FULL PRESCRIBING INFORMATION
WARNING: LIFE-THREATENING (INCLUDING FATAL) HEPATOTOXICITY and SKIN REACTIONS
HEPATOTOXICITY:
Severe, life-threatening, and in some cases fatal hepatotoxicity, particularly in the first 18 weeks, has been reported in patients treated with VIRAMUNE. In some cases, patients presented with non-specific prodromal signs or symptoms of hepatitis and progressed to hepatic failure. These events are often associated with rash. Female gender and higher CD4+ cell counts at initiation of therapy place patients at increased risk; women with CD4+ cell counts >250 cells/mm3, including pregnant women receiving VIRAMUNE in combination with other antiretrovirals for the treatment of HIV-1 infection, are at the greatest risk. However, hepatotoxicity associated with VIRAMUNE use can occur in both genders, all CD4+ cell counts and at any time during treatment. Patients with signs or symptoms of hepatitis, or with increased transaminases combined with rash or other systemic symptoms, must discontinue VIRAMUNE and seek medical evaluation immediately [see Warnings and Precautions (5.1)].
SKIN REACTIONS:
Severe, life-threatening skin reactions, including fatal cases, have occurred in patients treated with VIRAMUNE. These have included cases of Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions characterized by rash, constitutional findings, and organ dysfunction. Patients developing signs or symptoms of severe skin reactions or hypersensitivity reactions must discontinue VIRAMUNE and seek medical evaluation immediately. Transaminase levels should be checked immediately for all patients who develop a rash in the first 18 weeks of treatment. The 14-day lead-in period with VIRAMUNE 200 mg daily dosing has been observed to decrease the incidence of rash and must be followed [see Warnings and Precautions (5.2)].
MONITORING:
Patients must be monitored intensively during the first 18 weeks of therapy with VIRAMUNE to detect potentially life-threatening hepatotoxicity or skin reactions. Extra vigilance is warranted during the first 6 weeks of therapy, which is the period of greatest risk of these events. Do not restart VIRAMUNE following severe hepatic, skin or hypersensitivity reactions. In some cases, hepatic injury has progressed despite discontinuation of treatment.
1 INDICATIONS AND USAGE
VIRAMUNE is indicated for use in combination with other antiretroviral agents for the treatment of HIV-1 infection. This indication is based on one principal clinical trial (BI 1090) that demonstrated prolonged suppression of HIV-1 RNA and two smaller supportive studies, one of which (BI 1046) is described below.
Additional important information regarding the use of VIRAMUNE for the treatment of HIV-1 infection:
- Based on serious and life-threatening hepatotoxicity observed in controlled and uncontrolled studies, VIRAMUNE should not be initiated in adult females with CD4+ cell counts greater than 250 cells/mm3 or in adult males with CD4+ cell counts greater than 400 cells/mm3 unless the benefit outweighs the risk [see Boxed Warning and Warnings and Precautions (5.1)].
- The 14-day lead-in period with VIRAMUNE 200 mg daily dosing has been demonstrated to reduce the frequency of rash [see Dosage and Administration (2.4) and Warnings and Precautions (5.2)].
- If rash persists beyond the 14-day lead-in period, do not dose escalate to 200 mg twice daily. The 200 mg once-daily dosing regimen should not be continued beyond 28 days, at which point an alternative regimen should be sought.
2 DOSAGE AND ADMINISTRATION
2.1 Adults
The recommended dose for VIRAMUNE is one 200 mg tablet daily for the first 14 days, followed by one 200 mg tablet twice daily, in combination with other antiretroviral agents. The lead-in period has been observed to decrease the incidence of rash. For concomitantly administered antiretroviral therapy, the manufacturer’s recommended dosage and monitoring should be followed.
2.2 Pediatric Patients
The recommended oral dose for pediatric patients 15 days and older is 150 mg/m2 once daily for 14 days followed by 150 mg/m2 twice daily thereafter. The total daily dose should not exceed 400 mg for any patient.
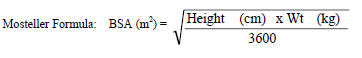
| BSA range (m2) | Volume (mL) |
| 0.06 – 0.12 | 1.25 |
| 0.12 – 0.25 | 2.5 |
| 0.25 – 0.42 | 5 |
| 0.42 – 0.58 | 7.5 |
| 0.58 – 0.75 | 10 |
| 0.75 – 0.92 | 12.5 |
| 0.92 – 1.08 | 15 |
| 1.08 – 1.25 | 17.5 |
| 1.25+ | 20 |
VIRAMUNE suspension should be shaken gently prior to administration. It is important to administer the entire measured dose of suspension by using an oral dosing syringe or dosing cup. An oral dosing syringe is recommended, particularly for volumes of 5 mL or less. If a dosing cup is used, it should be thoroughly rinsed with water and the rinse should also be administered to the patient.
2.3 Monitoring of Patients
Intensive clinical and laboratory monitoring, including liver enzyme tests, is essential at baseline and during the first 18 weeks of treatment with VIRAMUNE. The optimal frequency of monitoring during this period has not been established. Some experts recommend clinical and laboratory monitoring more often than once per month, and in particular, would include monitoring of liver enzyme tests at baseline, prior to dose escalation, and at two weeks post-dose escalation. After the initial 18-week period, frequent clinical and laboratory monitoring should continue throughout VIRAMUNE treatment [see Warnings and Precautions (5)]. In some cases, hepatic injury has progressed despite discontinuation of treatment.
2.4 Dosage Adjustment
Patients with Rash
VIRAMUNE should be discontinued if a patient experiences severe rash or any rash accompanied by constitutional findings [see Boxed Warning, Warnings and Precautions (5.2), and Patient Counseling Information (17.1)]. A patient experiencing mild to moderate rash without constitutional symptoms during the 14-day lead-in period of 200 mg/day (150 mg/m2/day in pediatric patients) should not have their VIRAMUNE dose increased until the rash has resolved [see Warnings and Precautions (5.2) and Patient Counseling Information (17.1)]. The total duration of the once daily lead-in dosing period should not exceed 28 days at which point an alternative regimen should be sought.
Patients with Hepatic Events
If a clinical (symptomatic) hepatic event occurs, VIRAMUNE should be permanently discontinued. Do not restart VIRAMUNE after recovery [see Warnings and Precautions (5.1)].
Patients with Dose Interruption
Patients who interrupt VIRAMUNE dosing for more than 7 days should restart the recommended dosing, using one 200 mg tablet daily (150 mg/m2/day in pediatric patients) for the first 14 days (lead-in) followed by one 200 mg tablet twice daily (150 mg/m2 twice daily for pediatric patients).
Patients on Dialysis
An additional 200 mg dose of VIRAMUNE following each dialysis treatment is indicated in patients requiring dialysis. Nevirapine metabolites may accumulate in patients receiving dialysis; however, the clinical significance of this accumulation is not known [see Clinical Pharmacology (12.3)]. Patients with CrCL ≥20 mL/min do not require an adjustment in VIRAMUNE dosing.
3 DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg, white, oval, biconvex, tablets embossed with 54 193 on one side
Oral suspension: 50 mg/5 mL, white to off-white oral suspension
4 CONTRAINDICATIONS
VIRAMUNE is contraindicated in patients with moderate or severe (Child-Pugh Class B or C, respectively) hepatic impairment [see Warnings and Precautions (5.1) and Use in Specific Populations (8.7)].
5 WARNINGS AND PRECAUTIONS
The most serious adverse reactions associated with VIRAMUNE are hepatitis/hepatic failure, Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions. Hepatitis/hepatic failure may be associated with signs of hypersensitivity which can include severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema, eosinophilia, granulocytopenia, lymphadenopathy, or renal dysfunction.
The first 18 weeks of therapy with VIRAMUNE are a critical period during which intensive clinical and laboratory monitoring of patients is required to detect potentially life-threatening hepatic events and skin reactions. The optimal frequency of monitoring during this time period has not been established. Some experts recommend clinical and laboratory monitoring more often than once per month, and in particular, would include monitoring of liver enzyme tests at baseline, prior to dose escalation and at two weeks post-dose escalation. After the initial 18-week period, frequent clinical and laboratory monitoring should continue throughout VIRAMUNE treatment. In addition, the 14-day lead-in period with VIRAMUNE 200 mg daily dosing has been demonstrated to reduce the frequency of rash [see Dosage and Administration (2.1)].
5.1 Hepatotoxicity and Hepatic Impairment
Severe, life-threatening, and in some cases fatal hepatotoxicity, including fulminant and cholestatic hepatitis, hepatic necrosis and hepatic failure, have been reported in patients treated with VIRAMUNE. In controlled clinical trials, symptomatic hepatic events regardless of severity occurred in 4% (range 0% to 11.0%) of patients who received VIRAMUNE and 1.2% of patients in control groups.
The risk of symptomatic hepatic events regardless of severity was greatest in the first 6 weeks of therapy. The risk continued to be greater in the VIRAMUNE groups compared to controls through 18 weeks of treatment. However, hepatic events may occur at any time during treatment. In some cases, patients presented with non-specific, prodromal signs or symptoms of fatigue, malaise, anorexia, nausea, jaundice, liver tenderness or hepatomegaly, with or without initially abnormal serum transaminase levels. Rash was observed in approximately half of the patients with symptomatic hepatic adverse events. Fever and flu-like symptoms accompanied some of these hepatic events. Some events, particularly those with rash and other symptoms, have progressed to hepatic failure with transaminase elevation, with or without hyperbilirubinemia, hepatic encephalopathy, prolonged partial thromboplastin time, or eosinophilia. Rhabdomyolysis has been observed in some patients experiencing skin and/or liver reactions associated with VIRAMUNE use. Patients with signs or symptoms of hepatitis must be advised to discontinue VIRAMUNE and immediately seek medical evaluation, which should include liver enzyme tests.
Transaminases should be checked immediately if a patient experiences signs or symptoms suggestive of hepatitis and/or hypersensitivity reaction. Transaminases should also be checked immediately for all patients who develop a rash in the first 18 weeks of treatment. Physicians and patients should be vigilant for the appearance of signs or symptoms of hepatitis, such as fatigue, malaise, anorexia, nausea, jaundice, bilirubinuria, acholic stools, liver tenderness or hepatomegaly. The diagnosis of hepatotoxicity should be considered in this setting, even if transaminases are initially normal or alternative diagnoses are possible [see Boxed Warning, Dosage and Administration (2.3), and Patient Counseling Information (17.1)].
