FULL PRESCRIBING INFORMATION
WARNING: EMBRYOFETAL TOXICITY, MALIGNANCIES AND SERIOUS INFECTIONS
- Use during pregnancy is associated with increased risks of first trimester pregnancy loss and congenital malformations. Avoid if safer treatment options are available. Females of reproductive potential must be counseled regarding pregnancy prevention and planning [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
- Increased risk of development of lymphoma and other malignancies, particularly of the skin [see Warnings and Precautions (5.2)].
- Increased susceptibility to bacterial, viral, fungal and protozoal infections, including opportunistic infections and viral reactivation of hepatitis B and C, which may lead to hospitalizations and fatal outcomes [see Warnings and Precautions (5.3)].
1 INDICATIONS AND USAGE
Mycophenolate mofetil (MMF) is indicated for the prophylaxis of organ rejection, in recipients of allogeneic kidney [see Clinical Studies (14.1)], heart [see Clinical Studies (14.2)] or liver transplants [see Clinical Studies (14.3)], in combination with other immunosuppressants.
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Mycophenolate mofetil should not be used without the supervision of a physician with experience in immunosuppressive therapy.
Mycophenolate Mofetil for Injection
Mycophenolate mofetil for injection is recommended for patients unable to take oral mycophenolate mofetil. Mycophenolate mofetil for injection should be administered within 24 hours following transplant. Mycophenolate mofetil for injection can be administered for up to 14 days; however, patients should be switched to oral mycophenolate mofetil as soon as they can tolerate oral medication.
Mycophenolate mofetil for injection must be reconstituted before use [see Dosage and Administration (2.6)]. Mycophenolate mofetil for injection is incompatible with other intravenous infusion solutions and should not be mixed or administered concurrently via the same infusion catheter with other intravenous drugs or infusion admixtures.
Mycophenolate mofetil for injection must not be administered as a bolus. Following reconstitution, mycophenolate mofetil for injection must be administered by slow intravenous infusion over a period of no less than 2 hours by either peripheral or central vein, as rapid infusion increases the risk of local adverse reactions such as phlebitis and thrombosis [see Adverse Reactions (6.1)].
2.2 Dosing for Kidney Transplant Patients: Adults and Pediatrics
Adults
The recommended dose for adult kidney transplant patients is 1 g orally or intravenously infused over no less than 2 hours, twice daily (daily dose of 2 g).
Pediatrics (3 months and older)
Pediatric dosing is based on body surface area (BSA). The recommended dose of mycophenolate mofetil oral suspension for pediatric kidney transplant patients 3 months and older is 600 mg/m2, administered twice daily (maximum daily dose of 2 g or 10 mL of the oral suspension). Pediatric patients with BSA ≥ 1.25 m2 may be dosed with capsules or tablets as follows:
| Body Surface Area | Dosing |
| 1.25 m2 to < 1.5 m2 | Mycophenolate mofetil capsule 750 mg twice daily (1.5 g daily dose) |
| ≥ 1.5 m2 | Mycophenolate mofetil capsules or tablets 1 g twice daily (2 g daily dose) |
2.3 Dosing for Heart Transplant Patients: Adults
The recommended dose of mycophenolate mofetil for adult heart transplant patients is 1.5 g orally or intravenously infused over no less than 2 hours administered twice daily (daily dose of 3 g).
2.4 Dosing for Liver Transplant Patients: Adults
The recommended dose of mycophenolate mofetil for adult liver transplant patients is 1.5 g administered orally twice daily (daily dose of 3 g) or 1 g infused intravenously over no less than 2 hours, twice daily (daily dose of 2 g).
2.5 Dosing Adjustments: Patients with Renal Impairment, Neutropenia
Renal Impairment
No dose adjustments are needed in kidney transplant patients with delayed graft function postoperatively [see Clinical Pharmacology (12.3)]. In kidney transplant patients with severe chronic impairment of the graft (GFR < 25 mL/min/1.73 m2), do not administer doses of mycophenolate mofetil greater than 1 g twice a day. These patients should be carefully monitored [see Clinical Pharmacology (12.3)].
2.6 Preparation Instructions of Intravenous for Pharmacists
General Preparation Instructions Before Handling the Formulations
Mycophenolate mofetil (MMF) has demonstrated teratogenic effects in humans. Follow applicable special handling and disposal procedures1 [see Warnings and Precautions (5.1), Adverse Reactions (6.2), Use in Specific Populations (8.1, 8.3), How Supplied/Storage and Handling (16.1)].
Care should be taken to avoid inhalation or direct contact with skin or mucous membranes of the dry powder or the constituted suspension because MMF has demonstrated teratogenic effects in humans. Wearing disposable gloves is recommended during reconstitution and when wiping the outer surface of the bottle/cap and the table surface after reconstitution. If such contact occurs, wash hands thoroughly with soap and water; rinse eyes with water.
Alert patients that they and others should also avoid inhalation or contact of the skin or mucous membranes with the oral suspension. Advise them to wash the area thoroughly with soap and water if such contact occurs; if ocular contact occurs, rinse eyes with plain water.
Mycophenolate Mofetil for Injection
Before proceeding with the preparation steps for mycophenolate mofetil for injection read the general preparation instructions [see General Preparation Instructions Before Handling the Formulations] and note the following:
Mycophenolate mofetil for injection does not contain an antibacterial preservative; therefore, reconstitution and dilution of the product must be performed under aseptic conditions.
This product is sealed under vacuum and should retain a vacuum throughout its shelf life. If a lack of vacuum in the vial is noted while adding the diluent, the vial should not be used.
Mycophenolate mofetil for injection must be reconstituted and further diluted. A detailed description of the preparation is given below.
| Preparation of the 1 g dose |
|
| Preparation of the 1.5 g dose |
|
The administration of the infusion should be initiated within 4 hours of reconstitution and dilution of the drug product. Keep solutions at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F). Discard unused portion of the reconstituted solutions.
Mycophenolate mofetil for injection should not be mixed or administered concurrently via the same infusion catheter with other intravenous drugs or infusion admixtures.
3 DOSAGE FORMS AND STRENGTHS
Mycophenolate Mofetil for Injection, USP is available in the following dosage form and strength:
- For injection: 500 mg mycophenolate mofetil white to off-white lyophilized powder, in a single-dose vial for reconstitution
4 CONTRAINDICATIONS
Allergic reactions to mycophenolate mofetil have been observed; therefore, mycophenolate mofetil is contraindicated in patients with a hypersensitivity to mycophenolate mofetil (MMF), mycophenolic acid (MPA) or any component of the drug product. Mycophenolate mofetil for injection is contraindicated in patients who are allergic to polysorbate 80 (TWEEN).
5 WARNINGS AND PRECAUTIONS
5.1 Embryofetal Toxicity
Use of MMF during pregnancy is associated with an increased risk of first trimester pregnancy loss and an increased risk of congenital malformations, especially external ear and other facial abnormalities including cleft lip and palate, and anomalies of the distal limbs, heart, esophagus, kidney and nervous system. Females of reproductive potential must be made aware of these risks and must be counseled regarding pregnancy prevention and planning. Avoid use of MMF during pregnancy if safer treatment options are available [see Use in Specific Populations (8.1, 8.3)].
