FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
YONDELIS® is indicated for the treatment of patients with unresectable or metastatic liposarcoma or leiomyosarcoma who received a prior anthracycline-containing regimen [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose is 1.5 mg/m2 administered as an intravenous infusion over 24 hours through a central venous line every 21 days (3 weeks), until disease progression or unacceptable toxicity.
Hepatic Impairment: The recommended dose is 0.9 mg/m2 in patients with moderate hepatic impairment (bilirubin levels greater than 1.5 times to 3 times the upper limit of normal, and AST and ALT less than 8 times the upper limit of normal). Do not administer YONDELIS to patients with severe hepatic impairment (bilirubin levels above 3 times the upper limit of normal, and any AST and ALT) [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.2 Premedication
Administer dexamethasone 20 mg intravenously 30 minutes prior to each dose of YONDELIS.
2.3 Dose Modifications for Adverse Reactions
Permanently discontinue YONDELIS for:
- Persistent adverse reactions requiring a delay in dosing of more than 3 weeks.
- Adverse reactions requiring dose reduction following YONDELIS administered at 1.0 mg/m2 for patients with normal hepatic function or at 0.3 mg/m2 for patients with pre-existing moderate hepatic impairment.
- Severe liver dysfunction: bilirubin two times the upper limit of normal, and AST or ALT three times the upper limit of normal, and alkaline phosphatase less than two times the upper limit of normal in the prior treatment cycle for patients with normal liver function at baseline.
- Exacerbation of liver dysfunction in patients with pre-existing moderate hepatic impairment.
- Capillary leak syndrome.
- Rhabdomyolysis.
- Grade 3 or 4 cardiac adverse events (AEs) indicative of cardiomyopathy or for subjects with an LVEF that decreases below the lower limit of normal.
The recommended dose modifications for adverse reactions are listed in Table 1. Once reduced, the dose of YONDELIS should not be increased in subsequent treatment cycles.
| Laboratory Result or Adverse Reaction | DELAY next dose of YONDELIS for up to 3 weeks | REDUCE next dose of YONDELIS by one dose level for adverse reaction(s) during prior cycle |
|---|---|---|
| Platelets | Less than 100,000 platelets/microliter | Less than 25,000 platelets/microliter |
| Absolute neutrophil count | Less than 1,500 neutrophils/microliter |
|
| Total bilirubin | Greater than the upper limit of normal | Greater than the upper limit of normal |
| Aspartate aminotransferase (AST) or alanine aminotransferase (ALT) | More than 2.5 times the upper limit of normal | More than 5 times the upper limit of normal |
| Alkaline phosphatase (ALP) | More than 2.5 times the upper limit of normal | More than 2.5 times the upper limit of normal |
| Creatine phosphokinase | More than 2.5 times the upper limit of normal | More than 5 times the upper limit of normal |
| Other non-hematologic adverse reactions | Grade 3 or 4 | Grade 3 or 4 |
The recommended starting doses and dose reductions for YONDELIS are listed in Table 2:
| Starting Dose and Dose Reduction | For patients with normal hepatic function or mild hepatic impairment* prior to initiation of YONDELIS treatment | For patients with moderate hepatic impairment† prior to initiation of YONDELIS treatment |
|---|---|---|
| Starting Dose | 1.5 mg/m2 | 0.9 mg/m2 |
| Dose Reduction | ||
| First dose reduction | 1.2 mg/m2 | 0.6 mg/m2 |
| Second dose reduction | 1.0 mg/m2 | 0.3 mg/m2 |
2.4 Preparation for Administration
- YONDELIS is a cytotoxic drug. Follow applicable special handling and disposal procedures.1
- Using aseptic technique, inject 20 mL of Sterile Water for Injection, USP into the vial. Shake the vial until complete dissolution. The reconstituted solution is clear, colorless to pale brownish-yellow, and contains 0.05 mg/mL of trabectedin.
- Inspect for particulate matter and discoloration prior to further dilution. Discard vial if particles or discoloration are observed.
- Immediately following reconstitution, withdraw the calculated volume of trabectedin and further dilute in 500 mL of 0.9% Sodium Chloride, USP or 5% Dextrose Injection, USP.
- Do not mix YONDELIS with other drugs.
- Discard any remaining solution within 30 hours of reconstituting the lyophilized powder.
