DESCRIPTION
Nitrofurantoin, a synthetic chemical, is a stable, yellow, crystalline compound. Nitrofurantoin Oral Suspension, USP is an antibacterial agent for specific urinary tract infections. Nitrofurantoin Oral Suspension, USP is available in 25 mg/5 mL liquid suspension for oral administration.
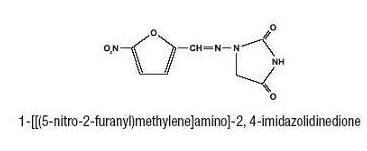
Nitrofurantoin Oral Suspension, USP contains carboxymethylcellulose sodium, citric acid, flavors, glycerin, magnesium aluminum silicate, methylparaben, propylparaben, purified water, saccharin sodium, sodium citrate, and sorbitol.
CLINICAL PHARMACOLOGY
Orally administered Nitrofurantoin is readily absorbed and rapidly excreted in urine. Blood concentrations at therapeutic dosage are usually low. It is highly soluble in urine, to which it may impart a brown color.
Following a dose regimen of 100 mg q.i.d. for 7 days, average urinary drug recoveries (0-24 hours) on day 1 and day 7 were 42.7% and 43.6%.
Unlike many drugs, the presence of food or agents delaying gastric emptying can increase the bioavailability of Nitrofurantoin, presumably by allowing better dissolution in gastric juices.
Mode of Action
Nitrofurantoin is reduced by a wide range of enzymes including bacterial flavoproteins to reactive intermediates which are damaging to macromolecules such as DNA and proteins.
Cross-Resistance
Although cross-resistance with other antimicrobials may occur, cross resistance with sulfonamides has not been observed.
Interaction with Other Antimicrobials
Antagonism has been demonstrated in vitro between nitrofurantoin and quinolone antimicrobial agents.
Nitrofurantoin, in the form of nitrofurantoin oral suspension, has been shown to be active against most strains of the following bacteria both in vitro and in clinical infections: (See INDICATIONS AND USAGE).
Gram-positive Aerobes
Staphylococcus aureus
Enterococcus Species
Gram-Negative Aerobes
Escherichia coli
NOTE: Some strains of Enterobacter species and Klebsiella species are resistant to nitrofurantoin.
The following in vitro data are available, but their clinical significance is unknown. Nitrofurantoin exhibits in vitro activity against the following bacteria; however, the safety and effectiveness of nitrofurantoin in treating clinical infections due to these bacteria have not been established in adequate and well controlled clinical trials.
Gram-Positive Aerobes Coagulase-negative staphylococci (including Staphylococcus epidermidis and Staphylococcus saprophyticus)
Streptococcus agalactiae
Viridans group streptococci
Gram-Negative Aerobes
Citrobacter Koseri
Citrobacter freundii
Klebsiella oxytoca
Nitrofurantoin is not active against most strains of Proteus species or Serratia species. It has no activity against Pseudomonas species.
Susceptibility Tests Methods
When available, the clinical microbiology laboratory should provide the results of in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas to the physician as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting an antibacterial drug product for treatment.
Dilution Techniques
Quantitative methods are used to determine antimicrobial minimal inhibitory concentrations (MIC's). These MIC's provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MIC's should be determined using a standardized test method1,3(broth or agar). The MIC values should be interpreted according to the criteria in Table 1.
Diffusion Techniques
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. The zone size provides an estimate of the susceptibility of bacteria to antimicrobial compounds. The zone size should be determined using a standardized method.2This procedure uses paper disks impregnated with 300 mcg of nitrofurantoin to test the susceptibility of bacteria to nitrofurantoin. The disk diffusion interpretive criteria are provided in Table 1.
| Pathogen
| Minimum Inhibitory Concentrations (mcg/ml)
| Disk Diffusion Zone (mm)
|
||||
|
| S
| I
| R
| S
| I
| R
|
| Enterobacteriaceae
| ≤32 | 64 | ≥128 | ≥17 | 15-16 | ≤14 |
| Staphylococcusaureus | ≤32 | 64 | ≥128 | ≥17 | 15-16 | ≤14 |
| Enterococcus species | ≤32 | 64 | ≥128 | ≥17 | 15-16 | ≤14 |
| S=susceptible, I=intermediate, R=resistant |
||||||
A report of "Susceptible" indicates that the antimicrobial is likely to inhibit growth of the pathogen if the antimicrobial compound reaches the concentration at the infection site necessary to inhibit growth of the pathogen. A report of "Intermediate" indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body site where the drug is physiologically concentrated. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the antimicrobial is not likely to inhibit growth of the pathogen if the antimicrobial compound reaches the concentrations usually achievable at the infection site; other therapy should be selected.