5.2 Lymphoma and Other Malignancies
Patients receiving immunosuppressants, including mycophenolate mofetil, are at increased risk of developing lymphomas and other malignancies, particularly of the skin [see Adverse Reactions (6.1)]. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent. For patients with increased risk for skin cancer, exposure to sunlight and UV light should be limited by wearing protective clothing and using a sunscreen with a high protection factor.
Post-transplant lymphoproliferative disorder (PTLD) developed in 0.4% to 1% of patients receiving mycophenolate mofetil (2 g or 3 g) with other immunosuppressive agents in controlled clinical trials of kidney, heart and liver transplant patients [see Adverse Reactions (6.1)]. The majority of PTLD cases appear to be related to Epstein Barr Virus (EBV) infection. The risk of PTLD appears greatest in those individuals who are EBV seronegative, a population which includes many young children. In pediatric patients, no other malignancies besides PTLD were observed in clinical trials [see Adverse Reactions (6.1)].
5.3 Serious Infections
Patients receiving immunosuppressants, including mycophenolate mofetil, are at increased risk of developing bacterial, fungal, protozoal and new or reactivated viral infections, including opportunistic infections. The risk increases with the total immunosuppressive load. These infections may lead to serious outcomes, including hospitalizations and death [see Adverse Reactions (6.1, 6.2)].
Serious viral infections reported include:
- Polyomavirus-associated nephropathy (PVAN), especially due to BK virus infection
- JC virus-associated progressive multifocal leukoencephalopathy (PML), and
- Cytomegalovirus (CMV) infections: CMV seronegative transplant patients who receive an organ from a CMV seropositive donor are at highest risk of CMV viremia and CMV disease.
- Viral reactivation in patients infected with Hepatitis B and C
Consider reducing immunosuppression in patients who develop new infections or reactivate viral infections, weighing the risk that reduced immunosuppression represents to the functioning allograft.
PVAN, especially due to BK virus infection, is associated with serious outcomes, including deteriorating renal function and renal graft loss [see Adverse Reactions (6.2)]. Patient monitoring may help detect patients at risk for PVAN.
PML, which is sometimes fatal, commonly presents with hemiparesis, apathy, confusion, cognitive deficiencies, and ataxia [see Adverse Reactions (6.2)]. In immunosuppressed patients, physicians should consider PML in the differential diagnosis in patients reporting neurological symptoms.
The risk of CMV viremia and CMV disease is highest among transplant recipients seronegative for CMV at time of transplant who receive a graft from a CMV seropositive donor. Therapeutic approaches to limiting CMV disease exist and should be routinely provided. Patient monitoring may help detect patients at risk for CMV disease.
Viral reactivation has been reported in patients infected with HBV or HCV. Monitoring infected patients for clinical and laboratory signs of active HBV or HCV infection is recommended.
5.4 Blood Dyscrasias: Neutropenia and Pure Red Cell Aplasia (PRCA)
Severe neutropenia [absolute neutrophil count (ANC) < 0.5 x 103/mcL] developed in transplant patients receiving mycophenolate mofetil 3 g daily [see Adverse Reactions (6.1)]. Patients receiving mycophenolate mofetil should be monitored for neutropenia. Neutropenia has been observed most frequently in the period from 31 to 180 days post-transplant in patients treated for prevention of kidney, heart and liver rejection. The development of neutropenia may be related to mycophenolate mofetil itself, concomitant medications, viral infections, or a combination of these causes. If neutropenia develops (ANC < 1.3 x 103/mcL), dosing with mycophenolate mofetil should be interrupted or the dose reduced, appropriate diagnostic tests performed, and the patient managed appropriately [see Dosage and Administration (2.5)].
Patients receiving mycophenolate mofetil should be instructed to report immediately any evidence of infection, unexpected bruising, bleeding or any other manifestation of bone marrow depression.
Consider monitoring with complete blood counts weekly for the first month, twice monthly for the second and third months, and monthly for the remainder of the first year.
Cases of pure red cell aplasia (PRCA) have been reported in patients treated with mycophenolate mofetil in combination with other immunosuppressive agents. In some cases, PRCA was found to be reversible with dose reduction or cessation of mycophenolate mofetil therapy. In transplant patients, however, reduced immunosuppression may place the graft at risk.
5.5 Gastrointestinal Complications
Gastrointestinal bleeding requiring hospitalization, ulceration and perforations were observed in clinical trials. Physicians should be aware of these serious adverse effects particularly when administering mycophenolate mofetil to patients with a gastrointestinal disease.
5.6 Patients with Hypoxanthine-Guanine Phosphoribosyl-Transferase Deficiency (HGPRT)
Mycophenolate mofetil is an inosine monophosphate dehydrogenase (IMPDH) inhibitor; therefore it should be avoided in patients with hereditary deficiencies of hypoxanthine-guanine phosphoribosyl-transferase (HGPRT) such as Lesch-Nyhan and Kelley-Seegmiller syndromes because it may cause an exacerbation of disease symptoms characterized by the overproduction and accumulation of uric acid leading to symptoms associated with gout such as acute arthritis, tophi, nephrolithiasis or urolithiasis and renal disease including renal failure.
5.7 Immunizations
During treatment with mycophenolate mofetil, the use of live attenuated vaccines should be avoided (e.g., intranasal influenza, measles, mumps, oral polio, BCG, yellow fever, varicella, and TY21a typhoid vaccines) and patients should be advised that vaccinations may be less effective. Advise patients to discuss with the physician before seeking any immunizations.
5.8 Local Reactions with Rapid Intravenous Administration
Mycophenolate mofetil for injection solution must not be administered by rapid or bolus intravenous injection as rapid infusion increases the risk of local adverse reactions such as phlebitis and thrombosis [see Adverse Reactions (6.1)].
5.9 Risks in Patients with Phenylketonuria
Phenylalanine can be harmful to patients with phenylketonuria (PKU). Mycophenolate mofetil oral suspension contains aspartame, a source of phenylalanine (0.56 mg phenylalanine/mL suspension). Before prescribing mycophenolate mofetil oral suspension to a patient with PKU, consider the combined daily amount of phenylalanine from all sources, including mycophenolate mofetil.
5.10 Blood Donation
Patients should not donate blood during therapy and for at least 6 weeks following discontinuation of mycophenolate mofetil because their blood or blood products might be administered to a female of reproductive potential or a pregnant woman.
5.11 Semen Donation
Based on animal data, men should not donate semen during therapy and for 90 days following discontinuation of mycophenolate mofetil [see Use in Specific Populations (8.3)].
5.12 Effect of Concomitant Medications on Mycophenolic Acid Concentrations
A variety of drugs have potential to alter systemic MPA exposure when co-administered with mycophenolate mofetil. Therefore, determination of MPA concentrations in plasma before and after making any changes to immunosuppressive therapy, or when adding or discontinuing concomitant medications, may be appropriate to ensure MPA concentrations remain stable.
5.13 Potential Impairment of Ability to Drive or Operate Machinery
Mycophenolate mofetil may impact the ability to drive and use machines. Patients should avoid driving or using machines if they experience somnolence, confusion, dizziness, tremor, or hypotension during treatment with mycophenolate mofetil [see Adverse Reactions (6.1)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Embryofetal Toxicity [see Warnings and Precautions (5.1)]
- Lymphomas and Other Malignancies [see Warnings and Precautions (5.2)]
- Serious Infections [see Warnings and Precautions (5.3)]
- Blood Dyscrasias: Neutropenia, Pure Red Cell Aplasia [see Warnings and Precautions (5.4)]
- Gastrointestinal Complications [see Warnings and Precautions (5.5)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
An estimated total of 1557 patients received mycophenolate mofetil during pivotal clinical trials in the prevention of acute organ rejection. Of these, 991 were included in the three renal studies, 277 were included in one hepatic study, and 289 were included in one cardiac study. Patients in all study arms also received cyclosporine and corticosteroids.