- YONDELIS diluted solution is compatible with Type I colorless glass vials, polyvinylchloride (PVC) and polyethylene (PE) bags and tubing, PE and polypropylene (PP) mixture bags, polyethersulfone (PES) in-line filters, titanium, platinum or plastic ports, silicone and polyurethane catheters, and pumps having contact surfaces made of PVC, PE, or PE/PP.
2.5 Administration
- Infuse the reconstituted, diluted solution over 24 hours through a central venous line using an infusion set with a 0.2 micron polyethersulfone (PES) in-line filter to reduce the risk of exposure to adventitious pathogens that may be introduced during solution preparation.
- Complete infusion within 30 hours of initial reconstitution. Discard any unused portion of the reconstituted product or of the infusion solution.
3 DOSAGE FORMS AND STRENGTHS
For injection: 1 mg, lyophilized powder in single-dose vial for reconstitution.
4 CONTRAINDICATIONS
YONDELIS is contraindicated in patients with known severe hypersensitivity, including anaphylaxis, to trabectedin.
5 WARNINGS AND PRECAUTIONS
5.1 Neutropenic Sepsis
Neutropenic sepsis, including fatal cases, can occur with YONDELIS. In Trial ET743-SAR-3007, the incidence of Grade 3 or 4 neutropenia, based on laboratory values, in patients receiving YONDELIS was 43% (161/378). The median time to the first occurrence of Grade 3 or 4 neutropenia was 16 days (range: 8 days to 9.7 months); the median time to complete resolution of neutropenia was 13 days (range: 3 days to 2.3 months). Febrile neutropenia (fever ≥38.5°C with Grade 3 or 4 neutropenia) occurred in 18 patients (5%) treated with YONDELIS. Ten patients (2.6%) experienced neutropenic sepsis, 5 of whom had febrile neutropenia, which was fatal in 4 patients (1.1%).
Assess neutrophil count prior to administration of each dose of YONDELIS and periodically throughout the treatment cycle. Withhold or reduce dose of YONDELIS based on severity of adverse reaction [see Dosage and Administration (2.3)].
5.2 Rhabdomyolysis
YONDELIS can cause rhabdomyolysis and musculoskeletal toxicity. In Trial ET743-SAR-3007, rhabdomyolysis leading to death occurred in 3 (0.8%) of the 378 patients receiving YONDELIS. Elevations in creatine phosphokinase (CPK) occurred in 122 (32%) of the 378 patients receiving YONDELIS, including Grade 3 or 4 CPK elevation in 24 patients (6%), compared to 15 (9%) of the 172 patients receiving dacarbazine with any CPK elevation, including 1 patient (0.6%) with Grade 3 CPK elevation. Among the 24 patients receiving YONDELIS with Grade 3 or 4 CPK elevation, renal failure occurred in 11 patients (2.9%); rhabdomyolysis with the complication of renal failure occurred in 4 of these 11 patients (1.1%). The median time to first occurrence of Grade 3 or 4 CPK elevations was 2 months (range: 1 to 11.5 months). The median time to complete resolution was 14 days (range: 5 days to 1 month).
Assess CPK levels prior to each administration of YONDELIS. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3)].
5.3 Hepatotoxicity
Hepatotoxicity, including hepatic failure, can occur with YONDELIS. Patients with serum bilirubin levels above the upper limit of normal or AST or ALT levels >2.5 × upper limit of normal were not enrolled in Trial ET743-SAR-3007. In Trial ET743-SAR-3007, the incidence of Grade 3–4 elevated liver function tests (LFTs; defined as elevations in ALT, AST, total bilirubin, or alkaline phosphatase) was 35% (134/378) in patients receiving YONDELIS. The median time to development of Grade 3–4 elevation in ALT or AST was 29 days (range: 3 days to 11.5 months). Of the 134 patients with Grade 3–4 elevations in LFTs, 114 (85%) experienced complete resolution with the median time to complete resolution of 13 days (range: 4 days to 4.4 months).
In Trial ET743-SAR-3007, the incidence of drug-induced liver injury (defined as concurrent elevation in ALT or AST of more than three times the upper limit of normal, alkaline phosphatase less than two times the upper limit of normal, and total bilirubin at least two times the upper limit of normal) was 1.3% (5/378) in patients receiving YONDELIS. ALT or AST elevation greater than eight times the upper limit of normal occurred in 18% (67/378) of patients receiving YONDELIS.
Assess LFTs prior to each administration of YONDELIS and as clinically indicated based on underlying severity of pre-existing hepatic impairment. Manage elevated LFTs with treatment interruption, dose reduction, or permanent discontinuation based on severity and duration of LFT abnormality [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)].