If clinical hepatitis or transaminase elevations combined with rash or other systemic symptoms occur, VIRAMUNE should be permanently discontinued. Do not restart VIRAMUNE after recovery. In some cases, hepatic injury progresses despite discontinuation of treatment.
The patients at greatest risk of hepatic events, including potentially fatal events, are women with high CD4+ cell counts. In general, during the first 6 weeks of treatment, women have a three-fold higher risk than men for symptomatic, often rash-associated, hepatic events (5.8% versus 2.2%), and patients with higher CD4+ cell counts at initiation of VIRAMUNE therapy are at higher risk for symptomatic hepatic events with VIRAMUNE. In a retrospective review, women with CD4+ cell counts >250 cells/mm3 had a 12-fold higher risk of symptomatic hepatic adverse events compared to women with CD4+ cell counts <250 cells/mm3 (11.0% versus 0.9%). An increased risk was observed in men with CD4+ cell counts >400 cells/mm3 (6.3% versus 1.2% for men with CD4+ cell counts <400 cells/mm3). However, all patients, regardless of gender, CD4+ cell count, or antiretroviral treatment history, should be monitored for hepatotoxicity since symptomatic hepatic adverse events have been reported at all CD4+ cell counts. Co-infection with hepatitis B or C and/or increased transaminase elevations at the start of therapy with VIRAMUNE are associated with a greater risk of later symptomatic events (6 weeks or more after starting VIRAMUNE) and asymptomatic increases in AST or ALT.
In addition, serious hepatotoxicity (including liver failure requiring transplantation in one instance) has been reported in HIV-1 uninfected individuals receiving multiple doses of VIRAMUNE in the setting of post-exposure prophylaxis, an unapproved use.
Increased nevirapine trough concentrations have been observed in some patients with hepatic fibrosis or cirrhosis. Therefore, patients with either hepatic fibrosis or cirrhosis should be monitored carefully for evidence of drug-induced toxicity. Nevirapine should not be administered to patients with moderate or severe (Child-Pugh Class B or C, respectively) hepatic impairment [see Contraindications (4), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
5.2 Skin Reactions
Severe and life-threatening skin reactions, including fatal cases, have been reported, occurring most frequently during the first 6 weeks of therapy. These have included cases of Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions characterized by rash, constitutional findings, and organ dysfunction including hepatic failure. Rhabdomyolysis has been observed in some patients experiencing skin and/or liver reactions associated with VIRAMUNE use. In controlled clinical trials, Grade 3 and 4 rashes were reported during the first 6 weeks in 1.5% of VIRAMUNE recipients compared to 0.1% of placebo subjects.
Patients developing signs or symptoms of severe skin reactions or hypersensitivity reactions (including, but not limited to, severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema, and/or hepatitis, eosinophilia, granulocytopenia, lymphadenopathy, and renal dysfunction) must permanently discontinue VIRAMUNE and seek medical evaluation immediately [see Boxed Warning and Patient Counseling Information (17.1)]. Do not restart VIRAMUNE following severe skin rash, skin rash combined with increased transaminases or other symptoms, or hypersensitivity reaction.
If patients present with a suspected VIRAMUNE-associated rash, transaminases should be measured immediately. Patients with rash-associated transaminase elevations should be permanently discontinued from VIRAMUNE [see Warnings and Precautions (5.1)].
Therapy with VIRAMUNE must be initiated with a 14-day lead-in period of 200 mg/day (150 mg/m2/day in pediatric patients), which has been shown to reduce the frequency of rash. VIRAMUNE should be discontinued if a patient experiences severe rash or any rash accompanied by constitutional findings. A patient experiencing a mild to moderate rash without constitutional symptoms during the 14-day lead-in period of 200 mg/day (150 mg/m2/day in pediatric patients) should not have their VIRAMUNE dose increased until the rash has resolved. The total duration of the once-daily lead-in dosing period should not exceed 28 days at which point an alternative regimen should be sought [see Dosage and Administration (2.4)]. Patients should be monitored closely if isolated rash of any severity occurs. Delay in stopping VIRAMUNE treatment after the onset of rash may result in a more serious reaction.
Women appear to be at higher risk than men of developing rash with VIRAMUNE.
In a clinical trial, concomitant prednisone use (40 mg/day for the first 14 days of VIRAMUNE administration) was associated with an increase in incidence and severity of rash during the first 6 weeks of VIRAMUNE therapy. Therefore, use of prednisone to prevent VIRAMUNE-associated rash is not recommended.
5.3 Resistance
VIRAMUNE must not be used as a single agent to treat HIV-1 or added on as a sole agent to a failing regimen. Resistant virus emerges rapidly when nevirapine is administered as monotherapy. The choice of new antiretroviral agents to be used in combination with nevirapine should take into consideration the potential for cross resistance. When discontinuing an antiretroviral regimen containing VIRAMUNE, the long half-life of nevirapine should be taken into account; if antiretrovirals with shorter half-lives than VIRAMUNE are stopped concurrently, low plasma concentrations of nevirapine alone may persist for a week or longer and virus resistance may subsequently develop [see Clinical Pharmacology (12.4)].
5.4 Drug Interactions
See Table 4 for listings of established and potential drug interactions [see Drug Interactions (7)].
Concomitant use of St. John's wort (Hypericum perforatum) or St. John's wort-containing products and VIRAMUNE is not recommended. Co-administration of St. John’s wort with non-nucleoside reverse transcriptase inhibitors (NNRTIs), including VIRAMUNE, is expected to substantially decrease NNRTI concentrations and may result in sub-optimal levels of VIRAMUNE and lead to loss of virologic response and possible resistance to VIRAMUNE or to the class of NNRTIs. Co-administration of VIRAMUNE and efavirenz is not recommended as this combination has been associated with an increase in adverse reactions and no improvement in efficacy.
5.5 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including VIRAMUNE. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia (PCP), or tuberculosis), which may necessitate further evaluation and treatment.
5.6 Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
6 ADVERSE REACTIONS
6.1 Clinical Trials in Adults
The most serious adverse reactions associated with VIRAMUNE are hepatitis, hepatic failure, Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions. Hepatitis/hepatic failure may be isolated or associated with signs of hypersensitivity which may include severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema, eosinophilia, granulocytopenia, lymphadenopathy, or renal dysfunction [see Boxed Warning and Warnings and Precautions (5.1, 5.2)].
Hepatic Reaction
In controlled clinical trials, symptomatic hepatic events regardless of severity occurred in 4.0% (range 0% to 11.0%) of patients who received VIRAMUNE and 1.2% of patients in control groups. Female gender and higher CD4+ cell counts (>250 cells/mm3 in women and >400 cells/mm3 in men) place patients at increased risk of these events [see Boxed Warning and Warnings and Precautions (5.1)].
Asymptomatic transaminase elevations (AST or ALT >5X ULN) were observed in 5.8% (range 0% to 9.2%) of patients who received VIRAMUNE and 5.5% of patients in control groups. Co-infection with hepatitis B or C and/or increased transaminase elevations at the start of therapy with VIRAMUNE are associated with a greater risk of later symptomatic events (6 weeks or more after starting VIRAMUNE) and asymptomatic increases in AST or ALT.
Liver enzyme abnormalities (AST, ALT, GGT) were observed more frequently in patients receiving VIRAMUNE than in controls (see Table 3).
Skin Reaction
The most common clinical toxicity of VIRAMUNE is rash, which can be severe or life-threatening [see Boxed Warning and Warnings and Precautions (5.2)]. Rash occurs most frequently within the first 6 weeks of therapy. Rashes are usually mild to moderate, maculopapular erythematous cutaneous eruptions, with or without pruritus, located on the trunk, face and extremities. In controlled clinical trials (Trials 1037, 1038, 1046, and 1090), Grade 1 and 2 rashes were reported in 13.3% of patients receiving VIRAMUNE compared to 5.8% receiving placebo during the first 6 weeks of therapy. Grade 3 and 4 rashes were reported in 1.5% of VIRAMUNE recipients compared to 0.1% of subjects receiving placebo. Women tend to be at higher risk for development of VIRAMUNE-associated rash [see Boxed Warning and Warnings and Precautions (5.2)].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Treatment-related, adverse experiences of moderate or severe intensity observed in >2% of patients receiving VIRAMUNE in placebo-controlled trials are shown in Table 2.
| 1 Background therapy included 3TC for all patients and combinations of NRTIs and PIs. Patients had CD4+ cell counts <200 cells/mm3. 2 Background therapy included ZDV and ZDV+ddI; VIRAMUNE monotherapy was administered in some patients. Patients had CD4+ cell count ≥200 cells/mm3. |
||||
| Trial 10901 | Trials 1037, 1038, 10462 | |||
| VIRAMUNE | Placebo | VIRAMUNE | Placebo | |
| (n=1121) | (n=1128) | (n=253) | (n=203) | |
| Median exposure (weeks) | 58 | 52 | 28 | 28 |
| Any adverse event | 14.5% | 11.1% | 31.6% | 13.3% |
| Rash | 5.1 | 1.8 | 6.7 | 1.5 |
| Nausea | 0.5 | 1.1 | 8.7 | 3.9 |
| Granulocytopenia | 1.8 | 2.8 | 0.4 | 0 |
| Headache | 0.7 | 0.4 | 3.6 | 0.5 |
| Fatigue | 0.2 | 0.3 | 4.7 | 3.9 |
| Diarrhea | 0.2 | 0.8 | 2.0 | 0.5 |
| Abdominal pain | 0.1 | 0.4 | 2.0 | 0 |
| Myalgia | 0.2 | 0 | 1.2 | 2.0 |
Laboratory Abnormalities
Liver enzyme test abnormalities (AST, ALT) were observed more frequently in patients receiving VIRAMUNE than in controls (Table 3). Asymptomatic elevations in GGT occur frequently but are not a contraindication to continue VIRAMUNE therapy in the absence of elevations in other liver enzyme tests. Other laboratory abnormalities (bilirubin, anemia, neutropenia, thrombocytopenia) were observed with similar frequencies in clinical trials comparing VIRAMUNE and control regimens (see Table 3).
