DESCRIPTION
Megestrol acetate oral suspension, USP contains megestrol acetate, a synthetic derivative of the steroid hormone, progesterone. Megestrol acetate is a white, crystalline solid chemically designated as 17-Hydroxy-6-methylpregna-4,6-diene-3,20-dione acetate. Solubility at 37°C in water is 2 mcg per mL, solubility in plasma is 24 mcg per mL. Its molecular weight is 384.52.
The chemical formula is C 24H 32O 4 and the structural formula is represented as follows:
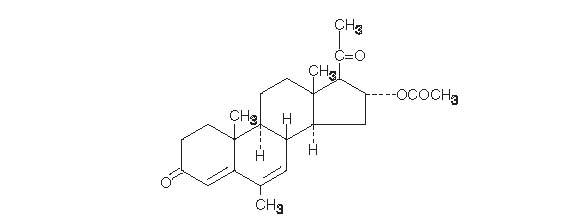
megestrol acetate, USP
Megestrol acetate oral suspension is supplied as an oral suspension containing 40 mg of micronized megestrol acetate per mL.
Megestrol acetate oral suspension contains the following inactive ingredients: alcohol (max 0.06% v/v from flavor), artificial lime flavor, citric acid monohydrate, docusate sodium, glycerin, natural and artificial lemon flavor, purified water, sodium benzoate, sodium citrate dihydrate, sucrose and xanthan gum.
Megestrol acetate oral suspension, 40 mg/mL complies with USP Dissolution Test 2.
CLINICAL PHARMACOLOGY
The precise mechanism by which megestrol acetate produces effects in anorexia and cachexia is unknown at the present time.
There are several analytical methods used to estimate megestrol acetate plasma concentrations, including gas chromatography-mass fragmentography (GC-MF), high pressure liquid chromatography (HPLC) and radioimmunoassay (RIA). The GC-MF and HPLC methods are specific for megestrol acetate and yield equivalent concentrations. The RIA method reacts to megestrol acetate metabolites and is, therefore, non-specific and indicates higher concentrations than the GC-MF and HPLC methods. Plasma concentrations are dependent, not only on the method used, but also on intestinal and hepatic inactivation of the drug, which may be affected by factors such as intestinal tract motility, intestinal bacteria, antibiotics administered, body weight, diet and liver function.
The major route of drug elimination in humans is urine. When radiolabeled megestrol acetate was administered to humans in doses of 4 to 90 mg, the urinary excretion within 10 days ranged from 56.5 to 78.4% (mean 66.4%) and fecal excretion ranged from 7.7 to 30.3% (mean 19.8%). The total recovered radioactivity varied between 83.1 and 94.7% (mean 86.2%). Megestrol acetate metabolites which were identified in urine constituted 5 to 8% of the dose administered. Respiratory excretion as labeled carbon dioxide and fat storage may have accounted for at least part of the radioactivity not found in urine and feces.
Plasma steady state pharmacokinetics of megestrol acetate were evaluated in 10 adult, cachectic male patients with acquired immunodeficiency syndrome (AIDS) and an involuntary weight loss greater than 10% of baseline. Patients received single oral doses of 800 mg/day of megestrol acetate oral suspension for 21 days. Plasma concentration data obtained on day 21 were evaluated for up to 48 hours past the last dose.
Mean (±1SD) peak plasma concentration (C max) of megestrol acetate was 753 (±539) ng/mL. Mean area under the concentration time-curve (AUC) was 10476 (±7788) ng x hr/mL. Median T max value was five hours. Seven of 10 patients gained weight in three weeks.
Additionally, 24 adult, asymptomatic HIV seropositive male subjects were dosed once daily with 750 mg of megestrol acetate oral suspension. The treatment was administered for 14 days. Mean C max and AUC values were 490 (±238) ng/mL and 6779 (±3048) ng x hr/mL, respectively. The median T max value was three hours. The mean C min value was 202 (±101) ng/mL. The mean % of fluctuation value was 107 (±40).
The effect of food on the bioavailability of megestrol acetate oral suspension has not been evaluated.