5.4 Cardiomyopathy
Cardiomyopathy including cardiac failure, congestive heart failure, ejection fraction decreased, diastolic dysfunction, or right ventricular dysfunction can occur with YONDELIS. In Trial ET743-SAR-3007, a significant decrease in LVEF was defined as an absolute decrease of ≥15% or below the lower limit of normal with an absolute decrease of ≥5%. Patients with a history of New York Heart Association Class II to IV heart failure or abnormal left ventricular ejection fraction (LVEF) at baseline were ineligible. In Trial ET743-SAR-3007, cardiomyopathy occurred in 23 patients (6%) receiving YONDELIS and in four patients (2.3%) receiving dacarbazine. Grade 3 or 4 cardiomyopathy occurred in 15 patients (4%) receiving YONDELIS and 2 patients (1.2%) receiving dacarbazine; cardiomyopathy leading to death occurred in 1 patient (0.3%) receiving YONDELIS and in none of the patients receiving dacarbazine. The median time to development of Grade 3 or 4 cardiomyopathy in patients receiving YONDELIS was 5.3 months (range: 26 days to 15.3 months).
Patients with LVEF < lower limit of normal, prior cumulative anthracycline dose of ≥300 mg/m2, age ≥65 years, or a history of cardiovascular disease may be at increased risk of cardiac dysfunction. Assess LVEF by echocardiogram (ECHO) or multigated acquisition (MUGA) scan before initiation of YONDELIS and at 2- to 3-month intervals thereafter until YONDELIS is discontinued. Discontinue treatment with YONDELIS based on severity of adverse reaction [see Dosage and Administration (2.3)].
5.5 Capillary Leak Syndrome
Capillary leak syndrome (CLS) characterized by hypotension, edema, and hypoalbuminemia has been reported with YONDELIS, including serious CLS resulting in death. Monitor for signs and symptoms of CLS. Discontinue YONDELIS and promptly initiate standard management for patients with CLS, which may include a need for intensive care [see Adverse Reactions (6.2)].
5.6 Extravasation Resulting in Tissue Necrosis
Extravasation of YONDELIS, resulting in tissue necrosis requiring debridement, can occur. Evidence of tissue necrosis can occur more than 1 week after the extravasation. There is no specific antidote for extravasation of YONDELIS. Administer YONDELIS through a central venous line [see Dosage and Administration (2.5)].
5.7 Embryo-Fetal Toxicity
Based on its mechanism of action, YONDELIS can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during therapy and for at least 2 months after the last dose of YONDELIS. Advise males with female partners of reproductive potential to use effective contraception during therapy and for at least 5 months after the last dose of YONDELIS [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Anaphylaxis [see Contraindications (4)]
- Neutropenic Sepsis [see Warnings and Precautions (5.1)]
- Rhabdomyolysis [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Cardiomyopathy [see Warnings and Precautions (5.4)]
- Capillary Leak Syndrome [see Warnings and Precautions (5.5)]
- Extravasation Resulting in Tissue Necrosis [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to YONDELIS in 755 patients with soft tissue sarcoma including 197 (26%) patients exposed to YONDELIS for greater than or equal to 6 months and 57 (8%) patients exposed to YONDELIS for greater than or equal to 1 year. The safety of YONDELIS was evaluated in six open-label, single-arm trials, in which 377 patients received YONDELIS and one open-label, randomized, active-controlled clinical trial in which 378 patients received YONDELIS (Trial ET743-SAR-3007). All patients received YONDELIS at the recommended dosing regimen of 1.5 mg/m2 administered as an intravenous infusion over 24 hours once every 3 weeks (q3wk, 24-h). The median age was 54 years (range: 18 to 81 years), 63% were female, and all patients had metastatic soft tissue sarcoma.
Tables 3 and 4 present selected adverse reactions and laboratory abnormalities, respectively, observed in Trial ET743-SAR-3007, an open-label, randomized (2:1), active-controlled trial in which 550 patients with previously treated leiomyosarcoma or liposarcoma (dedifferentiated, myxoid round cell, or pleomorphic) received YONDELIS 1.5 mg/m2 intravenous infusion over 24 hours once every 3 weeks (n=378) or dacarbazine 1000 mg/m2 intravenous infusion over 20 to 120 minutes once every 3 weeks (n=172) [see Clinical Studies (14)]. All patients treated with YONDELIS were required to receive dexamethasone 20 mg intravenous injection 30 minutes prior to start of the YONDELIS infusion.