The data described below primarily derive from five randomized, active-controlled double-blind 12-month trials of mycophenolate mofetil in de novo kidney (3), heart (1), and liver (1) transplant patients [see Clinical Studies (14.1, 14.2 and 14.3)].
Mycophenolate Mofetil Oral
The incidence of adverse reactions for mycophenolate mofetil was determined in five randomized, comparative, double-blind trials in the prevention of rejection in kidney, heart and liver transplant patients (two active- and one placebo-controlled trials, one active-controlled trial, and one active-controlled trial, respectively) [see Clinical Studies (14.1, 14.2 and 14.3)].
The three de novo kidney studies with 12-month duration compared two dose levels of oral mycophenolate mofetil (1 g twice daily and 1.5 g twice daily) with azathioprine (2 studies) or placebo (1 study) when administered in combination with cyclosporine (Sandimmune®) and corticosteroids to prevent acute rejection episodes. One study also included anti-thymocyte globulin (ATGAM®) induction therapy.
In the de novo heart transplantation study with 12-month duration, patients received mycophenolate mofetil 1.5 g twice daily (n=289) or azathioprine 1.5 to 3 mg/kg/day (n=289), in combination with cyclosporine (Sandimmune® or Neoral®) and corticosteroids as maintenance immunosuppressive therapy.
In the de novo liver transplantation study with 12-month duration, patients received mycophenolate mofetil 1 g twice daily intravenously for up to 14 days followed by mycophenolate mofetil 1.5 g twice daily orally or azathioprine 1 to 2 mg/kg/day intravenously followed by azathioprine 1 to 2 mg/kg/day orally, in combination with cyclosporine (Neoral®) and corticosteroids as maintenance immunosuppressive therapy. The total number of patients enrolled was 565.
Approximately 53% of the kidney transplant patients, 65% of the heart transplant patients, and 48% of the liver transplant patients were treated for more than 1 year. Adverse reactions reported in ≥ 20% of patients in the mycophenolate mofetil treatment groups are presented below. The safety data of three kidney transplantation studies are pooled together.
|
a : “-“ Indicates that the incidence was below the cutoff value of 20% for inclusion in the table. |
|||||||
|
b : “Edema” includes peripheral edema, facial edema, scrotal edema. |
|||||||
|
c : “Pain” includes musculoskeletal pain (myalgia, neck pain, back pain). |
|||||||
| Adverse drug reaction
(MedDRA) System Organ Class | Kidney Studies | Heart Study | Liver Study | ||||
| Mycophenolate
Mofetil 2 g/day (n=501) or 3 g/day (n=490) | AZA
1 to 2 mg/kg/day or 100 to 150 mg/day | Placebo | Mycophenolate
Mofetil 3 g/day | AZA
1.5 to 3 mg/kg/day | Mycophenolate
Mofetil 3 g/day | AZA 1 to 2 mg/kg/day | |
| (n=991) | (n=326) | (n=166) | (n=289) | (n=289) | (n=277) | (n=287) | |
| % | % | % | % | % | % | % | |
| Infections and infestations | |||||||
| Bacterial infections | 39.9 | 33.7 | 37.3 | – | – | 27.4 | 26.5 |
| Viral infections | – a | – | – | 31.1 | 24.9 | – | – |
| Blood and lymphatic system disorders | |||||||
| Anemia | 20.0 | 23.6 | 2.4 | 45.0 | 47.1 | 43.0 | 53.0 |
| Ecchymosis | – | – | – | 20.1 | 9.7 | – | – |
| Leukocytosis | – | – | – | 42.6 | 37.4 | 22.4 | 21.3 |
| Leukopenia | 28.6 | 24.8 | 4.2 | 34.3 | 43.3 | 45.8 | 39.0 |
| Thrombocytopenia | – | – | – | 24.2 | 28.0 | 38.3 | 42.2 |
| Metabolism and nutrition disorders | |||||||
| Hypercholesterolemia | – | – | – | 46.0 | 43.9 | – | – |
| Hyperglycemia | – | – | – | 48.4 | 53.3 | 43.7 | 48.8 |
| Hyperkalemia | – | – | – | – | – | 22.0 | 23.7 |
| Hypocalcemia | – | – | – | – | – | 30.0 | 30.0 |
| Hypokalemia | – | – | – | 32.5 | 26.3 | 37.2 | 41.1 |
| Hypomagnesemia | – | – | – | 20.1 | 14.2 | 39.0 | 37.6 |
| Psychiatric disorders | |||||||
| Depression | – | – | – | 20.1 | 15.2 | – | – |
| Insomnia | – | – | – | 43.3 | 39.8 | 52.3 | 47.0 |
| Nervous system diorders | |||||||
| Dizziness | – | – | – | 34.3 | 33.9 | – | – |
| Headache | – | – | – | 58.5 | 55.4 | 53.8 | 49.1 |
| Tremor | – | – | – | 26.3 | 25.6 | 33.9 | 35.5 |
| Cardiac disorders | |||||||
| Tachycardias | – | – | – | 22.8 | 21.8 | 22.0 | 15.7 |
| Vascular disorders | |||||||
| Hypertension | 27.5 | 32.2 | 19.3 | 78.9 | 74.0 | 62.1 | 59.6 |
| Hypotension | – | – | – | 34.3 | 40.1 | – | – |
| Respiratory, thoracic and mediastinal disorders | |||||||
| Cough | – | – | – | 40.5 | 32.2 | – | – |
| Dyspnea | – | – | – | 44.3 | 44.3 | 31.0 | 30.3 |
| Pleural effusion | – | – | – | – | – | 34.3 | 35.9 |
| Gastrointestinal disorders | |||||||
| Abdominal pain | 22.4 | 23.0 | 11.4 | 41.9 | 39.4 | 62.5 | 51.2 |
| Constipation | – | – | – | 43.6 | 38.8 | 37.9 | 38.3 |
| Decreased appetite | – | – | – | – | – | 25.3 | 17.1 |
| Diarrhea | 30.4 | 20.9 | 13.9 | 52.6 | 39.4 | 51.3 | 49.8 |
| Dyspepsia | – | – | – | 22.1 | 22.1 | 22.4 | 20.9 |
| Nausea | – | – | – | 56.1 | 60.2 | 54.5 | 51.2 |
| Vomiting | – | – | – | 39.1 | 34.6 | 32.9 | 33.4 |
| Hepatobiliary disorders | |||||||
| Blood lactate dehydrogenase increased | – | – | – | 23.5 | 18.3 | – | – |
| Hepatic enzyme increased | – | – | – | – | – | 24.9 | 19.2 |
| Skin and subcutaneous tissues disorders | |||||||
| Rash | – | – | – | 26.0 | 20.8 | – | – |
| Renal and urinary disorders | |||||||
| Blood creatinine increased | – | – | – | 42.2 | 39.8 | – | – |
| Blood urea increased | – | – | – | 36.7 | 34.3 | – | – |
| General disorders and administration site conditions | |||||||
| Asthenia | – | – | – | 49.1 | 41.2 | 35.4 | 33.8 |
| Edemab | 21.0 | 28.2 | 8.4 | 67.5 | 55.7 | 48.4 | 47.7 |
| Painc | 24.8 | 32.2 | 9.6 | 79.2 | 77.5 | 74.0 | 77.5 |
| Pyrexia | – | – | – | 56.4 | 53.6 | 52.3 | 56.1 |
In the three de novo kidney studies, patients receiving 2 g/day of mycophenolate mofetil had an overall better safety profile than did patients receiving 3 g/day of mycophenolate mofetil.