DESCRIPTION OF CLINICAL STUDIES
The clinical efficacy of megestrol acetate oral suspension was assessed in two clinical trials. One was a multicenter, randomized, double-blind, placebo-controlled study comparing megestrol acetate (MA) at doses of 100 mg, 400 mg, and 800 mg per day versus placebo in AIDS patients with anorexia/cachexia and significant weight loss. Of the 270 patients entered on study, 195 met all inclusion/exclusion criteria, had at least two additional post baseline weight measurements over a 12 week period or had one post baseline weight measurement but dropped out for therapeutic failure. The percent of patients gaining five or more pounds at maximum weight gain in 12 study weeks was statistically significantly greater for the 800 mg (64%) and 400 mg (57%) MA-treated groups than for the placebo group (24%). Mean weight increased from baseline to last evaluation in 12 study weeks in the 800 mg MA-treated group by 7.8 pounds, the 400 mg MA group by 4.2 pounds, the 100 mg MA group by 1.9 pounds and decreased in the placebo group by 1.6 pounds. Mean weight changes at 4, 8 and 12 weeks for patients evaluable for efficacy in the two clinical trials are shown graphically. Changes in body composition during the 12 study weeks as measured by bioelectrical impedance analysis showed increases innon-water body weight in the MA-treated groups (see CLINICAL STUDIES table). In addition, edema developed or worsened in only 3 patients.
Greater percentages of MA-treated patients in the 800 mg group (89%), the 400 mg group (68%) and the 100 mg group (72%), than in the placebo group (50%), showed an improvement in appetite at last evaluation during the 12 study weeks. A statistically significant difference was observed between the 800 mg MA-treated group and the placebo group in the change in caloric intake from baseline to time of maximum weight change. Patients were asked to assess weight change, appetite, appearance, and overall perception of well-being in a 9 question survey. At maximum weight change only the 800 mg MA-treated group gave responses that were statistically significantly more favorable to all questions when compared to the placebo-treated group. A dose response was noted in the survey with positive responses correlating with higher dose for all questions.
The second trial was a multicenter, randomized, double-blind, placebo-controlled study comparing megestrol acetate 800 mg/day versus placebo in AIDS patients with anorexia/cachexia and significant weight loss. Of the 100 patients entered on study, 65 met all inclusion/exclusion criteria, had at least two additional post baseline weight measurements over a 12 week period or had one post baseline weight measurement but dropped out for therapeutic failure. Patients in the 800 mg MA-treated group had a statistically significantly larger increase in mean maximum weight change than patients in the placebo group. From baseline to study week 12, mean weight increased by 11.2 pounds in the MA-treated group and decreased 2.1 pounds in the placebo group. Changes in body composition as measured by bioelectrical impedance analysis showed increases in non-water weight in the MA-treated group (see CLINICAL STUDIES table). No edema was reported in the MA-treated group. A greater percentage of MA-treated patients (67%) than placebo-treated patients (38%) showed an improvement in appetite at last evaluation during the 12 study weeks; this difference was statistically significant. There were no statistically significant differences between treatment groups in mean caloric change or in daily caloric intake at time to maximum weight change. In the same 9 question survey referenced in the first trial, patients’ assessments of weight change, appetite, appearance, and overall perception of well-being showed increases in mean scores in MA-treated patients as compared to the placebo group.
In both trials, no statistically significant differences were seen between the treatment groups with regard to laboratory abnormalities, , new opportunistic infections, lymphocyte counts, T 4 counts, T 8 counts, or skin reactivity tests ( see ADVERSE REACTIONS).
| Megestrol Acetate Oral Suspension Clinical Efficacy Trials | ||||||
| Trial 1 Study Accrual Dates 11/88 to 12/90 | Trial 2 Study Accrual Dates 5/89 to 4/91 | |||||
| Megestrol Acetate, mg/day | 0 | 100 | 400 | 800 | 0 | 800 |
| Entered Patients | 38 | 82 | 75 | 75 | 48 | 52 |
| Evaluable Patients | 28 | 61 | 53 | 53 | 29 | 36 |
| Mean Change in Weight (lb) | ||||||
| Baseline to 12 Weeks | 0.0 | 2.9 | 9.3 | 10.7 | -2.1 | 11.2 |
| % Patients ≥ 5 Pound Gain at Last Evaluation in 12 Weeks | 21 | 44 | 57 | 64 | 28 | 47 |
| Mean Changes in Body Composition* | ||||||
| Fat Body Mass (lb.) | 0.0 | 2.2 | 2.9 | 5.5 | 1.5 | 5.7 |
| Lean Body Mass (lb.) | -1.7 | -0.3 | 1.5 | 2.5 | -1.6 | -0.6 |
| Water (liters) | -1.3 | -0.3 | 0.0 | 0.0 | -0.1 | -0.1 |
| % Patients With Improved Appetite: | ||||||
| At Time of Maximum Weight Change | 50 | 72 | 72 | 93 | 48 | 69 |
| At Last Evaluation in 12 Weeks | 50 | 72 | 68 | 89 | 38 | 67 |
| Mean Change in Daily Caloric Intake: | ||||||
| Baseline to Time of Maximum Weight Change | -107 | 326 | 308 | 646 | 30 | 464 |
| *Based on bioelectrical impedance analysis determinations at last evaluation in 12 weeks. | ||||||
.