In Trial ET743-SAR-3007, patients had been previously treated with an anthracycline- and ifosfamide-containing regimen or with an anthracycline-containing regimen and one additional cytotoxic chemotherapy regimen. The trial excluded patients with known central nervous system metastasis, elevated serum bilirubin or significant chronic liver disease, such as cirrhosis or active hepatitis, and history of myocardial infarction within 6 months, history of New York Heart Association Class II to IV heart failure, or abnormal left ventricular ejection fraction at baseline. The median age of patients in Trial ET743-SAR-3007 was 57 years (range: 17 to 81 years), with 69% female, 77% White, 12% Black or African American, 4% Asian, and <1% American Indian or Alaska Native. The median duration of exposure to trabectedin was 13 weeks (range: 1 to 127 weeks) with 30% of patients exposed to YONDELIS for greater than 6 months and 7% of patients exposed to YONDELIS for greater than 1 year.
In Trial ET743-SAR-3007, adverse reactions resulting in permanent discontinuation of YONDELIS occurred in 26% (98/378) of patients; the most common were increased liver tests (defined as ALT, AST, alkaline phosphatase, bilirubin) (5.6%), thrombocytopenia (3.4%), fatigue (1.6%), increased creatine phosphokinase (1.1%), and decreased ejection fraction (1.1%). Adverse reactions that led to dose reductions occurred in 42% (158/378) of patients treated with YONDELIS; the most common were increased liver tests (24%), neutropenia (including febrile neutropenia) (8%), thrombocytopenia (4.2%), fatigue (3.7%), increased creatine phosphokinase (2.4%), nausea (1.1%), and vomiting (1.1%). Adverse reactions led to dose interruptions in 52% (198/378) of patients treated with YONDELIS; the most common were neutropenia (31%), thrombocytopenia (15%), increased liver tests (6%), fatigue (2.9%), anemia (2.6%), increased creatinine (1.1%), and nausea (1.1%).
The most common adverse reactions (≥20%) were nausea, fatigue, vomiting, constipation, decreased appetite, diarrhea, peripheral edema, dyspnea, and headache. The most common laboratory abnormalities (≥20%) were increases in AST or ALT, increased alkaline phosphatase, hypoalbuminemia, increased creatinine, increased creatine phosphokinase, anemia, neutropenia, and thrombocytopenia.
| YONDELIS (N=378) | Dacarbazine (N=172) |
|||
|---|---|---|---|---|
| System Organ Class Adverse Reaction | All Grades†
(%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
||||
| Gastrointestinal disorders | ||||
| Nausea | 75 | 7 | 50 | 1.7 |
| Vomiting | 46 | 6 | 22 | 1.2 |
| Constipation | 37 | 0.8 | 31 | 0.6 |
| Diarrhea | 35 | 1.6 | 23 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue‡ | 69 | 8 | 52 | 1.7 |
| Peripheral edema | 28 | 0.8 | 13 | 0.6 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 37 | 1.9 | 21 | 0.6 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Dyspnea | 25 | 4.2 | 20 | 1.2 |
| Nervous system disorders | ||||
| Headache | 25 | 0.3 | 19 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 15 | 0 | 8 | 1.2 |
| Myalgia | 12 | 0 | 6 | 0 |
| Psychiatric disorders | ||||
| Insomnia | 15 | 0.3 | 9 | 0 |
Other clinically important adverse reactions observed in <10% of patients (N=755) with soft tissue sarcoma receiving YONDELIS were:
Nervous system disorders: peripheral neuropathy, paresthesia, hypoesthesia.
Respiratory, thoracic, and mediastinal disorders: pulmonary embolism.