Quality Control
Standardized susceptibility test procedures require the use of laboratory controls to monitor and ensure the accuracy and precision of supplies and reagents used in the assay, and the techniques of the individuals performing the test.1,2,3 Standard nitrofurantoin powder should provide the following MIC values provided in Table 2. For the diffusion technique using the 300-mcg nitrofurantoin disk the criteria provided in Table 2 should be achieved.
| Quality Control Organism
| Minimum Inhibitory
Concentrations(mcg/ml) | Disk Diffusion(zone
diameters in mm) |
| Escherichia coli (ATCC 25922) | 4-16 | 20-25 |
| Staphylococcus aureus (ATCC 25923) | N/A* | 18-22 |
| Staphylococcus aureus (ATCC 29213) | 8-32 | N/A* |
| Streptococcus pneumonia
(ATCC 49619) | 4-16 | 23-29 |
| Enterococcus faecalis (ATCC 29212) | 4-16 | N/A* |
| * Not Applicable |
||
INDICATIONS AND USAGE
Nitrofurantoin Oral Suspension, USP is specifically indicated for the treatment of urinary tract infections when due to susceptible strains of Escherichia coli, enterococci, Staphylococcus aureus, and certain susceptible strains of Klebsiella and Enterobacter species.
Nitrofurantoin is not indicated for the treatment of pyelonephritis or perinephric abscesses.
Nitrofurantoins lack the broader tissue distribution of other therapeutic agents approved for urinary tract infections. Consequently, many patients who are treated with nitrofurantoin oral suspension, USP are predisposed to persistence or reappearance of bacteriuria. Urine specimens for culture and susceptibility testing should be obtained before and after completion of therapy. If persistence or reappearance of bacteriuria occurs after treatment with nitrofurantoin oral suspension, USP, other therapeutic agents with broader tissue distribution should be selected. In considering the use of nitrofurantoin oral suspension, USP, lower eradication rates should be balanced against the increased potential for systemic toxicity and for the development of antimicrobial resistance when agents with broader tissue distribution are utilized.
CONTRAINDICATIONS
Anuria, oliguria, or significant impairment of renal function (creatinine clearance under 60 mL per minute or clinically significant elevated serum creatinine) are contraindications. Treatment of this type of patient carries an increased risk of toxicity because of impaired excretion of the drug.
Because of the possibility of hemolytic anemia due to immature erythrocyte enzyme systems (glutathione instability), the drug is contraindicated in pregnant patients at term (38-42 weeks gestation), during labor and delivery, or when the onset of labor is imminent. For the same reason, the drug is contraindicated in neonates under one month of age.
Nitrofurantoin Oral Suspension, USP is contraindicated in patients with a previous history of cholestatic jaundice/hepatic dysfunction associated with nitrofurantoin. Nitrofurantoin Oral Suspension, USP is also contraindicated in those patients with known hypersensitivity to nitrofurantoin.
WARNINGS
ACUTE, SUBACUTE, OR CHRONIC PULMONARY REACTIONS HAVE BEEN OBSERVED IN PATIENTS TREATED WITH NITROFURANTOIN. IF THESE REACTIONS OCCUR, NITROFURANTOIN ORAL SUSPENSION, USP SHOULD BE DISCONTINUED AND APPROPRIATED MEASURES TAKEN. REPORTS HAVE CITED PULMONARY REACTIONS AS A CONTRIBUTING CAUSE OF DEATH.
CHRONIC PULMONARY REACTIONS (DIFFUSE INTERSTITIAL PNEUMONITIS OR PULMONARY FIBROSIS, OR BOTH) CAN DEVELOP INSIDIOUSLY. THESE REACTIONS OCCUR RARELY AND GENERALLY IN PATIENTS RECEIVING THERAPY FOR SIX MONTHS OR LONGER. CLOSE MONITORING OF THE PULMONARY CONDITION OF PATIENTS RECEIVING LONG-TERM THERAPY IS WARRANTED AND REQUIRES THAT THE BENEFITS OF THERAPY BE WEIGHED AGAINST POTENTIAL RISKS.(see RESPIRATORY REACTIONS.)