The following figures are the results of mean weight changes for patients evaluable for efficacy in trials 1 and 2.
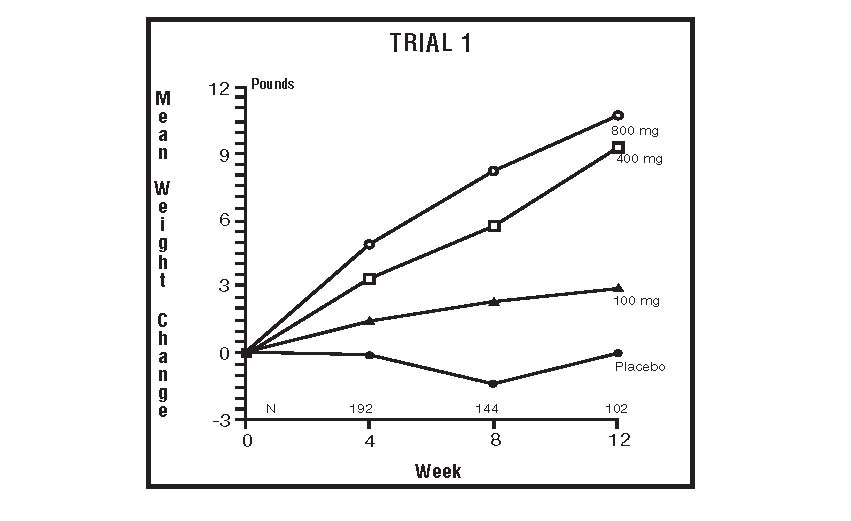
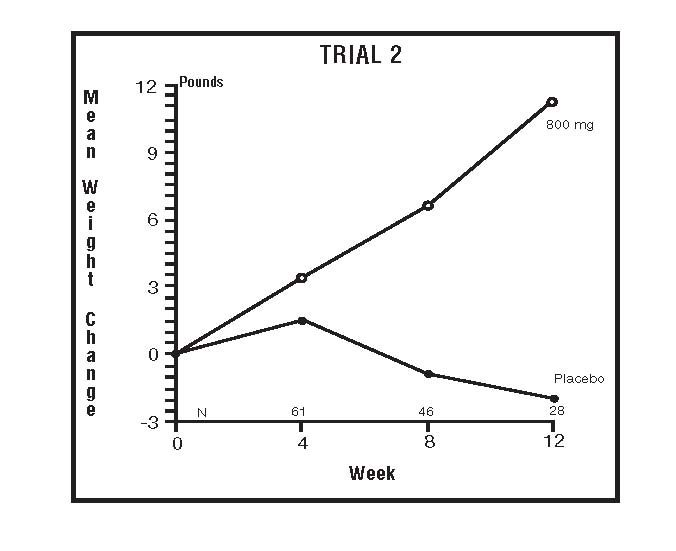
INDICATIONS AND USAGE
Megestrol acetate oral suspension, USP is indicated for the treatment of anorexia, cachexia, or an unexplained, significant weight loss in patients with a diagnosis of acquired immunodeficiency syndrome (AIDS).
CONTRAINDICATIONS
History of hypersensitivity to megestrol acetate or any component of the formulation. Known or suspected pregnancy.
WARNINGS
Megestrol acetate may cause fetal harm when administered to a pregnant woman. For animal data on fetal effects, (see PRECAUTIONS: Carcinogenesis, Mutagenesis, Impairment of Fertility: Impairment of Fertility). There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking (receiving) this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Megestrol acetate is not intended for prophylactic use to avoid weight loss.
(See also PRECAUTIONS: Carcinogenesis, Mutagenesis, and Impairment of Fertility section.)