Post-transplant lymphoproliferative disease (PTLD) developed in 0.4% to 1% of patients receiving mycophenolate mofetil (2 g or 3 g daily) with other immunosuppressive agents in controlled clinical trials of kidney, heart and liver transplant patients followed for at least 1 year [see Warnings and Precautions (5.2)]. Non-melanoma skin carcinomas occurred in 1.6% to 4.2% of patients, other types of malignancy in 0.7% to 2.1% of patients. Three-year safety data in kidney and heart transplant patients did not reveal any unexpected changes in incidence of malignancy compared to the 1-year data. In pediatric patients, PTLD was observed in 1.35% (2/148) by 12 months post-transplant.
Cytopenias, including leukopenia, anemia, thrombocytopenia and pancytopenia are a known risk associated with mycophenolate and may lead or contribute to the occurrence of infections and hemorrhages [see Warnings and Precautions (5.3)]. Severe neutropenia (ANC <0.5 x 103/mcL) developed in up to 2% of kidney transplant patients, up to 2.8% of heart transplant patients and up to 3.6% of liver transplant patients receiving mycophenolate mofetil 3 g daily [see Warnings and Precautions (5.4) and Dosage and Administration (2.5)].
The most common opportunistic infections in patients receiving mycophenolate mofetil with other immunosuppressants were mucocutaneous candida, CMV viremia/syndrome, and herpes simplex. The proportion of patients with CMV viremia/syndrome was 13.5%. In patients receiving mycophenolate mofetil (2 g or 3 g) in controlled studies for prevention of kidney, heart or liver rejection, fatal infection/sepsis occurred in approximately 2% of kidney and heart patients and in 5% of liver patients [see Warnings and Precautions (5.3)].
The most serious gastrointestinal disorders reported were ulceration and hemorrhage, which are known risk associated with mycophenolate mofetil. Mouth, esophageal, gastric, duodenal, and intestinal ulcers often complicated by hemorrhage, as well as hematemesis, melena, and hemorrhagic form of gastritis and colitis were commonly reported during the pivotal clinical trials, while the most common gastrointestinal disorders were diarrhea, nausea and vomiting. Endoscopic investigation of patients with mycophenolate mofetil-related diarrhea revealed isolated cases of intestinal villous atrophy [see Warning and Precautions (5.5)].
The following adverse reactions were reported with 3% to <20% incidence in kidney, heart, and liver transplant patients treated with mycophenolate mofetil, in combination with cyclosporine and corticosteroids.
| System Organ Class | Adverse Reactions |
| Body as a Whole | cellulitis, chills, hernia, malaise |
| Infections and Infestations | fungal infections |
| Hematologic and Lymphatic | coagulation disorder, ecchymosis, pancytopenia |
| Urogenital | hematuria |
| Cardiovascular | hypotension |
| Metabolic and Nutritional | acidosis, alkaline phosphatase increased, hyperlipemia, hypophosphatemia, weight loss |
| Digestive | esophagitis, flatulence, gastritis, gastrointestinal hemorrhage, hepatitis, ileus, nausea and vomiting, stomach ulcer, stomatitis |
| Neoplasm benign, malignant and unspecified | neoplasm |
| Skin and Appendages | skin benign neoplasm, skin carcinoma |
| Psychiatric | confusional state |
| Nervous | hypertonia, paresthesia, somnolence |
| Musculoskeletal | arthralgia, myasthenia |
Pediatric Study
The type and frequency of adverse events in a clinical study for prevention of kidney allograft rejection in 100 pediatric patients 3 months to 18 years of age dosed with mycophenolate mofetil oral suspension 600 mg/m2 twice daily (up to 1 g twice daily) were generally similar to those observed in adult patients dosed with mycophenolate mofetil capsules at a dose of 1 g twice daily with the exception of abdominal pain, fever, infection, pain, sepsis, diarrhea, vomiting, pharyngitis, respiratory tract infection, hypertension, leukopenia, and anemia, which were observed in a higher proportion in pediatric patients.
Geriatrics
Elderly patients (≥65 years), particularly those who are receiving mycophenolate mofetil as part of a combination immunosuppressive regimen, may be at increased risk of certain infections (including cytomegalovirus [CMV] tissue invasive disease) and possibly gastrointestinal hemorrhage and pulmonary edema, compared to younger individuals [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Mycophenolate Mofetil for Injection
The safety profile of mycophenolate mofetil for injection was determined from a single, double-blind, controlled comparative study of the safety of 2 g/day of intravenous and oral mycophenolate mofetil in kidney transplant patients in the immediate post-transplant period (administered for the first 5 days). The potential venous irritation of mycophenolate mofetil for injection was evaluated by comparing the adverse reactions attributable to peripheral venous infusion of mycophenolate mofetil for injection with those observed in the intravenous placebo group; patients in the placebo group received active medication by the oral route.
Adverse reactions attributable to peripheral venous infusion were phlebitis and thrombosis, both observed at 4% in patients treated with mycophenolate mofetil for injection.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of mycophenolate mofetil. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- Embryo-Fetal Toxicity: Congenital malformations and spontaneous abortions, mainly in the first trimester, have been reported following exposure to mycophenolate mofetil (MMF) in combination with other immunosuppressants during pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1), (8.3)]. Congenital malformations include:
- -
- Facial malformations: cleft lip, cleft palate, micrognathia, hypertelorism of the orbits
- -
- Abnormalities of the ear and eye: abnormally formed or absent external/middle ear, coloboma, microphthalmos
- -
- Malformations of the fingers: polydactyly, syndactyly, brachydactyly
- -
- Cardiac abnormalities: atrial and ventricular septal defects
- -
- Esophageal malformations: esophageal atresia
- -
- Nervous system malformations: such as spina bifida
- Cardiovascular: Venous thrombosis has been reported in patients treated with mycophenolate mofetil administered intravenously.
- Digestive: Colitis, pancreatitis.
- Hematologic and Lymphatic: Bone marrow failure, cases of pure red cell aplasia (PRCA) and hypogammaglobulinemia have been reported in patients treated with mycophenolate mofetil in combination with other immunosuppressive agents [see Warnings and Precautions (5.4)].
- Immune: Hypersensitivity, hypogammaglobinemia.
- Infections: Meningitis, infectious endocarditis, tuberculosis, atypical mycobacterial infection, progressive multifocal leukoencephalopathy, BK virus infection, viral reactivation of hepatitis B and hepatitis C, protozoal infections [see Warnings and Precautions (5.3)].
- Respiratory: Bronchiectasis, interstitial lung disease, fatal pulmonary fibrosis, have been reported rarely and should be considered in the differential diagnosis of pulmonary symptoms ranging from dyspnea to respiratory failure in post-transplant patients receiving mycophenolate mofetil.
- Vascular: Lymphocele.