General disorders and administration site conditions: mucosal inflammation.
| Laboratory Abnormalities | YONDELIS | Dacarbazine | ||
|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
| YONDELIS group (range: 373 to 377 patients) and dacarbazine group (range: 166 to 168 patients). | ||||
|
||||
| Chemistry | ||||
| Increased ALT | 90 | 31 | 33 | 0.6 |
| Increased AST | 84 | 17 | 32 | 1.2 |
| Increased alkaline phosphatase | 70 | 1.6 | 60 | 0.6 |
| Hypoalbuminemia | 63 | 3.7 | 51 | 3.0 |
| Increased creatinine | 46 | 4.2 | 29 | 1.2 |
| Increased creatine phosphokinase | 33 | 6.4 | 9 | 0.6 |
| Hyperbilirubinemia | 13 | 1.9 | 5 | 0.6 |
| Hematology | ||||
| Anemia | 96 | 19 | 79 | 12 |
| Neutropenia | 66 | 43 | 47 | 26 |
| Thrombocytopenia | 59 | 21 | 57 | 20 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of YONDELIS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular disorders: capillary leak syndrome
7 DRUG INTERACTIONS
7.1 Effect of Cytochrome CYP3A Inhibitors
Coadministration of YONDELIS with ketoconazole, a strong CYP3A inhibitor, increased systemic exposure of trabectedin by 66%. Avoid using strong CYP3A inhibitors (e.g., oral ketoconazole, itraconazole, posaconazole, voriconazole, clarithromycin, telithromycin, indinavir, lopinavir, ritonavir, boceprevir, nelfinavir, saquinavir, telaprevir, nefazodone, conivaptan) in patients taking YONDELIS. If a strong CYP3A inhibitor for short-term use (i.e., less than 14 days) must be used, administer the strong CYP3A inhibitor 1 week after the YONDELIS infusion, and discontinue it the day prior to the next YONDELIS infusion [see Clinical Pharmacology (12.3)].
7.2 Effect of Cytochrome CYP3A Inducers
Coadministration of YONDELIS with rifampin, a strong CYP3A inducer, decreased systemic exposure of trabectedin by 31%. Avoid using strong CYP3A inducers (e.g., rifampin, phenobarbital, St. John's wort) in patients taking YONDELIS [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, trabectedin can cause fetal harm when administered during pregnancy [see Clinical Pharmacology (12.1)]. There are no available data with the use of YONDELIS during pregnancy. Animal reproductive and developmental studies at relevant doses have not been conducted with trabectedin; however, placental transfer of trabectedin was demonstrated in pregnant rats. Advise pregnant woman of the potential risk to a fetus. The background risk of major birth defects and miscarriage for the indicated population are unknown; however, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
8.2 Lactation
Risk Summary
There are no data on the presence of trabectedin in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions from YONDELIS in breastfed infants, advise a nursing woman to discontinue nursing during treatment with YONDELIS.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating YONDELIS [see Use in Specific Populations (8.1)].
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during and for 2 months after the last dose of YONDELIS [see Use in Specific Populations (8.1)].
Males
YONDELIS may damage spermatozoa, resulting in possible genetic and fetal abnormalities. Advise males with a female sexual partner of reproductive potential to use effective contraception during and for 5 months after the last dose of YONDELIS [see Nonclinical Toxicology (13.1)].
Infertility
YONDELIS may result in decreased fertility in males and females [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Safety (n=61) and efficacy (n=58) were assessed across five open-label studies (NCT00006463, NCT01453283, NCT00005625, NCT00070109, and ET-B-023-00) in pediatric patients (aged 2 to <17 years) with pediatric histotypes of sarcoma (predominantly rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, and non-rhabdomyosarcoma soft tissue sarcoma). No new safety signals were observed in pediatric patients across these studies.
Pharmacokinetic parameters in 17 pediatric patients (aged 3 to 17 years) were within the range of values previously observed in adults given the same dose per body surface area.
8.5 Geriatric Use
Clinical studies of YONDELIS did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
The mean trabectedin exposure was (97%) higher in patients with moderate (bilirubin levels greater than 1.5 to 3 times the upper limit of normal, and AST and ALT less than 8 times the upper limit of normal) hepatic impairment compared to patients with normal (total bilirubin ≤ the upper limit of normal, and AST and ALT ≤ the upper limit of normal) liver function. Reduce YONDELIS dose in patients with moderate hepatic impairment [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Do not administer YONDELIS to patients with severe hepatic impairment (bilirubin levels above 3 times the upper limit of normal, and any AST and ALT) [see Warnings and Precautions (5.3)].
8.7 Renal Impairment
No dose adjustment is recommended in patients with mild [creatinine clearance (CLcr) 60–89 mL/min] or moderate (CLcr of 30–59 mL/min) renal impairment.
The pharmacokinetics of trabectedin has not been evaluated in patients with severe renal impairment (CLcr <30 mL/min) or end stage renal disease [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no specific antidote for YONDELIS. Hemodialysis is not expected to enhance the elimination of YONDELIS because trabectedin is highly bound to plasma proteins (97%) and not significantly renally excreted.