The glucocorticoid activity of megestrol acetate oral suspension has not been fully evaluated. Clinical cases of new onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and overt Cushing’s Syndrome have been reported in association with the chronic use of megestrol acetate. In addition, clinical cases of adrenal insufficiency have been observed in patients receiving or being withdrawn from chronic megestrol acetate therapy in the stressed and non-stressed state. Furthermore, adrenocorticotropin (ACTH) stimulation testing has revealedthe frequent occurrence of asymptomatic pituitary-adrenal suppression in patients treated with chronic megestrol acetate therapy. Therefore, the possibility of adrenal insufficiency should be considered in any patient receiving or being withdrawn from chronic megestrol acetate therapy who presents with symptoms and/or signs suggestive of hypoadrenalism (e.g., hypotension, nausea, vomiting, dizziness, or weakness) in either the stressed or non-stressed state. Laboratory evaluation for adrenal insufficiency and consideration of replacement or stress doses of a rapidly acting glucocorticoid are strongly recommended in such patients. Failure to recognize inhibition of the hypothalamic-pituitary-adrenal axis may result in death. Finally, in patients who are receiving or being withdrawn from chronic megestrol acetate therapy, consideration should be given to the use of empiric therapy with stress doses of a rapidly acting glucocorticoid in conditions of stress or serious intercurrent illness (e.g., surgery, infection).
PRECAUTIONS
General
Therapy with megestrol acetate oral suspension for weight loss should only be instituted after treatable causes of weight loss are sought and addressed. These treatable causes include possible malignancies, systemic infections, gastrointestinal disorders affecting absorption, endocrine disease and renal or psychiatric diseases.
Effects on HIV viral replication have not been determined.
Use with caution in patients with a history of thromboembolic disease.
Use in Diabetics
Exacerbation of pre-existing diabetes with increased insulin requirements has been reported in association with the use of megestrol acetate.
Information for Patients
Patients using megestrol acetate should receive the following instructions:
- This medication is to be used as directed by the physician.
- Report any adverse reaction experiences while taking this medication.
- Use contraception while taking this medication if you are a woman capable of becoming pregnant.
- Notify your physician if you become pregnant while taking this medication.
Drug Interactions
Pharmacokinetic studies show that there are no significant alterations in pharmacokinetic parameters of zidovudine or rifabutin to warrant dosage adjustment when megestrol acetate is administered with these drugs. The effects of zidovudine or rifabutin on the pharmacokinetics of megestrol acetate were not studied.
Megestrol acetate may interact with warfarin and increase International Normalized Ratio (INR). Closely monitor INR in patients taking megestrol acetate and warfarin.
Animal Toxicology
Long-term treatment with megestrol acetate may increase the risk of respiratory infections. A trend toward increased frequency of respiratory infections, decreased lymphocyte counts and increased neutrophil counts was observed in a two-year chronic toxicity/carcinogenicity study of megestrol acetate conducted in rats.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Data on carcinogenesis were obtained from studies conducted in dogs, monkeys and rats treated with megestrol acetate at doses 53.2, 26.6 and 1.3 times lower than the proposed dose (13.3 mg/kg/day) for humans. No males were used in the dog and monkey studies. In female beagles, megestrol acetate (0.01, 0.1 or 0.25 mg/kg/day) administered for up to 7 years induced both benign and malignant tumors of the breast. In female monkeys, no tumors were found following 10 years of treatment with 0.01, 0.1 or 0.5 mg/kg/day megestrol acetate. Pituitary tumors were observed in female rats treated with 3.9 or 10 mg/kg/day of megestrol acetate for 2 years. The relationship of these tumors in rats and dogs to humans is unknown but should be considered in assessing the risk-to-benefit ratio when prescribing megestrol acetate oral suspension and in surveillance of patients on therapy. (See WARNINGS section)
Impairment of Fertility
Perinatal/postnatal (segment III) toxicity studies were performed in rats at doses (0.05 to 12.5 mg/kg) less than that indicated for humans (13.3 mg/kg); in these low dose studies, the reproductive capability of male offspring of megestrol acetate-treated females was impaired. Similar results were obtained in dogs. Pregnant rats treated with megestrol acetate showed a reduction in fetal weight and number of live births, and feminization of male fetuses. No toxicity data are currently available on male reproduction (spermatogenesis).
Pregnancy
No adequate animal teratology information is available at clinically relevant doses. (See WARNINGS and PRECAUTIONS: Carcinogenesis, Mutagenesis, impairment of Fertility: Impairment of Fertility.)
Nursing Mothers
Because of the potential for adverse effects on the newborn, nursing should be discontinued if megestrol acetate oral suspension is required.
Use in Women
Breakthrough bleeding was observed in all 10 female patients participating in the clinical trials. Megestrol acetate oral suspension is a progesterone derivative, which may induce vaginal bleeding in women.
All 10 women in the clinical trials reported breakthrough bleeding.
Geriatric Use
Clinical studies of megestrol acetate oral suspension in the treatment of anorexia, cachexia, or an unexplained, significant weight loss in patients with AIDS did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently than younger patients. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease of other drug therapy.