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Mycophenolate Mofetil
| Antacids with Magnesium or Aluminum Hydroxide | |
| Clinical Impact | Concomitant use with an antacid containing magnesium or aluminum hydroxide decreases MPA systemic exposure [see Clinical Pharmacology (12.3)], which may reduce mycophenolate mofetil efficacy. |
| Prevention or Management | Administer magnesium or aluminum hydroxide containing antacids at least 2h after mycophenolate mofetil administration. |
| Proton Pump Inhibitors (PPIs) | |
| Clinical Impact | Concomitant use with PPIs decreases MPA systemic exposure [see Clinical Pharmacology (12.3)], which may reduce mycophenolate mofetil efficacy. |
| Prevention or Management | Monitor patients for alterations in efficacy when PPIs are co-administered with mycophenolate mofetil. |
| Examples | Lansoprazole, pantoprazole |
| Drugs that Interfere with Enterohepatic Recirculation | |
| Clinical Impact | Concomitant use with drugs that directly interfere with enterohepatic recirculation, or indirectly interfere with enterohepatic recirculation by altering the gastrointestinal flora, can decrease MPA systemic exposure [see Clinical Pharmacology (12.3)], which may reduce mycophenolate mofetil efficacy. |
| Prevention or Management | Monitor patients for alterations in efficacy or mycophenolate mofetil related adverse reactions when these drugs are co-administered with mycophenolate mofetil. |
| Examples | Trimethoprim/sulfamethoxazole, bile acid sequestrants (cholestyramine), rifampin as well as aminoglycoside, cephalosporin, fluoroquinolone and penicillin classes of antimicrobials |
| Drugs Modulating Glucuronidation | |
| Clinical Impact | Concomitant use with drugs inducing glucuronidation decreases MPA systemic exposure, potentially reducing mycophenolate mofetil efficacy, while use with drugs inhibiting glucuronidation increases MPA systemic exposure [see Clinical Pharmacology (12.3)], which may increase the risk of mycophenolate mofetil related adverse reactions. |
| Prevention or Management | Monitor patients for alterations in efficacy or mycophenolate mofetil related adverse reactions when these drugs are co-administered with mycophenolate mofetil. |
| Examples | Telmisartan (induces glucuronidation); isavuconazole (inhibits glucuronidation). |
| Calcium Free Phosphate Binders | |
| Clinical Impact | Concomitant use with calcium free phosphate binders decrease MPA systemic exposure [see Clinical Pharmacology (12.3)], which may reduce mycophenolate mofetil efficacy. |
| Prevention or Management | Administer calcium free phosphate binders at least 2 hours after mycophenolate mofetil. |
| Examples | Sevelamer |
7.2 Effect of Mycophenolate Mofetil on Other Drugs
| Drugs that Undergo Renal Tubular Secretion | |
| Clinical Impact | When concomitantly used with mycophenolate mofetil, its metabolite MPAG, may compete with drugs eliminated by renal tubular secretion which may increase plasma concentrations and/or adverse reactions associated with these drugs. |
| Prevention or Management | Monitor for drug-related adverse reactions in patients with renal impairment. |
| Examples | Acyclovir, ganciclovir, probenecid, valacyclovir, valganciclovir |
| Combination Oral Contraceptives | |
| Clinical Impact | Concomitant use with mycophenolate mofetil decreased the systemic exposure to levonorgestrel, but did not affect the systemic exposure to ethinylestradiol [see Clinical Pharmacology (12.3)], which may result in reduced combination oral contraceptive effectiveness. |
| Prevention or Management | Use additional barrier contraceptive methods. |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to mycophenolate during pregnancy and those becoming pregnant within 6 weeks of discontinuing mycophenolate mofetil treatment. To report a pregnancy or obtain information about the registry, visit www.mycophenolateREMS.com or call 1-800-617-8191.
Risk Summary
Use of mycophenolate mofetil (MMF) during pregnancy is associated with an increased risk of first trimester pregnancy loss and an increased risk of multiple congenital malformations in multiple organ systems [see Human Data]. Oral administration of mycophenolate to rats and rabbits during the period of organogenesis produced congenital malformations and pregnancy loss at doses less than the recommended clinical dose (0.02 to 0.1 times the recommended clinical doses in kidney and heart transplant patients) [see Animal Data].
Consider alternative immunosuppressants with less potential for embryofetal toxicity. Risks and benefits of mycophenolate mofetil should be discussed with the pregnant woman.
The estimated background risk of pregnancy loss and congenital malformations in organ transplant populations is not clear. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
A spectrum of congenital malformations (including multiple malformations in individual newborns) has been reported in 23 to 27% of live births in MMF exposed pregnancies, based on published data from pregnancy registries. Malformations that have been documented include external ear, eye, and other facial abnormalities including cleft lip and palate, and anomalies of the distal limbs, heart, esophagus, kidney, and nervous system.
Based on published data from pregnancy registries, the risk of first trimester pregnancy loss has been reported at 45 to 49% following MMF exposure.
Animal Data
In animal reproductive toxicology studies, there were increased rates of fetal resorptions and malformations in the absence of maternal toxicity. Oral administration of MMF to pregnant rats from Gestational Day 7 to Day 16 produced increased embryofetal lethality and fetal malformations including anophthalmia, agnathia, and hydrocephaly at doses equivalent to 0.03 and 0.02 times the recommended human doses for renal and cardiac transplant patients, respectively, when corrected for BSA. Oral administration of MMF to pregnant rabbits from Gestational Day 7 to Day 19 produced increased embryofetal lethality and fetal malformations included ectopia cordis, ectopic kidneys, diaphragmatic hernia, and umbilical hernia at dose equivalents as low as 0.1 and 0.06 times the recommended human doses for renal and cardiac transplant patients, respectively, when corrected for BSA.
8.2 Lactation
Risk Summary
There are no data on the presence of mycophenolate in human milk, or the effects on milk production. There are limited data in the National Transplantation Pregnancy Registry on the effects of mycophenolate on a breastfed child [see Data]. Studies in rats treated with MMF have shown mycophenolic acid (MPA) to be present in milk. Because available data are limited, it is not possible to exclude potential risks to a breastfeeding infant.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for mycophenolate mofetil and any potential adverse effects on the breastfed infant from mycophenolate mofetil or from the underlying maternal condition.
Data
Limited information is available from the National Transplantation Pregnancy Registry. Of seven infants reported by the National Transplantation Pregnancy Registry to have been breastfed while the mother was taking mycophenolate, all were born at 34 to 40 weeks gestation, and breastfed for up to 14 months. No adverse events were reported.
8.3 Females and Males of Reproductive Potential
Females of reproductive potential must be made aware of the increased risk of first trimester pregnancy loss and congenital malformations and must be counseled regarding pregnancy prevention and planning.
Pregnancy Planning
For patients who are considering pregnancy, consider alternative immunosuppressants with less potential for embryofetal toxicity whenever possible. Risks and benefits of mycophenolate mofetil should be discussed with the patient.
Pregnancy Testing
To prevent unplanned exposure during pregnancy, all females of reproductive potential should have a serum or urine pregnancy test with a sensitivity of at least 25 mIU/mL immediately before starting mycophenolate mofetil. Another pregnancy test with the same sensitivity should be done 8 to 10 days later. Repeat pregnancy tests should be performed during routine follow-up visits. Results of all pregnancy tests should be discussed with the patient. In the event of a positive pregnancy test, consider alternative immunosuppressants with less potential for embryofetal toxicity whenever possible.