| 1 Background therapy included 3TC for all patients and combinations of NRTIs and PIs. Patients had CD4+ cell counts <200 cells/mm3. 2 Background therapy included ZDV and ZDV+ddI; VIRAMUNE monotherapy was administered in some patients. Patients had CD4+ cell count >200 cells/mm3. |
||||
| Trial 10901 | Trials 1037, 1038, 10462 | |||
| VIRAMUNE | Placebo | VIRAMUNE | Placebo | |
| Laboratory Abnormality | (n=1121) | (n=1128) | (n=253) | (n=203) |
| Blood Chemistry | ||||
| SGPT (ALT) >250 U/L | 5.3 | 4.4 | 14.0 | 4.0 |
| SGOT (AST) >250 U/L | 3.7 | 2.5 | 7.6 | 1.5 |
| Bilirubin >2.5 mg/dL | 1.7 | 2.2 | 1.7 | 1.5 |
| Hematology | ||||
| Hemoglobin <8.0 g/dL | 3.2 | 4.1 | 0 | 0 |
| Platelets <50,000/mm3 | 1.3 | 1.0 | 0.4 | 1.5 |
| Neutrophils <750/mm3 | 13.3 | 13.5 | 3.6 | 1.0 |
6.2 Clinical Trials in Pediatric Patients
Adverse events were assessed in BI Trial 1100.1032 (ACTG 245), a double-blind, placebo-controlled trial of VIRAMUNE (n=305) in which pediatric patients received combination treatment with VIRAMUNE. In this trial two patients were reported to experience Stevens-Johnson syndrome or Stevens-Johnson/toxic epidermal necrolysis transition syndrome. Safety was also assessed in trial BI 1100.882 (ACTG 180), an open-label trial of VIRAMUNE (n=37) in which patients were followed for a mean duration of 33.9 months (range: 6.8 months to 5.3 years, including long-term follow-up in 29 of these patients in trial BI 1100.892). The most frequently reported adverse events related to VIRAMUNE in pediatric patients were similar to those observed in adults, with the exception of granulocytopenia, which was more commonly observed in children receiving both zidovudine and VIRAMUNE. Cases of allergic reaction, including one case of anaphylaxis, were also reported.
The safety of VIRAMUNE was also examined in BI Trial 1100.1368, an open-label, randomized clinical study performed in South Africa in which 123 HIV-1 infected treatment-naïve patients between 3 months and 16 years of age received combination treatment with VIRAMUNE oral suspension, lamivudine and zidovudine for 48 weeks [see Use In Specific Populations (8.4) and Clinical Pharmacology (12.3)]. Rash (all causality) was reported in 21% of the patients, 4 (3%) of whom discontinued drug due to rash. All 4 patients experienced the rash early in the course of therapy (<4 weeks) and resolved upon nevirapine discontinuation. Other clinically important adverse events (all causality) include neutropenia (8.9%), anemia (7.3%) and hepatotoxicity (2.4%) [see Use in Specific Populations (8.4) and Clinical Studies (14.2)].
Safety information on use of VIRAMUNE in combination therapy in pediatric patients 2 weeks to <3 months of age was assessed in 36 patients from the BI 1100.1222 (PACTG 356) study. No unexpected safety findings were observed although granulocytopenia was reported more frequently in this age group compared to the older pediatric age groups and adults.
6.3 Post-Marketing Surveillance
In addition to the adverse events identified during clinical trials, the following adverse reactions have been identified during post-approval use of VIRAMUNE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Body as a Whole: fever, somnolence, drug withdrawal [see Drug Interactions (7)], redistribution/accumulation of body fat [see Warnings and Precautions (5.6)]
- Gastrointestinal: vomiting
- Liver and Biliary: jaundice, fulminant and cholestatic hepatitis, hepatic necrosis, hepatic failure
- Hematology: anemia, eosinophilia, neutropenia
- Musculoskeletal: arthralgia, rhabdomyolysis associated with skin and/or liver reactions
- Neurologic: paraesthesia
- Skin and Appendages: allergic reactions including anaphylaxis, angioedema, bullous eruptions, ulcerative stomatitis and urticaria have all been reported. In addition, hypersensitivity syndrome and hypersensitivity reactions with rash associated with constitutional findings such as fever, blistering, oral lesions, conjunctivitis, facial edema, muscle or joint aches, general malaise, fatigue or significant hepatic abnormalities [see Warnings and Precautions (5.1)] plus one or more of the following: hepatitis, eosinophilia, granulocytopenia, lymphadenopathy and/or renal dysfunction have been reported with the use of VIRAMUNE.
In post-marketing surveillance anemia has been more commonly observed in children although development of anemia due to concomitant medication use cannot be ruled out.
7 DRUG INTERACTIONS
Nevirapine is principally metabolized by the liver via the cytochrome P450 isoenzymes, 3A and 2B6. Nevirapine is known to be an inducer of these enzymes. As a result, drugs that are metabolized by these enzyme systems may have lower than expected plasma levels when co-administered with nevirapine.
The specific pharmacokinetic changes that occur with co-administration of nevirapine and other drugs are listed in Clinical Pharmacology, Table 5. Clinical comments about possible dosage modifications based on established drug interactions are listed in Table 4. The data in Tables 4 and 5 are based on the results of drug interaction studies conducted in HIV-1 seropositive subjects unless otherwise indicated. In addition to established drug interactions, there may be potential pharmacokinetic interactions between nevirapine and other drug classes that are metabolized by the cytochrome P450 system. These potential drug interactions are also listed in Table 4. Although specific drug interaction studies in HIV-1 seropositive subjects have not been conducted for some classes of drugs listed in Table 4, additional clinical monitoring may be warranted when co-administering these drugs.
The in vitro interaction between nevirapine and the antithrombotic agent warfarin is complex. As a result, when giving these drugs concomitantly, plasma warfarin levels may change with the potential for increases in coagulation time. When warfarin is co-administered with nevirapine, anticoagulation levels should be monitored frequently.
| Drug Name | Effect on Concentration of Nevirapine or Concomitant Drug | Clinical Comment |
| Atazanavir/Ritonavir | ↓ Atazanavir ↑ Nevirapine | Do not co-administer nevirapine with atazanavir because nevirapine substantially decreases atazanavir exposure. |
| Clarithromycin | ↓ Clarithromycin ↑ 14-OH clarithromycin | Clarithromycin exposure was significantly decreased by nevirapine; however, 14-OH metabolite concentrations were increased. Because clarithromycin active metabolite has reduced activity against Mycobacterium avium-intracellulare complex, overall activity against this pathogen may be altered. Alternatives to clarithromycin, such as azithromycin, should be considered. |
| Efavirenz | ↓ Efavirenz | There has been no determination of appropriate doses for the safe and effective use of this combination [see Warnings and Precautions (5.4)]. |
| Ethinyl estradiol and Norethindrone | ↓ Ethinyl estradiol ↓ Norethindrone | Oral contraceptives and other hormonal methods of birth control should not be used as the sole method of contraception in women taking nevirapine, since nevirapine may lower the plasma levels of these medications. An alternative or additional method of contraception is recommended. |
| Fluconazole | ↑Nevirapine | Because of the risk of increased exposure to nevirapine, caution should be used in concomitant administration, and patients should be monitored closely for nevirapine-associated adverse events. |
| Fosamprenavir | ↓ Amprenavir ↑ Nevirapine | Co-administration of nevirapine and fosamprenavir without ritonavir is not recommended. |
| Fosamprenavir/Ritonavir | ↓ Amprenavir ↑ Nevirapine | No dosing adjustments are required when nevirapine is co-administered with 700/100 mg of fosamprenavir/ritonavir twice daily. |
| Indinavir | ↓ Indinavir | Appropriate doses for this combination are not established, but an increase in the dosage of indinavir may be required. |
| Ketoconazole | ↓ Ketoconazole | Nevirapine and ketoconazole should not be administered concomitantly because decreases in ketoconazole plasma concentrations may reduce the efficacy of the drug. |
| Lopinavir/Ritonavir | ↓Lopinavir | Lopinavir/ritonavir 400/100 mg tablets can be used twice daily in combination with nevirapine with no dose adjustment in antiretroviral-naïve patients. A dose increase of lopinavir/ritonavir tablets to 600/150 mg (3 tablets) twice daily may be considered when used in combination with nevirapine in treatment-experienced patients where decreased susceptibility to lopinavir is clinically suspected (by treatment history or laboratory evidence). A dose increase of lopinavir/ritonavir oral solution to 533/133 mg twice daily with food is recommended in combination with nevirapine. In children 6 months to 12 years of age, consideration should be given to increasing the dose of lopinavir/ritonavir to 13/3.25 mg/kg for those 7 to <15 kg; 11/2.75 mg/kg for those 15 to 45 kg; and up to a maximum dose of 533/133 mg for those >45 kg twice daily when used in combination with nevirapine, particularly for patients in whom reduced susceptibility to lopinavir/ritonavir is suspected. |
| Methadone | ↓ Methadone | Methadone levels were decreased; increased dosages may be required to prevent symptoms of opiate withdrawal. Methadone-maintained patients beginning nevirapine therapy should be monitored for evidence of withdrawal and methadone dose should be adjusted accordingly. |
| Nelfinavir | ↓Nelfinavir M8 Metabolite ↓Nelfinavir Cmin | The appropriate dose for nelfinavir in combination with nevirapine, with respect to safety and efficacy, has not been established. |
| Rifabutin | ↑Rifabutin | Rifabutin and its metabolite concentrations were moderately increased. Due to high intersubject variability, however, some patients may experience large increases in rifabutin exposure and may be at higher risk for rifabutin toxicity. Therefore, caution should be used in concomitant administration. |
| Rifampin | ↓ Nevirapine | Nevirapine and rifampin should not be administered concomitantly because decreases in nevirapine plasma concentrations may reduce the efficacy of the drug. Physicians needing to treat patients co-infected with tuberculosis and using a nevirapine-containing regimen may use rifabutin instead. |
| Saquinavir/ritonavir | The interaction between VIRAMUNE and saquinavir/ritonavir has not been evaluated | The appropriate doses of the combination of nevirapine and saquinavir/ritonavir with respect to safety and efficacy have not been established. |
|
Potential Drug Interactions: | ||
| Drug Class | Examples of Drugs | |
| Antiarrhythmics | Amiodarone, disopyramide, lidocaine | Plasma concentrations may be decreased. |
| Anticonvulsants | Carbamazepine, clonazepam, ethosuximide | Plasma concentrations may be decreased. |
| Antifungals | Itraconazole | Plasma concentrations of some azole antifungals may be decreased. Nevirapine and itraconazole should not be administered concomitantly due to a potential decrease in itraconazole plasma concentrations. |
| Calcium channel blockers | Diltiazem, nifedipine, verapamil | Plasma concentrations may be decreased. |
| Cancer chemotherapy | Cyclophosphamide | Plasma concentrations may be decreased. |
| Ergot alkaloids | Ergotamine | Plasma concentrations may be decreased. |
| Immunosuppressants | Cyclosporin, tacrolimus, sirolimus | Plasma concentrations may be decreased. |
| Motility agents | Cisapride | Plasma concentrations may be decreased. |
| Opiate agonists | Fentanyl | Plasma concentrations may be decreased. |
| Antithrombotics | Warfarin | Plasma concentrations may be increased. Potential effect on anticoagulation. Monitoring of anticoagulation levels is recommended. |
8 USE IN SPECIFIC POPULATIONS
Teratogenic Effects, Pregnancy Category B.