11 DESCRIPTION
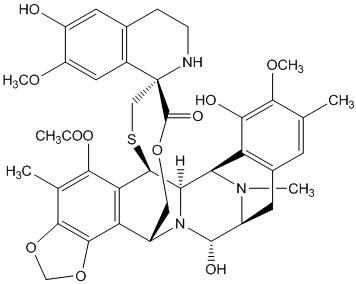
Trabectedin is an alkylating drug with the chemical name (1'R,6R,6aR,7R,13S,14S,16R)-5-(acetyloxy)-3',4',6,6a,7,13,14,16-octahydro-6',8,14-trihydroxy-7',9-dimethoxy-4,10,23-trimethyl-spiro[6,16-(epithiopropanoxymethano)-7,13-imino-12H-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1'(2'H)-isoquinolin]-19-one. The molecular formula is C39H43N3O11S. The molecular weight is 761.84 daltons. The chemical structure is shown below:
Trabectedin is hydrophobic and has a low solubility in water.
YONDELIS (trabectedin) for injection is supplied as a sterile lyophilized white to off-white powder/cake in a single-dose vial. Each single-dose vial contains 1 mg of trabectedin, 27.2 mg potassium dihydrogen phosphate, 400 mg sucrose, and phosphoric acid and potassium hydroxide (for pH adjustment to 3.6 – 4.2).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Trabectedin is an alkylating drug that binds guanine residues in the minor groove of DNA, forming adducts and resulting in a bending of the DNA helix towards the major groove. Adduct formation triggers a cascade of events that can affect the subsequent activity of DNA binding proteins, including some transcription factors, and DNA repair pathways, resulting in perturbation of the cell cycle and eventual cell death.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of trabectedin on the QT/QTc interval was evaluated in 75 patients who received placebo on day 1 and trabectedin (1.3 mg/m2) as a 3-hour intravenous infusion on day 2. No patients in the study showed a QTc interval exceeding 500 msec or more than 60 msec increase from baseline, and no large changes in the mean QTc interval (i.e., >20 msec) were observed.
12.3 Pharmacokinetics
The pharmacokinetics of trabectedin is characterized by a rapid decline phase at the end of the infusion and slower exponential phases. Population pharmacokinetic analyses suggest that the pharmacokinetics of trabectedin is dose-proportional (over the dose range of 0.024 to 1.8 mg/m2) and exposure is time-independent. No accumulation of trabectedin in plasma is observed upon repeated administrations every 3 weeks.
Distribution
Binding of trabectedin to human plasma proteins was approximately 97%, independent of trabectedin concentrations ranging from 10 ng/mL to 100 ng/mL. Steady state volume of distribution of trabectedin exceeds 5000 L.
Elimination
The estimated mean (% coefficient of variation) clearance of trabectedin is 31.5 L/hr (50%) and the terminal elimination half-life is approximately 175 hours.
Metabolism
CYP3A is the predominant CYP enzyme responsible for the hepatic metabolism of trabectedin.
Trabectedin was extensively metabolized with negligible unchanged drug in urine and feces following administration of trabectedin to humans.
Excretion
In patients with solid tumors, following a 3-hour or a 24-hour intravenous infusion of 14C-labeled trabectedin, 64% of the total administered radioactive dose was recovered in 24 days, with 58% in feces and 6% in urine.
Specific Populations
The following population characteristics are not associated with a clinically significant effect on the pharmacokinetics of trabectedin: sex, age (19 to 83 years), body weight (36 to 148 kg), body surface area (0.9 to 2.8 m2), mild hepatic impairment, or mild to moderate renal impairment. The effects of severe hepatic impairment, severe renal impairment or end stage renal disease on trabectedin exposure are unknown.
Hepatic Impairment
The geometric mean dose normalized trabectedin exposure (AUC) increased by 97% (90% CI: 20%, 222%) in patients with moderate hepatic impairment (bilirubin levels greater than 1.5 times to 3 times the upper limit of normal and AST and ALT less than 8 times the upper limit of normal) following administration of a single YONDELIS dose of 0.58 mg/m2 or 0.9 mg/m2 compared to patients with normal liver function following administration of a single YONDELIS dose of 1.3 mg/m2 [see Dosage and Administration (2.1) and Use in Specific Populations (8.6)].