Hepatotoxicity
Hepatic reactions, including hepatitis, cholestatic jaundice, chronic active hepatitis, and hepatic necrosis, occur rarely. Fatalities have been reported. The onset of chronic active hepatitis may be insidious, and patients should be monitored periodically for changes in biochemical tests that would indicate liver injury. If hepatitis occurs, the drug should be withdrawn immediately and appropriate measures should be taken.
Neuropathy
Peripheral neuropathy, which may become severe or irreversible, has occurred. Fatalities have been reported. Conditions such as renal impairment (creatinine clearance under 60 mL per minute or clinically significant elevated serum creatinine), anemia, diabetes mellitus, electrolyte imbalance, vitamin B deficiency, and debilitating disease may enhance the occurrence of peripheral neuropathy. Patients receiving long-term therapy should be monitored periodically for changes in renal function.
Optic neuritis has been reported rarely in postmarketing experience with nitrofurantoin formulations.
Hemolytic anemia
Cases of hemolytic anemia of the primaquine-sensitivity type have been induced by nitrofurantoin. Hemolysis appears to be linked to a glucose-6-phosphate dehydrogenase deficiency in the red blood cells of the affected patients. This deficiency is found in 10 percent of Blacks and a small percentage of ethnic groups of Mediterranean and Near-Eastern origin. Hemolysis is an indication for discontinuing Nitrofurantoin Oral Suspension, USP; hemolysis ceases when the drug is withdrawn.
Clostridium difficile-associated diarrhea
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including Nitrofurantoin Oral Suspension, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
PRECAUTIONS
Information for Patients
Patients should be advised to take Nitrofurantoin Oral Suspension, USP with food to further enhance tolerance and improve drug absorption. Patients should be instructed to complete the full course of therapy; however, they should be advised to contact their physician if any unusual symptoms should occur during therapy.
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Patients should be advised not to use antacid preparations containing magnesium trisilicate while taking Nitrofurantoin Oral Suspension, USP.
Drug Interactions
Antacids containing magnesium trisilicate, when administered concomitantly with nitrofurantoin, reduce both the rate and extent of absorption. The mechanism for this interaction probably is adsorption of nitrofurantoin onto the surface of magnesium trisilicate.
Uricosuric drugs, such as probenecid and sulfinpyrazone, can inhibit renal tubular secretion of nitrofurantoin. The resulting increase in nitrofurantoin serum levels may increase toxicity, and the decreased urinary levels could lessen its efficacy as a urinary tract antibacterial.
Drug/Laboratory Test Interactions
As a result of the presence of nitrofurantoin, a false-positive reaction for glucose in the urine may occur. This has been observed with Benedict's and Fehling's solutions but not with the glucose enzymatic test.
Carcinogenesis & Mutagenesis & Impairment of Fertility
Nitrofurantoin was not carcinogenic when fed to female Holtzman rats for 44.5 weeks or to female Sprague-Dawley rats for 75 weeks. Two chronic rodent bioassays utilizing male and female Sprague-Dawley rats and two chronic bioassays in Swiss mice and in BDF1 mice revealed no evidence of carcinogenicity.
Nitrofurantoin presented evidence of carcinogenic activity in female B6C3F1 mice as shown by increased incidences of tubular adenomas, benign mixed tumors, and granulosa cell tumors of the ovary. In male F344/N rats, there were increased incidences of uncommon kidney tubular cell neoplasms, osteosarcomas of the bone, and neoplasms of the subcutaneous tissue. In one study involving subcutaneous administration of 75 mg/kg nitrofurantoin to pregnant female mice, lung papillary adenomas of unknown significance were observed in the F1 generation.
Nitrofurantoin has been shown to induce point mutations in certain strains of Salmonella typhimurium and forward mutations L5178Y mouse lymphoma cells. Nitrofurantoin induced increased numbers of sister chromatid exchanges and chromosomal aberrations in Chinese hamster ovary cells but not in human cells in culture. Results of the sex-linked recessive lethal assay in Drosophila were negative after administration of nitrofurantoin by feeding or by injection. Nitrofurantoin did not induce heritable mutation in the rodent models examined.
The significance of carcinogenicity and mutagenicity findings relative to the therapeutic use of nitrofurantoin in humans is unknown.
The administration of high doses of nitrofurantoin to rats causes temporary spermatogenic arrest; this is reversible on discontinuing the drug. Doses of 10 mg/kg/day or greater in healthy human males may, in certain unpredictable instances, produce a slight to moderate spermatogenic arrest with a decrease in sperm count.