No observable teratogenicity was detected in reproductive studies performed in pregnant rats and rabbits. The maternal and developmental no-observable-effect level dosages produced systemic exposures approximately equivalent to or approximately 50% higher in rats and rabbits, respectively, than those seen at the recommended daily human dose (based on AUC). In rats, decreased fetal body weights were observed due to administration of a maternally toxic dose (exposures approximately 50% higher than that seen at the recommended human clinical dose).
There are no adequate and well-controlled studies of VIRAMUNE in pregnant women. The Antiretroviral Pregnancy Registry, which has been surveying pregnancy outcomes since January 1989, has not found an increased risk of birth defects following first trimester exposures to nevirapine. The prevalence of birth defects after any trimester exposure to nevirapine is comparable to the prevalence observed in the general population.
Severe hepatic events, including fatalities, have been reported in pregnant women receiving chronic VIRAMUNE therapy as part of combination treatment of HIV-1 infection. Regardless of pregnancy status, women with CD4+ cell counts >250 cells/mm3 should not initiate VIRAMUNE unless the benefit outweighs the risk. It is unclear if pregnancy augments the risk observed in non-pregnant women [see Boxed Warning].
VIRAMUNE should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Antiretroviral Pregnancy Registry
To monitor maternal-fetal outcomes of pregnant women exposed to VIRAMUNE, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling (800) 258-4263.
8.3 Nursing Mothers
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1. Nevirapine is excreted in breast milk. Because of both the potential for HIV-1 transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breastfeed if they are receiving VIRAMUNE.
8.4 Pediatric Use
The safety, pharmacokinetic profile, and virologic and immunologic responses of VIRAMUNE have been evaluated in HIV-1 infected pediatric patients age 3 months to 18 years [see Adverse Reactions (6.2) and Clinical Studies (14.2)]. The safety and pharmacokinetic profile of VIRAMUNE has been evaluated in HIV-1 infected pediatric patients age 15 days to <3 months [see Adverse Reactions (6.2) and Clinical Studies (14.2)].
The most frequently reported adverse events related to VIRAMUNE in pediatric patients were similar to those observed in adults, with the exception of granulocytopenia, which was more commonly observed in children receiving both zidovudine and VIRAMUNE [see Adverse Reactions (6.2) and Clinical Studies (14.2)].
8.5 Geriatric Use
Clinical studies of VIRAMUNE did not include sufficient numbers of subjects aged 65 and older to determine whether elderly subjects respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
In subjects with renal impairment (mild, moderate or severe), there were no significant changes in the pharmacokinetics of nevirapine. Nevirapine is extensively metabolized by the liver and nevirapine metabolites are extensively eliminated by the kidney. Nevirapine metabolites may accumulate in patients receiving dialysis; however, the clinical significance of this accumulation is not known. No adjustment in nevirapine dosing is required in patients with CrCL ≥20 mL/min. In patients undergoing chronic hemodialysis, an additional 200 mg dose following each dialysis treatment is indicated [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Because increased nevirapine levels and nevirapine accumulation may be observed in patients with serious liver disease, do not administer VIRAMUNE to patients with moderate or severe (Child-Pugh Class B or C, respectively) hepatic impairment [see Contraindications (4), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no known antidote for VIRAMUNE overdosage. Cases of VIRAMUNE overdose at doses ranging from 800 to 1800 mg per day for up to 15 days have been reported. Patients have experienced events including edema, erythema nodosum, fatigue, fever, headache, insomnia, nausea, pulmonary infiltrates, rash, vertigo, vomiting and weight decrease. All events subsided following discontinuation of VIRAMUNE.
11 DESCRIPTION
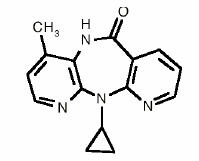
VIRAMUNE is the brand name for nevirapine, a non-nucleoside reverse transcriptase inhibitor (NNRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Nevirapine is structurally a member of the dipyridodiazepinone chemical class of compounds.
The chemical name of nevirapine is 11-cyclopropyl-5,11-dihydro-4-methyl-6H-dipyrido [3,2-b:2',3'-e][1,4] diazepin-6-one. Nevirapine is a white to off-white crystalline powder with the molecular weight of 266.30 and the molecular formula C15H14N4O. Nevirapine has the following structural formula:
VIRAMUNE Tablets are for oral administration. Each tablet contains 200 mg of nevirapine and the inactive ingredients microcrystalline cellulose, lactose monohydrate, povidone, sodium starch glycolate, colloidal silicon dioxide and magnesium stearate.
VIRAMUNE Oral Suspension is for oral administration. Each 5 mL of VIRAMUNE suspension contains 50 mg of nevirapine (as nevirapine hemihydrate). The suspension also contains the following excipients: carbomer 934P, methylparaben, propylparaben, sorbitol, sucrose, polysorbate 80, sodium hydroxide and purified water.
12 CLINICAL PHARMACOLOGY
Absorption and Bioavailability
Nevirapine is readily absorbed (>90%) after oral administration in healthy volunteers and in adults with HIV-1 infection. Absolute bioavailability in 12 healthy adults following single-dose administration was 93 ± 9% (mean ± SD) for a 50 mg tablet and 91 ± 8% for an oral solution. Peak plasma nevirapine concentrations of 2 ± 0.4 μg/mL (7.5 μM) were attained by 4 hours following a single 200 mg dose. Following multiple doses, nevirapine peak concentrations appear to increase linearly in the dose range of 200 to 400 mg/day. Steady-state trough nevirapine concentrations of 4.5 ± 1.9 μg/mL (17 ± 7 μM), (n = 242) were attained at 400 mg/day. Nevirapine tablets and suspension have been shown to be comparably bioavailable and interchangeable at doses up to 200 mg. When VIRAMUNE (200 mg) was administered to 24 healthy adults (12 female, 12 male), with either a high-fat breakfast (857 kcal, 50 g fat, 53% of calories from fat) or antacid (Maalox® 30 mL), the extent of nevirapine absorption (AUC) was comparable to that observed under fasting conditions. In a separate study in HIV-1 infected patients (n=6), nevirapine steady-state systemic exposure (AUCτ) was not significantly altered by didanosine, which is formulated with an alkaline buffering agent. VIRAMUNE may be administered with or without food, antacid or didanosine.
Distribution
Nevirapine is highly lipophilic and is essentially nonionized at physiologic pH. Following intravenous administration to healthy adults, the apparent volume of distribution (Vdss) of nevirapine was 1.21 ± 0.09 L/kg, suggesting that nevirapine is widely distributed in humans. Nevirapine readily crosses the placenta and is also found in breast milk [see Use In Specific Populations (8.3)]. Nevirapine is about 60% bound to plasma proteins in the plasma concentration range of 1-10 μg/mL. Nevirapine concentrations in human cerebrospinal fluid (n=6) were 45% (±5%) of the concentrations in plasma; this ratio is approximately equal to the fraction not bound to plasma protein.
Metabolism/Elimination
In vivo studies in humans and in vitro studies with human liver microsomes have shown that nevirapine is extensively biotransformed via cytochrome P450 (oxidative) metabolism to several hydroxylated metabolites. In vitro studies with human liver microsomes suggest that oxidative metabolism of nevirapine is mediated primarily by cytochrome P450 (CYP) isozymes from the CYP3A and CYP2B6 families, although other isozymes may have a secondary role. In a mass balance/excretion study in eight healthy male volunteers dosed to steady state with nevirapine 200 mg given twice daily followed by a single 50 mg dose of 14C-nevirapine, approximately 91.4 ± 10.5% of the radiolabeled dose was recovered, with urine (81.3 ± 11.1%) representing the primary route of excretion compared to feces (10.1 ± 1.5%). Greater than 80% of the radioactivity in urine was made up of glucuronide conjugates of hydroxylated metabolites. Thus cytochrome P450 metabolism, glucuronide conjugation, and urinary excretion of glucuronidated metabolites represent the primary route of nevirapine biotransformation and elimination in humans. Only a small fraction (<5%) of the radioactivity in urine (representing <3% of the total dose) was made up of parent compound; therefore, renal excretion plays a minor role in elimination of the parent compound.
Nevirapine is an inducer of hepatic cytochrome P450 (CYP) metabolic enzymes 3A and 2B6. Nevirapine induces CYP3A and CYP2B6 by approximately 20-25%, as indicated by erythromycin breath test results and urine metabolites. Autoinduction of CYP3A and CYP2B6 mediated metabolism leads to an approximately 1.5- to 2-fold increase in the apparent oral clearance of nevirapine as treatment continues from a single dose to two-to-four weeks of dosing with 200-400 mg/day. Autoinduction also results in a corresponding decrease in the terminal phase half-life of nevirapine in plasma, from approximately 45 hours (single dose) to approximately 25-30 hours following multiple dosing with 200-400 mg/day.
Renal Impairment
HIV-1 seronegative adults with mild (CrCL 50-79 mL/min; n=7), moderate (CrCL 30-49 mL/min; n=6), or severe (CrCL <30 mL/min; n=4) renal impairment received a single 200 mg dose of nevirapine in a pharmacokinetic study. These subjects did not require dialysis. The study included six additional subjects with renal failure requiring dialysis.