Contraception
Female Patients
Females of reproductive potential taking mycophenolate mofetil must receive contraceptive counseling and use acceptable contraception (see Table 7 for acceptable contraception methods). Patients must use acceptable birth control during the entire mycophenolate mofetil therapy, and for 6 weeks after stopping mycophenolate mofetil, unless the patient chooses abstinence.
Patients should be aware that mycophenolate mofetil reduces blood levels of the hormones from the oral contraceptive pill and could theoretically reduce its effectiveness [see Drug Interactions (7.2)].
| Option 1 | |
| Methods to Use Alone |
|
OR
| Option 2 | Hormone Methods choose 1 | Barrier Methods choose 1 | |
| Choose One
Hormone Method AND One Barrier Method | Estrogen and Progesterone
| AND |
|
OR
| Option 3 | Barrier Methods choose 1 | Barrier Methods choose 1 | |
| Choose One Barrier Method from each column
(must choose two methods) |
| AND |
|
Male Patients
Genotoxic effects have been observed in animal studies at exposures exceeding the human therapeutic exposures by approximately 2.5 times. Thus, the risk of genotoxic effects on sperm cells cannot be excluded. Based on this potential risk, sexually active male patients and/or their female partners are recommended to use effective contraception during treatment of the male patient and for at least 90 days after cessation of treatment. Also, based on the potential risk of genotoxic effects, male patients should not donate sperm during treatment with mycophenolate mofetil and for at least 90 days after cessation of treatment [see Use in Specific Populations (8.1), Nonclinical Toxicology (13.1), Patient Counseling Information (17.9)].
8.4 Pediatric Use
Safety and effectiveness of mycophenolate mofetil have been established in pediatric patients 3 months and older for the prophylaxis of kidney rejection after allogeneic kidney transplant. Use of mycophenolate mofetil in this population is supported by evidence from adequate and well-controlled studies of mycophenolate mofetil in adults with additional data from one open-label, pharmacokinetic and safety study of mycophenolate mofetil in pediatric patients after receiving allogeneic kidney transplant [see Dosage and Administration (2.2), Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.1)].
Safety and effectiveness in pediatric patients receiving allogeneic heart or liver transplants have not been established.
8.5 Geriatric Use
Clinical studies of mycophenolate mofetil did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should take into consideration the presence of decreased hepatic, renal or cardiac function and of concomitant drug therapies [see Adverse Reactions (6.1), Drug Interactions (7)].
8.6 Patients with Renal Impairment
Patients with Kidney Transplant
No dose adjustments are needed in kidney transplant patients experiencing delayed graft function postoperatively but patients should be carefully monitored [see Clinical Pharmacology (12.3)]. In kidney transplant patients with severe chronic impairment of the graft (GFR < 25 mL/min/1.73 m2), no dose adjustments are necessary; however, doses greater than 1 g administered twice a day should be avoided.
Patients with Heart and Liver Transplant
No data are available for heart or liver transplant patients with severe chronic renal impairment. Mycophenolate mofetil may be used for heart or liver transplant patients with severe chronic renal impairment if the potential benefits outweigh the potential risks.
8.7 Patients with Hepatic Impairment
Patients with Kidney Transplant
No dose adjustments are recommended for kidney transplant patients with severe hepatic parenchymal disease. However, it is not known whether dose adjustments are needed for hepatic disease with other etiologies [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Possible signs and symptoms of acute overdose include hematological abnormalities such as leukopenia and neutropenia, and gastrointestinal symptoms such as abdominal pain, diarrhea, nausea, vomiting, and dyspepsia.
The experience with overdose of mycophenolate mofetil in humans is limited. The reported effects associated with overdose fall within the known safety profile of the drug. The highest dose administered to kidney transplant patients in clinical trials has been 4 g/day. In limited experience with heart and liver transplant patients in clinical trials, the highest doses used were 4 g/day or 5 g/day. At doses of 4 g/day or 5 g/day, there appears to be a higher rate, compared to the use of 3 g/day or less, of gastrointestinal intolerance (nausea, vomiting, and/or diarrhea), and occasional hematologic abnormalities, particularly neutropenia [see Warnings and Precautions (5.4)].
Treatment and Management
MPA and the phenolic glucuronide metabolite of MPA (MPAG) are usually not removed by hemodialysis. However, at high MPAG plasma concentrations (>100 mcg/mL), small amounts of MPAG are removed. By increasing excretion of the drug, MPA can be removed by bile acid sequestrants, such as cholestyramine [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
Mycophenolate mofetil is an antimetabolite immunosuppressant. It is the 2-morpholinoethyl ester of mycophenolic acid (MPA), an immunosuppressive agent; inosine monophosphate dehydrogenase (IMPDH) inhibitor.
The chemical name for mycophenolate mofetil (MMF) is 2-morpholinoethyl (E)-6-(1,3-dihydro-4-hydroxy-6-methoxy-7-methyl-3-oxo-5-isobenzofuranyl)-4-methyl-4-hexenoate. It has an empirical formula of C23H31NO7, a molecular weight of 433.50, and the following structural formula:
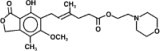
MMF is a white to off-white crystalline powder. It is slightly soluble in water (43 mcg/mL at pH 7.4); the solubility increases in acidic medium (4.27 mg/mL at pH 3.6). It is freely soluble in acetone, soluble in methanol, and sparingly soluble in ethanol. The apparent partition coefficient in 1-octanol/water (pH 7.4) buffer solution is 238. The pKa values for MMF are 5.6 for the morpholino group and 8.5 for the phenolic group.
MMF hydrochloride has a solubility of 65.8 mg/mL in 5% Dextrose Injection USP (D5W). The pH of the reconstituted solution is 2.4 to 4.1.
Mycophenolate Mofetil for Injection, USP is the hydrochloride salt of MMF. The chemical name for the hydrochloride salt of MMF is 2-morpholinoethyl (E)-6-(1,3-dihydro-4-hydroxy-6-methoxy-7-methyl-3-oxo-5-isobenzofuranyl)-4-methyl-4-hexenoate hydrochloride. It has an empirical formula of C23H31NO7 HCl and a molecular weight of 469.96.
Mycophenolate Mofetil for Injection, USP is available as a sterile white to off-white lyophilized powder in single-dose vials containing MMF hydrochloride for administration by intravenous infusion only. Each vial of Mycophenolate Mofetil for Injection, USP contains the equivalent of 500 mg MMF as the hydrochloride salt. The inactive ingredients are polysorbate 80, 25 mg, and citric acid, 5 mg. Sodium hydroxide may have been used in the manufacture of Mycophenolate Mofetil for Injection, USP to adjust the pH. Reconstitution and dilution with 5% Dextrose Injection, USP yields a slightly yellow solution of MMF, 6 mg/mL [see Dosage and Administration (2.6)].
