study in previously untreated patients with B-CLL, Rai Stage I–IV, with evidence of progressive disease requiring therapy. Patients received either CAMPATH 30 mg intravenously 3 times per week for a maximum of 12 weeks or chlorambucil 40 mg/m2 orally once every 28 days for a maximum of 12 cycles.
Of the 297 patients randomized, the median age was 60 years, 72% were male, 99% were Caucasian, 96% had a WHO performance status 0–1, 23% had maximum lymph node diameter ≥5 cm, 34% were Rai Stage III/IV, and 8% were treated in the U.S.
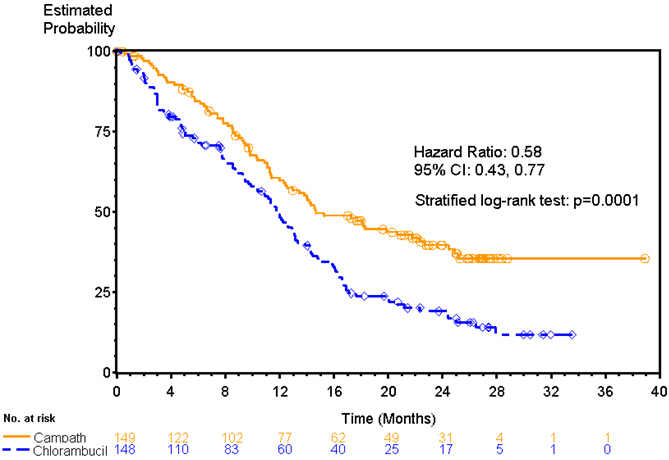
Patients randomized to receive CAMPATH experienced longer progression free survival (PFS) compared to those randomized to receive chlorambucil (median PFS 14.6 months vs. 11.7 months, respectively). The overall response rates were 83% and 55% (p <0.0001) and the complete response rates were 24% and 2% (p <0.0001) for CAMPATH and chlorambucil arms, respectively. The Kaplan-Meier curve for PFS is shown in Figure 1.
| Figure 1: Progression Free Survival in Previously Untreated B-CLL Patients* |
|---|
|
|
|
14.2 Previously Treated B-CLL Patients
CAMPATH was evaluated in three multicenter, open-label, single-arm studies of 149 patients with B-CLL previously treated with alkylating agents, fludarabine, or other chemotherapies. Patients were treated with the recommended dose of CAMPATH 30 mg intravenously 3 times per week for up to 12 weeks. Partial response rates of 21% to 31% and complete response rates of 0% to 2% were observed.
16 HOW SUPPLIED/STORAGE AND HANDLING
CAMPATH (alemtuzumab) is supplied in clear glass single-dose vial containing 30 mg of alemtuzumab in 1 mL of solution. Each carton contains three CAMPATH vials (NDC 58468-0357-3) or one CAMPATH vial (NDC 58468-0357-1).
17 PATIENT COUNSELING INFORMATION
Cytopenias
Advise patients to report any signs or symptoms such as bleeding, easy bruising, petechiae or purpura, pallor, weakness or fatigue [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Infusion-Related Reactions
Advise patients of the signs and symptoms of infusion-related reactions and of the need to take premedications as prescribed [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Immunosuppression/Infections
Advise patients to immediately report symptoms of infection (e.g., pyrexia) and to take prophylactic anti-infectives for PCP (trimethoprim/sulfamethoxazole DS or equivalent) and for herpes virus (famciclovir or equivalent) as prescribed [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Advise patients that irradiation of blood products is required [see Warnings and Precautions (5.3)].
Immunization
Advise patients that they should not be immunized with live viral vaccines if they have recently been treated with CAMPATH. Advise females with infants exposed to CAMPATH in utero to inform the pediatrician of the exposure [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1, 8.3)].
Advise female patients of reproductive potential to use effective contraception during treatment with CAMPATH and for 3 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise females not to breastfeed during treatment with CAMPATH and for 3 months after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that CAMPATH may impair fertility [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Glomerular Nephropathies
Advise patients on signs and symptoms of glomerular nephropathies that may occur months to years after receiving CAMPATH [see Adverse Reactions (6.2)].
Manufactured and distributed by:
Genzyme Corporation, Cambridge, MA 02141
A SANOFI COMPANY
US License Number: 1596
CAMPATH is a registered trademark of Genzyme Corporation.
©2023 Genzyme Corporation.