In subjects with renal impairment (mild, moderate or severe), there were no significant changes in the pharmacokinetics of nevirapine. However, subjects requiring dialysis exhibited a 44% reduction in nevirapine AUC over a one-week exposure period. There was also evidence of accumulation of nevirapine hydroxy-metabolites in plasma in subjects requiring dialysis. An additional 200 mg dose following each dialysis treatment is indicated [see Dosage and Administration (2.4) and Use in Specific Populations (8.6)].
Hepatic Impairment
In a steady-state study comparing 46 patients with mild (n=17; expansion of some portal areas; Ishak Score 1-2), moderate (n=20; expansion of most portal areas with occasional portal-to-portal and portal-to-central bridging; Ishak Score 3-4), or severe (n=9; marked bridging with occasional cirrhosis without decompensation indicating Child-Pugh A; Ishak Score 5-6) fibrosis as a measure of hepatic impairment, the multiple dose pharmacokinetic disposition of nevirapine and its five oxidative metabolites were not altered. However, approximately 15% of these patients with hepatic fibrosis had nevirapine trough concentrations above 9,000 μg/mL (2-fold the usual mean trough). Therefore, patients with hepatic impairment should be monitored carefully for evidence of drug-induced toxicity [see Warnings and Precautions (5.1)]. The patients studied were receiving antiretroviral therapy containing VIRAMUNE 200 mg twice daily for at least 6 weeks prior to pharmacokinetic sampling, with a median duration of therapy of 3.4 years.
In a pharmacokinetic study where HIV-1 negative cirrhotic patients with mild (Child-Pugh A; n=6) or moderate (Child-Pugh B; n=4) hepatic impairment received a single 200 mg dose of nevirapine, a significant increase in the AUC of nevirapine was observed in one patient with Child-Pugh B and ascites suggesting that patients with worsening hepatic function and ascites may be at risk of accumulating nevirapine in the systemic circulation. Because nevirapine induces its own metabolism with multiple dosing, this single-dose study may not reflect the impact of hepatic impairment on multiple-dose pharmacokinetics.
Do not administer nevirapine to patients with moderate or severe (Child-Pugh Class B or C, respectively) hepatic impairment [see Contraindications (4), Warnings and Precautions (5.1), and Use in Specific Populations (8.7)].
Gender
In the multinational 2NN study, a population pharmacokinetic substudy of 1077 patients was performed that included 391 females. Female patients showed a 13.8% lower clearance of nevirapine than did men. Since neither body weight nor Body Mass Index (BMI) had an influence on the clearance of nevirapine, the effect of gender cannot solely be explained by body size.
Race
An evaluation of nevirapine plasma concentrations (pooled data from several clinical trials) from HIV-1-infected patients (27 Black, 24 Hispanic, 189 Caucasian) revealed no marked difference in nevirapine steady-state trough concentrations (median Cminss = 4.7 μg/mL Black, 3.8 μg/mL Hispanic, 4.3 μg/mL Caucasian) with long-term nevirapine treatment at 400 mg/day. However, the pharmacokinetics of nevirapine have not been evaluated specifically for the effects of ethnicity.
Geriatric Patients
Nevirapine pharmacokinetics in HIV-1-infected adults do not appear to change with age (range 18–68 years); however, nevirapine has not been extensively evaluated in patients beyond the age of 55 years [see Use in Specific Populations (8.5)].
Pediatric Patients
Pharmacokinetic data for nevirapine have been derived from two sources: a 48-week pediatric trial in South Africa (BI Trial 1100.1368) involving 123 HIV-1 positive, antiretroviral-naïve patients aged 3 months to 16 years; and a consolidated analysis of five Pediatric AIDS Clinical Trials Group (PACTG) protocols comprising 495 patients aged 14 days to 19 years.
BI Trial 1100.1368 studied the safety, efficacy, and pharmacokinetics of a weight-based and a body surface area (BSA)-based dosing regimen of nevirapine. In the weight-based regimen, pediatric patients up to 8 years of age received a dose of 4 mg/kg once daily for two weeks followed by 7 mg/kg twice daily thereafter. Patients 8 years and older were dosed 4 mg/kg once daily for two weeks followed by 4 mg/kg twice daily thereafter. In the BSA regimen, all pediatric patients received 150 mg/m2 once daily for two weeks followed by 150 mg/m2 twice daily thereafter [see Use In Specific Populations (8.4) and Adverse Reactions (6.2)]. Dosing of nevirapine at 150 mg/m2 BID (after a two-week lead-in of 150 mg/m2 QD) produced geometric mean or mean trough nevirapine concentrations between 4-6 μg/mL (as targeted from adult data). In addition, the observed trough nevirapine concentrations were comparable between the two dosing regimens studied (BSA- and weight-based methods).
The consolidated analysis of Pediatric AIDS Clinical Trials Group (PACTG) protocols 245, 356, 366, 377, and 403 allowed for the evaluation of pediatric patients less than 3 months of age (n=17). The plasma nevirapine concentrations observed were within the range observed in adults and the remainder of the pediatric population, but were more variable between patients, particularly in the second month of age. For dose recommendations for pediatric patients [see Dosage and Administration (2.2)].
Drug Interactions [see Drug Interactions (7)]
Nevirapine induces hepatic cytochrome P450 metabolic isoenzymes 3A and 2B6. Co-administration of VIRAMUNE and drugs primarily metabolized by CYP3A or CYP2B6 may result in decreased plasma concentrations of these drugs and attenuate their therapeutic effects.
While primarily an inducer of cytochrome P450 3A and 2B6 enzymes, nevirapine may also inhibit this system. Among human hepatic cytochrome P450s, nevirapine was capable in vitro of inhibiting the 10-hydroxylation of (R)-warfarin (CYP3A). The estimated Ki for the inhibition of CYP3A was 270 μM, a concentration that is unlikely to be achieved in patients as the therapeutic range is <25 μM. Therefore, nevirapine may have minimal inhibitory effect on other substrates of CYP3A.
Nevirapine does not appear to affect the plasma concentrations of drugs that are substrates of other CYP450 enzyme systems, such as 1A2, 2D6, 2A6, 2E1, 2C9 or 2C19.
Table 5 (see below) contains the results of drug interaction studies performed with VIRAMUNE and other drugs likely to be co-administered. The effects of VIRAMUNE on the AUC, Cmax, and Cmin of co-administered drugs are summarized.
| Co-administered Drug | Dose of Co-administered Drug | Dose Regimen of VIRAMUNE | n | % Change of Co-administered Drug Pharmacokinetic Parameters (90% CI) | ||
|---|---|---|---|---|---|---|
| § = Cmin below detectable level of the assay ↑ = Increase, ↓ = Decrease, ⇔ = No Effect a For information regarding clinical recommendations, see Drug Interactions (7). b Pediatric subjects ranging in age from 6 months to 12 years c Parallel group design; n for VIRAMUNE+lopinavir/ritonavir, n for lopinavir/ritonavir alone. d Parallel group design; n=23 for atazanavir/ritonavir + nevirapine, n=22 for atazanavir/ritonavir without nevirapine. Changes in atazanavir PK are relative to atazanavir/ritonavir 300/100 mg alone. e Based on between-study comparison. f Based on historical controls. |
||||||
| Antiretrovirals | AUC | Cmax | Cmin | |||
| Atazanavir/Ritonavira,d | 300/100 mg QD day 4–13, then 400/100 mg QD, day 14–23 | 200 mg BID day 1-23. Subjects were treated with nevirapine prior to study entry. | 23 | Atazanavir 300/100 mg ↓42 (↓52 to ↓29) | Atazanavir 300/100 mg ↓28 (↓40 to ↓14) | Atazanavir 300/100 mg ↓72 (↓80 to ↓60) |
| Atazanavir 400/100 mg ↓19 (↓35 to ↑2) | Atazanavir 400/100 mg ↑2 (↓15 to ↑24) | Atazanavir 400/100 mg ↓59 (↓73 to ↓40) |
||||
| Darunavir/Ritonavire | 400/100 mg BID | 200 mg BID | 8 | ↑24 (↓3 to ↑57) | ↑40 (↑14 to ↑73) | ↑2 (↓21 to ↑32) |
| Didanosine | 100-150 mg BID | 200 mg QD x 14 days; 200 mg BID x 14 days | 18 | ⇔ | ⇔ | § |
| Efavirenza | 600 mg QD | 200 mg QD x 14 days; 400 mg QD x 14 days | 17 | ↓28 (↓34 to ↓14) | ↓12 (↓23 to ↑1) | ↓32 (↓35 to ↓19) |
| Fosamprenavir | 1400 mg BID | 200 mg BID. Subjects were treated with nevirapine prior to study entry. | 17 | ↓33 (↓45 to ↓20) | ↓25 (↓37 to ↓10) | ↓35 (↓50 to ↓15) |
| Fosamprenavir/Ritonavir | 700/100 mg BID | 200 mg BID. Subjects were treated with nevirapine prior to study entry | 17 | ↓11 (↓23 to ↑3) | ⇔ | ↓19 (↓32 to ↓4) |
| Indinavira | 800 mg q8H | 200 mg QD x 14 days; 200 mg BID x 14 days | 19 | ↓31 (↓39 to ↓22) | ↓15 (↓24 to ↓4) | ↓44 (↓53 to ↓33) |
| Lopinavira, b | 300/75 mg/m2 (lopinavir/ ritonavir) b | 7 mg/kg or 4 mg/kg QD x 2 weeks; BID x 1 week | 12, 15 c | ↓22 (↓44 to ↑9) | ↓14 (↓36 to ↑16) | ↓55 (↓75 to ↓19) |