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mycophenolate mofetil (MMF) is absorbed following oral administration and hydrolyzed to mycophenolic acid (MPA), the active metabolite. MPA is a selective, uncompetitive, and reversible inhibitor of inosine monophosphate dehydrogenase (IMPDH), and therefore inhibits the de novo pathway of guanosine nucleotide synthesis without incorporation into DNA. Because T- and B-lymphocytes are critically dependent for their proliferation on de novo synthesis of purines, whereas other cell types can utilize salvage pathways, MPA has potent cytostatic effects on lymphocytes. MPA inhibits proliferative responses of T- and B-lymphocytes to both mitogenic and allospecific stimulation. Addition of guanosine or deoxyguanosine reverses the cytostatic effects of MPA on lymphocytes. MPA also suppresses antibody formation by B-lymphocytes. MPA prevents the glycosylation of lymphocyte and monocyte glycoproteins that are involved in intercellular adhesion to endothelial cells and may inhibit recruitment of leukocytes into sites of inflammation and graft rejection. MMF did not inhibit early events in the activation of human peripheral blood mononuclear cells, such as the production of interleukin-1 (IL-1) and interleukin-2 (IL-2), but did block the coupling of these events to DNA synthesis and proliferation.
12.3 Pharmacokinetics
Absorption
Following oral and intravenous administration, MMF undergoes complete conversion to MPA, the active metabolite. In 12 healthy volunteers, the mean absolute bioavailability of oral MMF relative to intravenous MMF was 94%. Two 500 mg mycophenolate mofetil tablets have been shown to be bioequivalent to four 250 mg mycophenolate mofetil capsules. Five mL of the 200 mg/mL constituted mycophenolate mofetil oral suspension have been shown to be bioequivalent to four 250 mg capsules.
The mean (±SD) pharmacokinetic parameters estimates for MPA following the administration of MMF given as single doses to healthy volunteers, and multiple doses to kidney, heart, and liver transplant patients, are shown in Table 8. The area under the plasma-concentration time curve (AUC) for MPA appears to increase in a dose-proportional fashion in kidney transplant patients receiving multiple oral doses of MMF up to a daily dose of 3 g (1.5 g twice daily) (see Table 8).
|
a AUC(0-12h) values quoted are extrapolated from data from samples collected over 4 hours. |
||||
| Healthy Volunteers | Dose/Route | Tmax (h) | Cmax (mcg/mL) | Total AUC (mcg•h/mL) |
| Single dose | 1 g/oral | 0.80 (±0.36) (n=129) | 24.5 (±9.5) (n=129) | 63.9 (±16.2) (n=117) |
| Kidney Transplant Patients (twice daily dosing) Time After Transplantation | Dose/Route | Tmax (h) | Cmax (mcg/mL) | Interdosing Interval AUC(0- 12h) (mcg•h/mL) |
| 5 days | 1 g/IV | 1.58 (±0.46) (n=31) | 12.0 (±3.82) (n=31) | 40.8 (±11.4) (n=31) |
| 6 days | 1 g/oral | 1.33 (±1.05) (n=31) | 10.7 (±4.83) (n=31) | 32.9 (±15.0) (n=31) |
| Early (Less than 40 days) | 1 g/oral | 1.31 (±0.76) (n=25) | 8.16 (±4.50) (n=25) | 27.3 (±10.9) (n=25) |
| Early (Less than 40 days) | 1.5 g/oral | 1.21 (±0.81) (n=27) | 13.5 (±8.18) (n=27) | 38.4 (±15.4) (n=27) |
| Late (Greater than 3 months) | 1.5 g/oral | 0.90 (±0.24) (n=23) | 24.1 (±12.1) (n=23) | 65.3 (±35.4) (n=23) |
| Heart transplant Patients (twice daily dosing) Time After Transplantation | Dose/Route | Tmax (h) | Cmax (mcg/mL) | Interdosing Interval AUC(0- 12h) (mcg•h/mL) |
| Early (Day before discharge) | 1.5 g/oral | 1.8 (±1.3) (n=11) | 11.5 (±6.8) (n=11) | 43.3 (±20.8) (n=9) |
| Late (Greater than 6 months) | 1.5 g/oral | 1.1 (±0.7) (n=52) | 20.0 (±9.4) (n=52) | 54.1a
(±20.4) (n=49) |
| Liver transplant Patients (twice daily dosing) Time After Transplantation | Dose/Route | Tmax (h) | Cmax (mcg/mL) | Interdosing Interval AUC(0- 12h) (mcg•h/mL) |
| 4 to 9 days | 1 g/IV | 1.50 (±0.517) (n=22) | 17.0 (±12.7) (n=22) | 34.0 (±17.4) (n=22) |
| Early (5 to 8 days) | 1.5 g/oral | 1.15 (±0.432) (n=20) | 13.1 (±6.76) (n=20) | 29.2 (±11.9) (n=20) |
| Late (Greater than 6 months) | 1.5 g/oral | 1.54 (±0.51) (n=6) | 19.3 (±11.7) (n=6) | 49.3 (±14.8) (n=6) |
In the early post-transplant period (less than 40 days post-transplant), kidney, heart, and liver transplant patients had mean MPA AUCs approximately 20% to 41% lower and mean Cmax approximately 32% to 44% lower compared to the late transplant period (i.e., 3 to 6 months post-transplant) (non-stationarity in MPA pharmacokinetics).
Mean MPA AUC values following administration of 1 g twice daily intravenous mycophenolate mofetil over 2 hours to kidney transplant patients for 5 days were about 24% higher than those observed after oral administration of a similar dose in the immediate post-transplant phase.
In liver transplant patients, administration of 1 g twice daily intravenous mycophenolate mofetil followed by 1.5 g twice daily oral mycophenolate mofetil resulted in mean MPA AUC estimates similar to those found in kidney transplant patients administered 1 g mycophenolate mofetil twice daily.
Effect of Food
Food (27 g fat, 650 calories) had no effect on the extent of absorption (MPA AUC) of MMF when administered at doses of 1.5 g twice daily to kidney transplant patients. However, MPA Cmax was decreased by 40% in the presence of food [see Dosage and Administration (2.1)].
Distribution
The mean (±SD) apparent volume of distribution of MPA in 12 healthy volunteers was approximately 3.6 (±1.5) L/kg. At clinically relevant concentrations, MPA is 97% bound to plasma albumin. The phenolic glucuronide metabolite of MPA (MPAG) is 82% bound to plasma albumin at MPAG concentration ranges that are normally seen in stable kidney transplant patients; however, at higher MPAG concentrations (observed in patients with kidney impairment or delayed kidney graft function), the binding of MPA may be reduced as a result of competition between MPAG and MPA for protein binding. Mean blood to plasma ratio of radioactivity concentrations was approximately 0.6 indicating that MPA and MPAG do not extensively distribute into the cellular fractions of blood.
In vitro studies to evaluate the effect of other agents on the binding of MPA to human serum albumin (HSA) or plasma proteins showed that salicylate (at 25 mg/dL with human serum albumin) and MPAG (at ≥ 460 mcg/mL with plasma proteins) increased the free fraction of MPA. MPA at concentrations as high as 100 mcg/mL had little effect on the binding of warfarin, digoxin or propranolol, but decreased the binding of theophylline from 53% to 45% and phenytoin from 90% to 87%.
Elimination
Mean (±SD) apparent half-life and plasma clearance of MPA are 17.9 (±6.5) hours and 193 (±48) mL/min following oral administration and 16.6 (±5.8) hours and 177 (±31) mL/min following intravenous administration, respectively.
Metabolism
The parent drug, MMF, can be measured systemically during the intravenous infusion; however, approximately 5 minutes after the infusion is stopped or after oral administration, MMF concentrations are below the limit of quantitation (0.4 mcg/mL).