Megestrol acetate is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
ADVERSE REACTIONS
Adverse events which occurred in at least 5% of patients in any arm of the two clinical efficacy trials and the open trial are listed below by treatment group. All patients listed had at least one post baseline visit during the 12 study weeks. These adverse events should be considered by the physician when prescribing megestrol acetate oral suspension.
| ADVERSE EVENTS% of Patients Reporting | |||||||
| Trial 1 (N = 236) | Trail 2 (N=87) | Open Label Trial | |||||
| Megestrol Acetate mg/day No. of Patients | Placebo 0 N=34 | 100 N=68 | 400 N=69 | 800 N=65 | Placebo 0 N=38 | 800 N=49 | 1200 N=176 |
| Diarrhea | 15 | 13 | 8 | 15 | 8 | 6 | 10 |
| Impotence | 3 | 4 | 6 | 14 | 0 | 4 | 7 |
| Rash | 9 | 9 | 4 | 12 | 3 | 2 | 6 |
| Flatulence | 9 | 0 | 1 | 9 | 3 | 10 | 6 |
| Hypertension | 0 | 0 | 0 | 8 | 0 | 0 | 4 |
| Asthenia | 3 | 2 | 3 | 6 | 8 | 4 | 5 |
| Insomnia | 0 | 3 | 4 | 6 | 0 | 0 | 1 |
| Nausea | 9 | 4 | 0 | 5 | 3 | 4 | 5 |
| Anemia | 6 | 3 | 3 | 5 | 0 | 0 | 0 |
| Fever | 3 | 6 | 4 | 5 | 3 | 2 | 1 |
| Libido Decreased | 3 | 4 | 0 | 5 | 0 | 2 | 1 |
| Dyspepsia | 0 | 0 | 3 | 3 | 5 | 4 | 2 |
| Hyperglycemia | 3 | 0 | 6 | 3 | 0 | 0 | 3 |
| Headache | 6 | 10 | 1 | 3 | 3 | 0 | 3 |
| Pain | 6 | 0 | 0 | 2 | 5 | 6 | 4 |
| Vomiting | 9 | 3 | 0 | 2 | 3 | 6 | 4 |
| Pneumonia | 6 | 2 | 0 | 2 | 3 | 0 | 1 |
| Urinary Frequency | 0 | 0 | 1 | 2 | 5 | 2 | 1 |
Adverse events which occurred in 1 to 3% of all patients enrolled in the two clinical efficacy trials with at least one follow-up visit during the first 12 weeks of the study are listed below by body system. Adverse events occurring less than 1% are not included. There were no significant differences between incidence of these events in patients treated with megestrol acetate and patients treated with placebo.
Body as a Whole - abdominal pain, chest pain, infection, moniliasis and sarcoma
Cardiovascular System - cardiomyopathy and palpitation
Digestive System - constipation, dry mouth, hepatomegaly, increased salivation and oral moniliasis
Hemic and Lymphatic System - leukopenia
Metabolic and Nutritional - LDH increased, edema and peripheral edema
Nervous System - paresthesia, confusion, convulsion, depression, neuropathy, hypesthesia and abnormal thinking
Respiratory System - dyspnea, cough, pharyngitis and lung disorder
Skin and Appendages - alopecia, herpes, pruritus, vesiculobullous rash, sweating and skin disorder
Special Senses - amblyopia
Urogenital System - albuminuria, urinary incontinence, urinary tract infection and gynecomastia
Postmarketing - Postmarketing reports associated with megestrol acetate oral suspension include thromboembolic phenomena including thrombophlebitis and pulmonary embolism and glucose intolerance (see WARNINGS and PRECAUTIONS).
OVERDOSAGE
No serious unexpected side effects have resulted from studies involving megestrol acetate oral suspension administered in dosages as high as 1200 mg/day. In post-marketing experience, limited reports of overdose have been received. Signs and symptoms reported in the context of overdose included diarrhea, nausea, abdominal pain, shortness of breath, cough, unsteady gait, listlessness, and chest pain. There is no specific antidote for overdose with megestrol acetate oral suspension. In case of overdose, appropriate supportive measures should be taken. Megestrol acetate has not been tested for dialyzability, however, due to its low solubility it is postulated that dialysis would not be an effective means of treating overdose
DOSAGE AND ADMINISTRATION
The recommended adult initial dosage of megestrol acetate oral suspension, USP is 800 mg/day (20 mL/day). Shake container well before using.
In clinical trials evaluating different dose schedules, daily doses of 400 and 800 mg/day were found to be clinically effective.