| Lopinavira | 400/100 mg BID (lopinavir/ ritonavir) | 200 mg QD x 14 days; 200 mg BID >1 year | 22, 19 c | ↓27 (↓47 to ↓2) | ↓19 (↓38 to ↑5) | ↓51 (↓72 to ↓26) |
| Maravirocf | 300 mg SD | 200 mg BID | 8 | ↑1 (↓35 to ↑55) | ↑54 (↓6 to ↑151) | ⇔ |
| Nelfinavira | 750 mg TID | 200 mg QD x 14 days; 200 mg BID x 14 days | 23 | ⇔ | ⇔ | ↓32 (↓50 to ↑5) |
| Nelfinavir-M8 metabolite | ↓62 (↓70 to ↓53) | ↓59 (↓68 to ↓48) | ↓66 (↓74 to ↓55) |
|||
| Ritonavir | 600 mg BID | 200 mg QD x 14 days; 200 mg BID x 14 days | 18 | ⇔ | ⇔ | ⇔ |
| Stavudine | 30-40 mg BID | 200 mg QD x 14 days; 200 mg BID x 14 days | 22 | ⇔ | ⇔ | § |
| Zalcitabine | 0.125-0.25 mg TID | 200 mg QD x 14 days; 200 mg BID x 14 days | 6 | ⇔ | ⇔ | § |
| Zidovudine | 100-200 mg TID | 200 mg QD x 14 days; 200 mg BID x 14 days | 11 | ↓28 (↓40 to ↓4) | ↓30 (↓51 to ↑14) | § |
| Other Medications | AUC | Cmax | Cmin | |||
| Clarithromycina | 500 mg BID | 200 mg QD x 14 days; 200 mg BID x 14 days | 15 | ↓31 (↓38 to ↓24) | ↓23 (↓31 to ↓14) | ↓56 (↓70 to ↓36) |
| Metabolite 14-OH-clarithromycin | ↑42 (↑16 to ↑73) | ↑47 (↑21 to ↑80) | ⇔ | |||
| Ethinyl estradiola
and Norethindronea | 0.035 mg (as Ortho-Novum® 1/35) | 200 mg QD x 14 days; 200 mg BID x 14 days | 10 | ↓20 (↓33 to ↓3) | ⇔ | § |
| 1 mg (as Ortho-Novum® 1/35) | ↓19 (↓30 to ↓7) | ↓16 (↓27 to ↓3) | § | |||
| Depomedroxy-progesterone acetate | 150 mg every 3 months | 200 mg QD x 14 days; 200 mg BID x 14 days | 32 | ⇔ | ⇔ | ⇔ |
| Fluconazole | 200 mg QD | 200 mg QD x 14 days; 200 mg BID x 14 days | 19 | ⇔ | ⇔ | ⇔ |
| Ketoconazolea | 400 mg QD | 200 mg QD x 14 days; 200 mg BID x 14 days | 21 | ↓72 (↓80 to ↓60) | ↓44 (↓58 to ↓27) | § |
| Methadonea | Individual Patient Dosing | 200 mg QD x 14 days; 200 mg BID ≥7 days | 9 | In a controlled pharmacokinetic study with 9 patients receiving chronic methadone to whom steady-state nevirapine therapy was added, the clearance of methadone was increased by 3-fold, resulting in symptoms of withdrawal, requiring dose adjustments in 10 mg segments, in 7 of the 9 patients. Methadone did not have any effect on nevirapine clearance. | ||
| Rifabutina | 150 or 300 mg QD | 200 mg QD x 14 days; 200 mg BID x 14 days | 19 | ↑17 (↓2 to ↑40) | ↑28 (↑9 to ↑51) | ⇔ |
| Metabolite 25-O-desacetyl-rifabutin | ↑24 (↓16 to ↑84) | ↑29 (↓2 to ↑68) | ↑22 (↓14 to ↑74) |
|||
| Rifampina | 600 mg QD | 200 mg QD x 14 days; 200 mg BID x 14 days | 14 | ↑11 (↓4 to ↑28) | ⇔ | § |
Because of the design of the drug interaction trials (addition of 28 days of VIRAMUNE therapy to existing HIV-1 therapy), the effect of the concomitant drug on plasma nevirapine steady-state concentrations was estimated by comparison to historical controls.
Administration of rifampin had a clinically significant effect on nevirapine pharmacokinetics, decreasing AUC and Cmax by greater than 50%. Administration of fluconazole resulted in an approximate 100% increase in nevirapine exposure, based on a comparison to historic data [see Drug Interactions (7)]. The effect of other drugs listed in Table 5 on nevirapine pharmacokinetics was not significant. No significant interaction was observed when tipranavir was co-administered with low-dose ritonavir and nevirapine.
Mechanism of Action
Nevirapine is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of HIV-1. Nevirapine binds directly to reverse transcriptase (RT) and blocks the RNA-dependent and DNA-dependent DNA polymerase activities by causing a disruption of the enzyme's catalytic site. The activity of nevirapine does not compete with template or nucleoside triphosphates. HIV-2 RT and eukaryotic DNA polymerases (such as human DNA polymerases α, β, γ, or δ) are not inhibited by nevirapine.
Antiviral Activity
The antiviral activity of nevirapine has been measured in a variety of cell lines including peripheral blood mononuclear cells, monocyte-derived macrophages, and lymphoblastoid cell lines. In an assay using human embryonic kidney 293 cells, the median EC50 value (50% inhibitory concentration) of nevirapine was 90 nM against a panel of 2923 isolates of HIV-1 that were primarily (93%) clade B clinical isolates from the United States. The 99th percentile EC50 value was 470 nM in this study. The median EC50 value was 63 nM (range 14-302 nM, n=29) against clinical isolates of HIV-1 clades A, B, C, D, F, G, and H, and circulating recombinant forms CRF01_AE, CRF02_AG and CRF12_BF. Nevirapine had no antiviral activity in cell culture against group O HIV-1 isolates (n=3) or HIV-2 isolates (n=3) replicating in cord blood mononuclear cells. Nevirapine in combination with efavirenz exhibited strong antagonistic anti-HIV-1 activity in cell culture and was additive to antagonistic with the protease inhibitor ritonavir or the fusion inhibitor enfuvirtide. Nevirapine exhibited additive to synergistic anti-HIV-1 activity in combination with the protease inhibitors amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, saquinavir and tipranavir, and the NRTIs abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir and zidovudine. The anti-HIV-1 activity of nevirapine was antagonized by the anti-HBV drug adefovir and by the anti-HCV drug ribavirin in cell culture.
Resistance
HIV-1 isolates with reduced susceptibility (100- to 250-fold) to nevirapine emerge in cell culture. Genotypic analysis showed mutations in the HIV-1 RT gene encoding Y181C and/or V106A substitutions depending upon the virus strain and cell line employed. Time to emergence of nevirapine resistance in cell culture was not altered when selection included nevirapine in combination with several other NNRTIs.
Phenotypic and genotypic changes in HIV-1 isolates from treatment-naïve patients receiving either nevirapine (n=24) or nevirapine and ZDV (n=14) were monitored in Phase 1 and 2 trials over 1 to ≥12 weeks. After 1 week of nevirapine monotherapy, isolates from 3/3 patients had decreased susceptibility to nevirapine in cell culture. One or more of the RT mutations resulting in amino acid substitutions K103N, V106A, V108I, Y181C, Y188C and G190A were detected in HIV-1 isolates from some patients as early as 2 weeks after therapy initiation. By week eight of nevirapine monotherapy, 100% of the patients tested (n=24) had HIV-1 isolates with a >100-fold decrease in susceptibility to nevirapine in cell culture compared to baseline, and had one or more of the nevirapine-associated RT resistance substitutions. Nineteen of these patients (80%) had isolates with Y181C substitutions regardless of dose.
Genotypic analysis of isolates from antiretroviral-naïve patients experiencing virologic failure (n=71) receiving nevirapine once daily (n=25) or twice daily (n=46) in combination with lamivudine and stavudine (study 2NN) for 48 weeks showed that isolates from 8/25 and 23/46 patients, respectively, contained one or more of the following NNRTI resistance-associated substitutions: Y181C, K101E, G190A/S, K103N, V106A/M, V108I, Y188C/L, A98G, F227L and M230L.
Cross-resistance
Rapid emergence of HIV-1 strains which are cross-resistant to NNRTIs has been observed in cell culture. Nevirapine-resistant HIV-1 isolates were cross-resistant to the NNRTIs delavirdine and efavirenz. However, nevirapine-resistant isolates were susceptible to the NRTIs ddI and ZDV. Similarly, ZDV-resistant isolates were susceptible to nevirapine in cell culture.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies in mice and rats were carried out with nevirapine. Mice were dosed with 0, 50, 375 or 750 mg/kg/day for two years. Hepatocellular adenomas and carcinomas were increased at all doses in males and at the two high doses in females. In studies in which rats were administered nevirapine at doses of 0, 3.5, 17.5 or 35 mg/kg/day for two years, an increase in hepatocellular adenomas was seen in males at all doses and in females at the high dose. The systemic exposure (based on AUCs) at all doses in the two animal studies was lower than that measured in humans at the 200 mg BID dose. The mechanism of the carcinogenic potential is unknown. However, in genetic toxicology assays, nevirapine showed no evidence of mutagenic or clastogenic activity in a battery of in vitro and in vivo studies. These included microbial assays for gene mutation (Ames: Salmonella strains and E. coli), mammalian cell gene mutation assay (CHO/HGPRT), cytogenetic assays using a Chinese hamster ovary cell line and a mouse bone marrow micronucleus assay following oral administration. Given the lack of genotoxic activity of nevirapine, the relevance to humans of hepatocellular neoplasms in nevirapine-treated mice and rats is not known. In reproductive toxicology studies, evidence of impaired fertility was seen in female rats at doses providing systemic exposure, based on AUC, approximately equivalent to that provided with the recommended clinical dose of VIRAMUNE.