Pregnancy
Pregnancy Category B
Several reproduction studies have been performed in rabbits and rats at doses up to six times the human dose and have revealed no evidence of impaired fertility or harm to the fetus due to nitrofurantoin. In a single published study conducted in mice at 68 times the human dose (based on mg/kg administered to the dam), growth retardation and a low incidence of minor and common malformations were observed. However at 25 times the human dose, fetal malformations were not observed; the relevance of these findings to humans is uncertain. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Non-teratogenic effects
Nitrofurantoin has been shown in one published transplacental carcinogenicity study to induce lung papillary adenomas in the F1 generation mice at doses 19 times the human dose on a mg/kg basis. The relationship of this finding to potential human carcinogenesis is presently unknown. Because of the uncertainty regarding the human implications of these animal data, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Nitrofurantoin has been detected in human breast milk in trace amounts. Because of the potential for serious adverse reactions from nitrofurantoin in nursing infants under one month of age, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. (see CONTRAINDICATIONS)
Pediatric Use
Safety and effectiveness of Nitrofurantoin Oral Suspension, USP in neonates below the age of one month have not been established. (see CONTRAINDICATIONS)
ADVERSE REACTIONS
CHRONIC, SUBACUTE, OR ACUTE PULMONARY HYPERSENSITIVITY REACTIONS MAY OCCUR.
CHRONIC PULMONARY REACTIONS MAY OCCUR GENERALLY IN PATIENTS WHO HAVE RECEIVED CONTINUOUS TREATMENT FOR SIX MONTHS OR LONGER. MALAISE, DYSPNEA ON EXERTION, COUGH, AND ALTERED PULMONARY FUNCTION ARE COMMON MANIFESTATIONS WHICH CAN OCCUR INSIDIOUSLY. RADIOLOGIC AND HISTOLOGIC FINDINGS OF DIFFUSE INTERSTITIAL PNEUMONITIS OR FIBROSIS, OR BOTH, ARE ALSO COMMON MANIFESTATIONS OF THE CHRONIC PULMONARY REACTION. FEVER IS RARELY PROMINENT.
THE SEVERITY OF CHRONIC PULMONARY REACTIONS AND THEIR DEGREES OF RESOLUTION APPEAR TO BE RELATED TO THE DURATION OF THERAPY AFTER THE FIRST CLINICAL SIGNS APPEAR. PULMONARY FUNCTION MAY BE IMPAIRED PERMANENTLY, EVEN AFTER CESSATION OF THERAPY. THE RISK IS GREATER WHEN CHRONIC PULMONARY REACTIONS ARE NOT RECOGNIZED EARLY.
In subacute pulmonary reactions, fever and eosinophilia occur less often than in the acute form. Upon cessation of therapy, recovery may require several months. If the symptoms are not recognized as being drug-related and nitrofurantoin therapy is not stopped, the symptoms may become more severe.
Acute pulmonary reactions are commonly manifested by fever, chills, cough, chest pain, dyspnea, pulmonary infiltration with consolidation of pleural effusion on x-ray, and eosinophilia. Acute reactions usually occur within the first week of treatment and are reversible with cessation of therapy. Resolution often is dramatic. (see WARNINGS)
Changes in EKG (e.g., non-specific ST/T wave changes, bundle branch block) have been reported in association with pulmonary reactions.
Cyanosis has been reported rarely.
Hepatic
Hepatic reactions, including hepatitis, cholestatic jaundice, chronic active hepatitis, and hepatic neurosis, occur rarely. (see WARNINGS)
Neurologic
Peripheral neuropathy, which may become severe or irreversible, has occurred. Fatalities have been reported. Conditions such as renal impairment (creatinine clearance under 60 mL per minute or clinically significant elevated serum creatinine), anemia, diabetes mellitus, electrolyte imbalance, vitamin B deficiency, and debilitating diseases may increase the possibility of peripheral neuropathy (see WARNINGS)
Asthenia, vertigo, nystagmus, dizziness, headache, and drowsiness have also been reported with the use of nitrofurantoin.
Benign intracranial hypertension (pseudotumor cerebri), confusion, depression, optic neuritis, and psychotic reactions have been reported rarely. Bulging fontanels, as a sign of benign intracranial hypertension in infants, have been reported rarely.
Dermatologic
Exfoliative dermatitis and erythema multiforme (including Stevens-Johnson syndrome) have been reported rarely. Transient alopecia also has been reported.