Drug Interactions
Effect of Strong CYP3A Inhibitors on Trabectedin
Coadministration of multiple doses of ketoconazole (200 mg twice daily for 7.5 days) with a single dose of YONDELIS (0.58 mg/m2) on day 1 increased trabectedin dose-normalized AUC by 66% and Cmax by 22% compared to a single YONDELIS dose (1.3 mg/m2) given alone.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Trabectedin is genotoxic in both in vitro and in vivo studies. Long-term carcinogenicity studies have not been performed.
Fertility studies with trabectedin were not performed. In male rats there were limited histopathological signs of hemorrhage and degeneration in the testes following repeated administration of trabectedin at doses approximately 0.2 times the 1.5 mg/m2 human dose based on body surface area.
14 CLINICAL STUDIES
The clinical efficacy and safety of YONDELIS in patients with metastatic or recurrent leiomyosarcoma or liposarcoma were demonstrated in Trial ET743-SAR-3007 (NCT01343277), a randomized (2:1), open-label, active-controlled trial comparing treatment with YONDELIS 1.5 mg/m2 as a 24-hour continuous intravenous infusion once every 3 weeks to dacarbazine 1000 mg/m2 intravenous infusion (20 to 120 minutes) once every 3 weeks. Treatment continued in both arms until disease progression or unacceptable toxicity; all patients in the YONDELIS arm were required to receive dexamethasone 20 mg intravenous injection prior to each YONDELIS infusion. Patients were required to have unresectable, locally advanced or metastatic leiomyosarcoma or liposarcoma (dedifferentiated, myxoid round cell, or pleomorphic) and previous treatment with an anthracycline- and ifosfamide-containing regimen or an anthracycline-containing regimen and one additional cytotoxic chemotherapy regimen. Randomization was stratified by subtype of soft tissue sarcoma (leiomyosarcoma vs. liposarcoma), ECOG performance status (0 vs. 1), and number of prior chemotherapy regimens (1 vs. ≥2). The efficacy outcome measures were investigator-assessed progression-free survival (PFS) according to the Response Evaluation Criteria in Solid Tumors (RECIST v1.1), overall survival (OS), objective response rate (ORR), and duration of response (DOR). Patients in the dacarbazine arm were not offered YONDELIS at the time of disease progression.
A total of 518 patients were randomized, 345 to the YONDELIS arm and 173 patients to the dacarbazine arm. The median patient age was 56 years (range: 17 to 81); 30% were male; 76% White, 12% Black, and 4% Asian; 73% had leiomyosarcomas and 27% liposarcomas; 49% had an ECOG PS of 0; and 89% received ≥2 prior chemotherapy regimens. The most common (≥20%) pre-study chemotherapeutic agents administered were doxorubicin (90%), gemcitabine (81%), docetaxel (74%), and ifosfamide (59%). Approximately 10% of patients had received pazopanib.
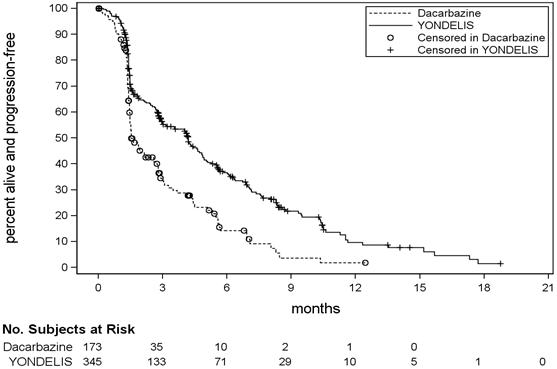
Trial ET743-SAR-3007 demonstrated a statistically significant improvement in PFS. An exploratory analysis of independent radiology committee-determined PFS, in a subgroup consisting of approximately 60% of the total population, provided similar results to the investigator-determined PFS. Efficacy results from Trial ET743-SAR-3007 are presented in the table below.