14 CLINICAL STUDIES
14.1 Clinical Studies in Adults
Trial BI 1090 was a placebo-controlled, double-blind, randomized trial in 2249 HIV-1 infected patients with <200 CD4+ cells/mm3 at screening. Initiated in 1995, BI 1090 compared treatment with VIRAMUNE + lamivudine + background therapy versus lamivudine + background therapy in NNRTI-naïve patients. Treatment doses were VIRAMUNE, 200 mg daily for two weeks followed by 200 mg twice daily or placebo, and lamivudine, 150 mg twice daily. Other antiretroviral agents were given at approved doses. Initial background therapy (in addition to lamivudine) was one NRTI in 1309 patients (58%), two or more NRTIs in 771 (34%), and PIs and NRTIs in 169 (8%). The patients (median age 36.5 years, 70% Caucasian, 79% male) had advanced HIV-1 infection, with a median baseline CD4+ cell count of 96 cells/mm3 and a baseline HIV-1 RNA of 4.58 log10 copies/mL (38,291 copies/mL). Prior to entering the trial, 45% had previously experienced an AIDS-defining clinical event. Eighty-nine percent had antiretroviral treatment prior to entering the trial. BI 1090 was originally designed as a clinical endpoint study. Prior to unblinding the trial, the primary endpoint was changed to proportion of patients with HIV-1 RNA <50 copies/mL and not previously failed at 48 weeks. Treatment response and outcomes are shown in Table 6.
| 1 including change to open-label nevirapine 2 includes withdrawal of consent, lost to follow-up, non-compliance with protocol, other administrative reasons |
||||||
| Outcome | VIRAMUNE (N=1121) % | Placebo (N=1128) % |
||||
| Responders at 48 weeks: HIV-1 RNA <50 copies/mL | 18.0 | 1.6 | ||||
| Treatment Failure | 82.0 | 98.4 | ||||
| Never suppressed viral load | 44.6 | 66.4 | ||||
| Virologic failure after response | 7.2 | 4.3 | ||||
| CDC category C event or death | 9.6 | 11.2 | ||||
| Added antiretroviral therapy1 while <50 copies/mL | 5.0 | 0.9 | ||||
| Discontinued trial therapy due to AE | 7.0 | 5.9 | ||||
| Discontinued trial <48 weeks2 | 8.5 | 9.8 | ||||
The change from baseline in CD4+ cell count through one year of therapy was significantly greater for the VIRAMUNE group compared to the placebo group for the overall study population (64 cells/mm3 vs 22 cells/mm3, respectively), as well as for patients who entered the trial as treatment naïve or having received only ZDV (85 cells/mm3 vs 25 cells/mm3, respectively).
At two years into the study, 16% of subjects on VIRAMUNE had experienced class C CDC events as compared to 21% of subjects on the control arm.
Trial BI 1046 (INCAS) was a double-blind, placebo-controlled, randomized, three-arm trial with 151 HIV-1 infected patients with CD4+ cell counts of 200-600 cells/mm3 at baseline. BI 1046 compared treatment with VIRAMUNE+zidovudine+didanosine to VIRAMUNE+zidovudine and zidovudine+didanosine. Treatment doses were VIRAMUNE at 200 mg daily for two weeks followed by 200 mg twice daily or placebo, zidovudine at 200 mg three times daily, and didanosine at 125 or 200 mg twice daily (depending on body weight). The patients had mean baseline HIV-1 RNA of 4.41 log10 copies/mL (25,704 copies/mL) and mean baseline CD4+ cell count of 376 cells/mm3. The primary endpoint was the proportion of patients with HIV-1 RNA <400 copies/mL and not previously failed at 48 weeks. The virologic responder rates at 48 weeks were 45% for patients treated with VIRAMUNE+zidovudine+didanosine, 19% for patients treated with zidovudine+didanosine, and 0% for patients treated with VIRAMUNE+zidovudine.
CD4+ cell counts in the VIRAMUNE+ZDV+ddI group increased above baseline by a mean of 139 cells/mm3 at one year, significantly greater than the increase of 87 cells/mm3 in the ZDV+ddI patients. The VIRAMUNE+ZDV group mean decreased by 6 cells/mm3 below baseline.
14.2 Clinical Studies in Pediatric Patients
The pediatric safety and efficacy of VIRAMUNE was examined in BI Trial 1100.1368, an open-label, randomized clinical study performed in South Africa in which 123 HIV-1 infected treatment-naïve patients between 3 months and 16 years of age received VIRAMUNE oral suspension for 48 weeks. Patients were divided into 4 age groups (3 months to <2 years, 2 to <7 years, 7 to <12 years, and 12 to ≤16 years) and randomized to receive one of two VIRAMUNE doses, determined by 2 different dosing methods [body surface area (150 mg/m2) and weight-based dosing (4 or 7 mg/kg)] in combination with zidovudine and lamivudine [see Adverse Reactions (6.2), Use in Specific Populations (8.4), and Clinical Pharmacology (12.3)]. The total daily dose of VIRAMUNE did not exceed 400 mg in either regimen. There were 66 patients in the body surface area (BSA) dosing group and 57 patients in the weight-based (BW) dosing group.
Baseline demographics included: 49% male; 81% Black and 19% Caucasian; 4% had previous exposure to ARVs. Patients had a median baseline HIV-1 RNA of 5.45 log10 copies/mL and a median baseline CD4+ cell count of 527 cells/mm3 (range 37-2279). One hundred and five (85%) completed the 48-week period while 18 (15%) discontinued prematurely. Of the patients who discontinued prematurely, 9 (7%) discontinued due to adverse reactions and 3 (2%) discontinued due to virologic failure. Overall the proportion of patients who achieved and maintained an HIV-1 RNA <400 copies/mL at 48 weeks was 47% (58/123).
For dose recommendations for pediatric patients [see Dosage and Administration (2.2)].
16 HOW SUPPLIED/STORAGE AND HANDLING
VIRAMUNE tablets, 200 mg, are white, oval, biconvex tablets, 9.3 mm x 19.1 mm. One side is embossed with "54 193", with a single bisect separating the "54" and "193". The opposite side has a single bisect.
Dispense in tight container as defined in the USP/NF.
VIRAMUNE oral suspension is a white to off-white preserved suspension containing 50 mg nevirapine (as nevirapine hemihydrate) in each 5 mL.
They are supplied by State of Florida DOH Central Pharmacy as follows:
| NDC | Strength | Quantity/Form | Color | Source Prod. Code |
| 53808-0808-1 | 200 mg | 30 Tablets in a Blister Pack | WHITE | 0597-0046 |
Storage
Store at 25°C (77°F); excursions permitted to 15°–30°C (59°–86°F) [see USP Controlled Room Temperature]. Store in a safe place out of the reach of children.
This product was Manufactured By:
Boehringer Ingelheim Pharmaceuticals, Inc.
900 Ridgebury Road
P.O. Box 368
Ridgefield, CT 06877
And Repackaged By:
State of Florida DOH Central Pharmacy
104-2 Hamilton Park Drive
Tallahassee, FL 32304
United States
17 PATIENT COUNSELING INFORMATION
See MEDICATION GUIDE
The Medication Guide provides written information for the patient, and should be dispensed with each new prescription and refill.
A Medication Guide is supplied as a tear-off following the full prescribing information.
ATTENTION PHARMACISTS: Detach "Medication Guide" and dispense with the product.
17.1 Hepatotoxicity and Skin Reactions
Patients should be informed of the possibility of severe liver disease or skin reactions associated with VIRAMUNE that may result in death. Patients developing signs or symptoms of liver disease or severe skin reactions should be instructed to discontinue VIRAMUNE and seek medical attention immediately, including performance of laboratory monitoring. Symptoms of liver disease include fatigue, malaise, anorexia, nausea, jaundice, acholic stools, liver tenderness or hepatomegaly. Symptoms of severe skin or hypersensitivity reactions include rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema and/or hepatitis.
Intensive clinical and laboratory monitoring, including liver enzymes, is essential during the first 18 weeks of therapy with VIRAMUNE to detect potentially life-threatening hepatotoxicity and skin reactions. However, liver disease can occur after this period; therefore, monitoring should continue at frequent intervals throughout VIRAMUNE treatment. Extra vigilance is warranted during the first 6 weeks of therapy, which is the period of greatest risk of hepatic events and skin reactions. Patients with signs and symptoms of hepatitis should discontinue VIRAMUNE and seek medical evaluation immediately. If VIRAMUNE is discontinued due to hepatotoxicity, do not restart it. Patients, particularly women, with increased CD4+ cell count at initiation of VIRAMUNE therapy (>250 cells/mm3 in women and >400 cells/mm3 in men) are at substantially higher risk for development of symptomatic hepatic events, often associated with rash. Patients should be advised that co-infection with hepatitis B or C and/or increased transaminases at the start of therapy with VIRAMUNE are associated with a greater risk of later symptomatic events (6 weeks or more after starting VIRAMUNE) and asymptomatic increases in AST or ALT [see Boxed Warning and Warnings and Precautions (5.1)].
The majority of rashes associated with VIRAMUNE occur within the first 6 weeks of initiation of therapy. Patients should be instructed that if any rash occurs during the two-week lead-in period, the VIRAMUNE dose should not be escalated until the rash resolves. The total duration of the once-daily lead-in dosing period should not exceed 28 days, at which point an alternative regimen may need to be started. Any patient experiencing a rash should have their liver enzymes (AST, ALT) evaluated immediately. Patients with severe rash or hypersensitivity reactions should discontinue VIRAMUNE immediately and consult a physician. VIRAMUNE should not be restarted following severe skin rash or hypersensitivity reaction. Women tend to be at higher risk for development of VIRAMUNE-associated rash [see Boxed Warning and Warnings and Precautions (5.2)].
17.2 Administration
Patients should be informed to take VIRAMUNE every day as prescribed. Patients should not alter the dose without consulting their doctor. If a dose is missed, patients should take the next dose as soon as possible. However, if a dose is skipped, the patient should not double the next dose. Patients should be advised to report to their doctor the use of any other medications.
Patients should be informed that VIRAMUNE therapy has not been shown to reduce the risk of transmission of HIV-1 to others through sexual contact or blood contamination. The long-term effects of VIRAMUNE are unknown at this time.
VIRAMUNE is not a cure for HIV-1 infection; patients may continue to experience illnesses associated with advanced HIV-1 infection, including opportunistic infections. Patients should be advised to remain under the care of a physician when using VIRAMUNE.
17.3 Drug Interactions
VIRAMUNE may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John's wort [see Warnings and Precautions (5.4) and Drug Interactions (7)].
17.4 Contraceptives
Hormonal methods of birth control, other than depomedroxy-progesterone acetate (DMPA), should not be used as the sole method of contraception in women taking VIRAMUNE, since VIRAMUNE may lower the plasma levels of these medications. Additionally, when oral contraceptives are used for hormonal regulation during VIRAMUNE therapy, the therapeutic effect of the hormonal therapy should be monitored [see Drug Interactions (7)].