Allergic
A lupus-like syndrome associated with pulmonary reactions to nitrofurantoin has been reported. Also, angioedema; maculopapular, erythematous, or eczematous eruptions; pruritus; urticaria; anaphylaxis; arthralgia; myalgia; drug fever; and chills have been reported. Hypersensitivity reactions present the most frequent spontaneously-reported adverse events in world-wide postmarketing experience with nitrofurantoin formulations.
Gastrointestinal
Nausea, emesis, and anorexia occur most often. Abdominal pain and diarrhea are less common gastrointestinal reactions. These dose-related reactions can be minimized by reduction of dosage. Sialadenitis and pancreatitis have been reported. There have been sporadic reports of pseudomembranous colitis with the use of nitrofurantoin. The onset of pseudomembranous colitis symptoms may occur during or after antimicrobial treatment. (see WARNINGS)
Hematologic
Cyanosis secondary to methemoglobinemia has been reported rarely.
Miscellaneous
As with other antimicrobial agents, superinfections caused by resistant organisms, e.g., Pseudomonas species or Candida species, can occur. There are sporadic reports of Clostridium difficile superinfections, or pseudomembranous colitis, with the use of nitrofurantoin.
The following laboratory adverse events have been reported with the use of nitrofurantoin; increased AST (SGOT), increased ALT (SGPT), decreased hemoglobin, increased serum phosphorus, eosinophilia, glucose-6-phosphate dehydrogenase deficiency anemia (see WARNINGS ), agranulocytosis, leukopenia, granulocytopenia, hemolytic anemia, thrombocytopenia, megaloblastic anemia. In most cases, these hematologic abnormalities resolved following cessation of therapy. Aplastic anemia has been reported rarely.
OVERDOSAGE
Occasional incidents of acute overdosage of Nitrofurantoin Oral Suspension, USP have not resulted in any specific symptoms other than vomiting. Induction of emesis is recommended. There is no specific antidote, but a high fluid intake should be maintained to promote urinary excretion of the drug. It is dialyzable.
DOSAGE AND ADMINISTRATION
Nitrofurantoin Oral Suspension, USP should be given with food to improve drug absorption and, in some patients, tolerance.
Adults
50 to100 mg four times a day -- the lower dosage level is recommended for uncomplicated urinary tract infections.
Pediatric Patients
5 to 7 mg/kg of body weight per 24 hours, given in four divided doses (contraindicated under one month of age).
The following table is based on an average weight in each range receiving 5 to 6 mg/kg of body weight per 24 hours, given in four divided doses. It can be used to calculate an average dose of Nitrofurantoin Oral Suspension, USP (25 mg/5 mL) for pediatric patients.
| Weight in Kilograms (kg)
| Pediatric Doses (milliliters and Frequency)
|
| 7 kg to 11 kg | 2.5 mL Four times Daily |
| 12 kg to 21 kg | 5 mL Four times Daily |
| 22 kg to 30 kg | 7.5 mL Four times Daily |
| 31 kg to 41 kg | 10 mL Four times Daily |
| 42 kg or greater | See Adult Dose |
Therapy should be continued for one week or for at least 3 days after sterility of the urine is obtained. Continued infection indicates the need for reevaluation.
For long-term suppressive therapy in adults, a reduction of dosage to 50 to100 mg at bedtime may be adequate. For long-term suppressive therapy in pediatric patients, doses as low as 1 mg/kg per 24 hours, given in a single dose or in two divided doses, may be adequate. SEE WARNINGSSECTION REGARDING RISKS ASSOCIATED WITH LONG TERM THERAPY.
HOW SUPPLIED
Nitrofurantoin Oral Suspension, USP is a yellow colored suspension with liquorish odor and flavor available in:
NDC 40032-450-11 glass amber bottle of 230 mL
Avoid exposure to strong light which may darken the drug. It is stable when stored between 20˚-25˚C (68˚-77˚F); excursions permitted to 15˚-30˚C (59˚-86˚F) [See USP Controlled Room Temperature]. Protect from freezing. Shake Vigorously. Dispense in tight, light-resistant amber bottles. Use within 30 days.
Keep out of reach of children.
Rx only
REFERENCES
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Ninth Edition. CLSI document M07-A9, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA, 2012.
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Disk Diffusion Susceptibility Tests; Approved Standard-Eleventh Edition. CLSI document M02-A11. Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087,USA, 2012
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-third Informational Supplement, CLSI document M100-S23. Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA, 2013.
Novel Laboratories, Inc.
Somerset, NJ 08873
NIN-450-01
Revised: 03/2014