| Efficacy Endpoint | YONDELIS N=345 | Dacarbazine N=173 |
|---|---|---|
| CR=Complete Response; PR=Partial Response; CI=Confidence Interval, HR=hazard ratio, NE=not estimable. | ||
| Progression-free survival | ||
| PFS Events, n (%) | 217 (63%) | 112 (65%) |
| Disease progression | 204 | 109 |
| Death | 13 | 3 |
| Median (95% CI) (months) | 4.2 (3.0, 4.8) | 1.5 (1.5, 2.6) |
| HR (95% CI)* | 0.55 (0.44, 0.70) | |
| p-value† | <0.001 | |
| Overall survival‡ | ||
| Events, n (%) | 258 (67%) | 123 (64%) |
| Median (95% CI) (months) | 13.7 (12.2, 16.0) | 13.1 (9.1, 16.2) |
| HR (95% CI)* | 0.93 (0.75, 1.15) | |
| p-value† | 0.49 | |
| Objective Response Rate (ORR: CR+PR) | ||
| Number of patients (%) | 23 (7%) | 10 (6%) |
| 95% CI§ | (4.3, 9.8) | (2.8, 10.4) |
| Duration of Response (CR+ PR) | ||
| Median (95% CI) (months) | 6.9 (4.5, 7.6) | 4.2 (2.9, NE) |
Figure 1: Kaplan-Meier Curves of Progression-Free Survival in Trial ET743-SAR-3007
16 HOW SUPPLIED/STORAGE AND HANDLING
YONDELIS is supplied in a single-dose glass vial containing 1 mg trabectedin. Each carton contains one vial (NDC: 59676-610-01).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Myelosuppression: Inform patients of the risks of myelosuppression. Instruct patients to immediately contact their healthcare provider for fever or unusual bruising, bleeding, tiredness, or paleness.
Rhabdomyolysis: Advise patients to contact their healthcare provider if they experience severe muscle pain or weakness.
Hepatotoxicity: Advise patients to contact their healthcare provider immediately for yellowing of skin and eyes (jaundice), pain in the upper right quadrant, severe nausea or vomiting, difficulty in concentrating, disorientation, or confusion.
Cardiomyopathy: Advise patients to contact their healthcare provider for new onset chest pain, shortness of breath, fatigue, lower extremity edema, or heart palpitations.
Hypersensitivity: Advise patients to seek immediate medical attention for symptoms of allergic reactions including difficulty breathing, chest tightness, wheezing, severe dizziness or light-headedness, swelling of the lips or skin rash.
Extravasation: Inform patients of the risks of extravasation and to notify their healthcare provider for redness, swelling, itchiness and discomfort or leakage at the injection site.
Capillary leak syndrome: Advise patients to report symptoms such as edema with or without hypotension [see Warnings and Precautions (5.5)].
Embryofetal toxicity: Advise pregnant women of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with YONDELIS [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1)].
Females and males of reproductive potential: Advise females of reproductive potential to use effective contraception during treatment with YONDELIS and for at least 2 months after last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with YONDELIS and for at least 5 months after the last dose [see Warnings and Precautions (5.7) and Use in Specific Populations (8.3)].
Lactation: Advise females not to breastfeed during treatment with YONDELIS [see Use in Specific Populations (8.2)].
Product of Spain
Manufactured by:
Baxter Oncology GmbH
Halle/Westfalen Germany
Manufactured for:
Janssen Products, LP
Horsham, PA
© 2015 Janssen Pharmaceutical Companies
Under license from Pharma Mar, S.A.
| PATIENT INFORMATION YONDELIS® (yon-DEL-ess) (trabectedin) for injection |
||
|---|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised 12/2017 | |
| What is YONDELIS?
YONDELIS is a prescription medicine used to treat people with liposarcoma or leiomyosarcoma that:
|
||
Who should not receive YONDELIS?
|
||
What should I tell my healthcare provider before receiving YONDELIS? Before receiving YONDELIS, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
||
How will I receive YONDELIS?
|
||
| What are the possible side effects of YONDELIS? YONDELIS may cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
| Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of YONDELIS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
| General information about the safe and effective use of YONDELIS
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your healthcare provider or pharmacist for information about YONDELIS that is written for health professionals. |
||
| What are the ingredients in YONDELIS? Active ingredient: trabectedin Inactive ingredients: potassium dihydrogen phosphate, sucrose, phosphoric acid and potassium hydroxide. Product of Spain Manufactured by: Baxter Oncology GmbH, Halle/Westfalen Germany Manufactured for: Janssen Products, LP, Horsham, PA For more information, call 1-800-526-7736 or go to www.YONDELIS.com. |
||
PRINCIPAL DISPLAY PANEL - 1 Vial Carton
NDC 59676-610-01
Yondelis®
(trabectedin)
for Injection
1 mg per vial
For Intravenous Infusion Only
Reconstitute before further dilution
Each vial contains 1 mg of trabectedin as a
sterile lyophilized powder.
Rx only
Single-dose vial
Discard any unused portion
Cytotoxic