17.5 Methadone
VIRAMUNE may decrease plasma concentrations of methadone by increasing its hepatic metabolism. Narcotic withdrawal syndrome has been reported in patients treated with VIRAMUNE and methadone concomitantly. Methadone-maintained patients beginning nevirapine therapy should be monitored for evidence of withdrawal and methadone dose should be adjusted accordingly [see Drug Interactions (7)].
17.6 Fat Redistribution
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time [see Warnings and Precautions (5.6)].
MEDICATION GUIDE
VIRAMUNE® (VIH-rah-mune)
(nevirapine)
Tablets
VIRAMUNE® (VIH-rah-mune)
(nevirapine)
Oral Suspension
Read this Medication Guide before you start taking VIRAMUNE and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
What is the most important information I should know about VIRAMUNE?
VIRAMUNE can cause serious side effects. These include severe liver and skin problems which can cause death. The risk of these problems is greatest during the first 18 weeks of treatment, but these problems can also happen at any time during treatment.
Severe liver problems: Anyone who takes VIRAMUNE may get severe liver problems. In some cases, these liver problems can lead to liver failure and the need for a liver transplant, or death. You may get a rash if you have liver problems. People with higher risk of these problems include women or anyone with higher CD4+ cell counts when they begin VIRAMUNE treatment. Women with CD4+ cell counts higher than 250 cells/mm3 at the start of treatment have the greatest risk for liver damage.
If you are a woman with CD4+ cell >250 cells/mm3 or a man with CD4+ cell >400 cells/mm3, you and your doctor will decide if VIRAMUNE is right for you.
People who have abnormal liver test results and people with hepatitis B or C have a greater chance of getting liver problems and further increases in liver test results during treatment.
If you get any of the following symptoms of liver problems, stop taking VIRAMUNE and call your doctor right away:
- fever
- "flu-like" symptoms or you do not feel well
- dark (tea colored) urine
- tiredness
- light-colored bowel movements (stools)
- nausea (feeling sick to your stomach)
- pain or tenderness on your right side below your ribs
- loss of appetite
- yellowing of your skin or whites of your eyes
Your doctor should see you and do blood tests often to check your liver function during the first 18 weeks of treatment with VIRAMUNE. You should continue to have your liver checked regularly during your treatment with VIRAMUNE. It is important for you to keep all of your doctor appointments.
Severe rash and skin reactions: Skin rash is the most common side effect of VIRAMUNE. Most rashes happen in the first 6 weeks of taking VIRAMUNE. Rashes and skin reactions may be severe, life-threatening, and in some people, may lead to death. If you get a rash with any of the following symptoms, stop using VIRAMUNE and call your doctor right away:
- "flu-like" symptoms or you do not feel well
- blisters
- fever
- mouth sores
- muscle or joint aches
- swelling of your face
- red or inflamed eyes, like "pink eye" (conjunctivitis)
- tiredness
- swollen glands (enlarged lymph nodes)
- liver problems (see symptoms of liver problems above)
If your doctor tells you to stop treatment with VIRAMUNE because you have had the serious liver or skin problems described above, you should never take VIRAMUNE again.
These are not all the side effects of VIRAMUNE. See the section "What are the possible side effects of VIRAMUNE?" for more information.
What is VIRAMUNE?
VIRAMUNE is a medicine used to treat Human Immunodeficiency Virus (HIV), the virus that causes AIDS (Acquired Immune Deficiency Syndrome).
VIRAMUNE is a type of anti-HIV medicine called a "non-nucleoside reverse transcriptase inhibitor" (NNRTI). VIRAMUNE works by lowering the amount of HIV in your blood ("viral load"). You must take VIRAMUNE with other anti-HIV medicines. When you take VIRAMUNE with other anti-HIV medicines, VIRAMUNE can lower your viral load and increase the number of CD4+ cells ("T cells"). CD4+ cells are a type of immune helper cell in the blood. VIRAMUNE may not have these effects in every person.
VIRAMUNE does not cure HIV or AIDS, and it is not known if it will help you live longer with HIV. People taking VIRAMUNE may still get infections common in people with HIV (opportunistic infections). It is very important that you stay under the care of your doctor.
Who should not take VIRAMUNE?
Tell your doctor if you have or have had liver problems. Your doctor may tell you not to take VIRAMUNE if you have certain liver problems.
What should I tell my doctor before taking VIRAMUNE?
Before you take VIRAMUNE, tell your doctor if you:
- have or have had hepatitis (inflammation of your liver) or problems with your liver. See "What is the most important information I should know about VIRAMUNE?" and "Who should not take VIRAMUNE?"
- receive dialysis
- have skin problems, such as a rash
- are pregnant or plan to become pregnant. It is not known if VIRAMUNE will harm your unborn baby.
Pregnancy Registry: There is a pregnancy registry for women who take antiviral medicines during pregnancy. The purpose of the registry is to collect information about the health of you and your baby. Talk to your doctor about how you can take part in this registry. - are breast-feeding or plan to breast-feed. VIRAMUNE can pass into your breast milk and may harm your baby. It is also recommended that HIV-positive women should not breast-feed their babies. Do not breast-feed during treatment with VIRAMUNE. Talk to your doctor about the best way to feed your baby.
Tell your doctor and pharmacist about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. VIRAMUNE may affect the way other medicines work, and other medicines may affect how VIRAMUNE works. Especially tell your doctor if you take:
- St. John’s Wort. You should not take products containing St. John’s Wort during treatment with VIRAMUNE because it can lower the amount of VIRAMUNE in your body.
- efavirenz (Sustiva®, Atripla®). You should not take efavirenz during treatment with VIRAMUNE. You may have an increased chance of side effects if this is taken together with VIRAMUNE.
- atazanavir (Reyataz®). You should not take atazanavir during treatment with VIRAMUNE.
- itraconazole (Sporanox®). You should not take itraconazole during treatment with VIRAMUNE.
- ketoconazole (Nizoral®). You should not take ketoconazole during treatment with VIRAMUNE.
- rifampin (Rifadin®, Rifamate®, Rifater®). You should not take rifampin during treatment with VIRAMUNE.
- Birth control pills. Birth control pills taken by mouth (oral contraceptives) and other hormone types of birth control may not work to prevent pregnancy if you use them during VIRAMUNE treatment. Talk with your doctor about other types of birth control that you can use to prevent pregnancy during treatment with VIRAMUNE.
Also tell your doctor if you are taking the following drugs:
- clarithromycin (Biaxin)
- fluconazole (Diflucan)
- methadone
- rifabutin (Mycobutin)
Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine.
How should I take VIRAMUNE?
- Take VIRAMUNE exactly as your doctor tells you to take it.
- The dose of VIRAMUNE for children is based on their size and thus may not be the same as in adults.
- You may take VIRAMUNE with or without food.
- If you or your child takes VIRAMUNE suspension (liquid), shake it gently before use. Use an oral dosing syringe or dosing cup to measure the right dose. The oral dosing syringe and dosing cup are not provided with VIRAMUNE Suspension.
- After drinking the medicine, fill the dosing cup with water and drink it to make sure you get all the medicine. If the dose is less than 1 teaspoon (5 mL), use the syringe instead of the dosing cup.
- Do not change your dose unless your doctor tells you to.
- Your doctor should start you with one dose each day to lower your chance of getting a serious rash. It is important that you only take one dose of VIRAMUNE each day for the first 14 days.
- Call your doctor right away if you get a skin rash during the first 14 days of VIRAMUNE treatment and do not increase your dose to 2 times a day.
- You should never take your starting dose (200 mg one time each day) for longer than 28 days. If after 28 days you are still receiving this starting dose because you have a rash, you and your doctor should discuss replacing VIRAMUNE with another HIV medicine. Never increase your dose to 2 times a day if you have a rash.
- Do not miss a dose of VIRAMUNE, because this could make HIV harder to treat. If you miss a dose of VIRAMUNE, take the missed dose as soon as you remember. If it is almost time for your next dose, do not take the missed dose, just take the next dose at your regular time.
- If you stop taking VIRAMUNE for more than 7 days, ask your doctor how much to take before you start taking it again. You may need to start with one time each day dosing again.
- If you take too much VIRAMUNE, call your local poison control center or emergency room right away.
What are the possible side effects of VIRAMUNE?
VIRAMUNE may cause serious side effects.
- See "What is the most important information I should know about VIRAMUNE?"
- Other common side effects of VIRAMUNE include nausea, fatigue, fever, headache, vomiting, diarrhea, abdominal pain, and muscle pain.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your doctor if you start having new symptoms after starting your HIV medicine.
- Changes in body fat can happen in some people who take antiretroviral therapy. These changes may include increased amount of fat in the upper back and neck ("buffalo hump"), breast, and around the middle of your body (trunk). Loss of fat from your legs, arms, and face can also happen. The cause and long-term health effects of these problems are not known at this time.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of VIRAMUNE. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VIRAMUNE?
- Store VIRAMUNE at 59° to 86°F (15° to 30°C).
- Throw away VIRAMUNE that is no longer needed or out-of-date.
Keep VIRAMUNE and all medicines out of the reach of children.
General information about VIRAMUNE
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use VIRAMUNE for a condition for which it was not prescribed. Do not give VIRAMUNE to other people, even if they have the same condition you have. It may harm them.
This Medication Guide summarizes the most important information about VIRAMUNE. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about VIRAMUNE that is written for health professionals.
For more information, call Boehringer Ingelheim Pharmaceuticals, Inc., at 1-800-542-6257, or (TTY) 1-800-459-9906.
What are the ingredients in VIRAMUNE?
Active Ingredient: nevirapine
Inactive ingredients:
Viramune Tablets: microcrystalline cellulose, lactose monohydrate, povidone, sodium starch glycolate, colloidal silicon dioxide, and magnesium stearate
Viramune Oral Suspension: carbomer 934P, methylparaben, propylparaben, sorbitol, sucrose, polysorbate 80, sodium hydroxide, and purified water
This Medication Guide has been approved by the US Food and Drug Administration
Atripla, Reyataz, and Sustiva are trademarks of Bristol Myers Squibb. Biaxin is a trademark of Abbott Laboratories. Diflucan is a trademark of Pfizer, Inc. Mycobutin is a trademark of Pharmacia & Upjohn Company. Nizoral and Sporanox are trademarks of Janssen Pharmaceutica. Rifadin, Rifamate, and Rifater are trademarks of Aventis Pharmaceuticals, Inc.