Metabolism to MPA occurs pre-systemically after oral dosing. MPA is metabolized principally by glucuronyl transferase to form MPAG, which is not pharmacologically active. In vivo, MPAG is converted to MPA during enterohepatic recirculation. The following metabolites of the 2-hydroxyethyl-morpholino moiety are also recovered in the urine following oral administration of MMF to healthy subjects: N-(2-carboxymethyl)-morpholine, N-(2-hydroxyethyl)-morpholine, and the N-oxide of N-(2-hydroxyethyl)-morpholine.
Due to the enterohepatic recirculation of MPAG/MPA, secondary peaks in the plasma MPA concentration-time profile are usually observed 6 to 12 hours post-dose. Bile sequestrants, such as cholestyramine, reduce MPA AUC by interfering with this enterohepatic recirculation of the drug [see Overdose (10) and Drug Interaction Studies below].
Excretion
Negligible amount of drug is excreted as MPA (less than 1% of dose) in the urine. Orally administered radiolabeled MMF resulted in complete recovery of the administered dose, with 93% of the administered dose recovered in the urine and 6% recovered in feces. Most (about 87%) of the administered dose is excreted in the urine as MPAG. At clinically encountered concentrations, MPA and MPAG are usually not removed by hemodialysis. However, at high MPAG plasma concentrations (> 100 mcg/mL), small amounts of MPAG are removed.
Increased plasma concentrations of MMF metabolites (MPA 50% increase and MPAG about a 3-fold to 6-fold increase) are observed in patients with renal insufficiency [see Specific Populations].
Specific Populations
Patients with Renal Impairment
The mean (±SD) pharmacokinetic parameters for MPA following the administration of oral MMF given as single doses to non-transplant subjects with renal impairment are presented in Table 9.
In a single-dose study, MMF was administered as a capsule or as an intravenous infusion over 40 minutes. Plasma MPA AUC observed after oral dosing to volunteers with severe chronic renal impairment (GFR < 25 mL/min/1.73 m2) was about 75% higher relative to that observed in healthy volunteers (GFR > 80 mL/min/1.73 m2). In addition, the single-dose plasma MPAG AUC was 3-fold to 6-fold higher in volunteers with severe renal impairment than in volunteers with mild renal impairment or healthy volunteers, consistent with the known renal elimination of MPAG. No data are available on the safety of long-term exposure to this level of MPAG.
Plasma MPA AUC observed after single-dose (1 g) intravenous dosing to volunteers (n=4) with severe chronic renal impairment (GFR < 25 mL/min/1.73 m2) was 62.4 mcg•h/mL (±19.3). Multiple dosing of MMF in patients with severe chronic renal impairment has not been studied.
Patients with Delayed Graft Function or Nonfunction
In patients with delayed renal graft function post-transplant, mean MPA AUC(0-12h) was comparable to that seen in post-transplant patients without delayed renal graft function. There is a potential for a transient increase in the free fraction and concentration of plasma MPA in patients with delayed renal graft function. However, dose adjustment does not appear to be necessary in patients with delayed renal graft function. Mean plasma MPAG AUC(0-12h) was 2-fold to 3-fold higher than in post-transplant patients without delayed renal graft function [see Dosage and Administration (2.5)].
In eight patients with primary graft non-function following kidney transplantation, plasma concentrations of MPAG accumulated about 6-fold to 8-fold after multiple dosing for 28 days. Accumulation of MPA was about 1-fold to 2-fold.
The pharmacokinetics of MMF are not altered by hemodialysis. Hemodialysis usually does not remove MPA or MPAG. At high concentrations of MPAG (> 100 mcg/mL), hemodialysis removes only small amounts of MPAG.
Patients with Hepatic Impairment
The mean (± SD) pharmacokinetic parameters for MPA following the administration of oral MMF given as single doses to non-transplant subjects with hepatic impairment is presented in Table 9.
In a single-dose (1 g oral) study of 18 volunteers with alcoholic cirrhosis and 6 healthy volunteers, hepatic MPA glucuronidation processes appeared to be relatively unaffected by hepatic parenchymal disease when pharmacokinetic parameters of healthy volunteers and alcoholic cirrhosis patients within this study were compared. However, it should be noted that for unexplained reasons, the healthy volunteers in this study had about a 50% lower AUC as compared to healthy volunteers in other studies, thus making comparisons between volunteers with alcoholic cirrhosis and healthy volunteers difficult. In a single-dose (1 g intravenous) study of 6 volunteers with severe hepatic impairment (aminopyrine breath test less than 0.2% of dose) due to alcoholic cirrhosis, MMF was rapidly converted to MPA. MPA AUC was 44.1 mcg•h/mL (±15.5).
| Pharmacokinetic Parameters for Renal Impairment | |||||
| Dose | Tmax (h) | Cmax (mcg/mL) | AUC(0-96h) (mcg•h/mL) | ||
| Healthy Volunteers GFR greater than 80 mL/min/1.73 m2 (n=6) | 1 g | 0.75 (±0.27) | 25.3 (±7.99) | 45.0 (±22.6) |
|
| Mild Renal Impairment GFR 50 to 80 mL/min/1.73 m2 (n=6) | 1 g | 0.75 (±0.27) | 26.0 (±3.82) | 59.9 (±12.9) |
|
| Moderate Renal Impairment GFR 25 to 49 mL/min/1.73 m2 (n=6) | 1 g | 0.75 (±0.27) | 19.0 (±13.2) | 52.9 (±25.5) |
|
| Severe Renal Impairment GFR less than 25 mL/min/1.73 m2 (n=7) | 1 g | 1.00 (±0.41) | 16.3 (±10.8) | 78.6 (±46.4) |
|
| Pharmacokinetic Parameters for Hepatic Impairment | |||||
| Dose | Tmax (h) | Cmax (mcg/mL) | AUC(0-48h) (mcg•h/mL) | ||
| Healthy Volunteers (n=6) | 1 g | 0.63 (±0.14) | 24.3 (±5.73) | 29.0 (±5.78) |
|
| Alcoholic Cirrhosis (n=18) | 1 g | 0.85 (±0.58) | 22.4 (±10.1) | 29.8 (±10.7) |
|
Pediatric Patients
The pharmacokinetic parameters of MPA and MPAG have been evaluated in 55 pediatric patients (ranging from 1 year to 18 years of age) receiving mycophenolate mofetil oral suspension at a dose of 600 mg/m2 twice daily (up to a maximum of 1 g twice daily) after allogeneic kidney transplantation. The pharmacokinetic data for MPA is provided in Table 10.
|
a adjusted to a dose of 600 mg/m2 |
||||||||
|
b n=20 |
||||||||
|
c n=16 |
||||||||
|
d a subset of 1 to <6 yr |
||||||||
| Age Group | (n) | Time | Tmax (h) | Dose Adjusteda Cmax (mcg/mL) | Dose Adjusteda AUC0-12 (mcg•h/mL) | |||
| Early (Day 7) | ||||||||
| 1 to less than 2 yr 1 to less than 6 yr 6 to less than 12 yr 12 to 18 yr | (6)d
(17) (16) (21) | 3.03 1.63 0.940 1.16 | (4.70) (2.85) (0.546) (0.830) | 10.3 13.2 13.1 11.7 | (5.80) (7.16) (6.30) (10.7) | 22.5 27.4 33.2 26.3 | (6.66) (9.54) (12.1) (9.14)b |
|
| Late (Month 3) | ||||||||
| 1 to less than 2 yr 1 to less than 6 yr 6 to less than 12 yr 12 to 18 yr | (4)d
(15) (14) (17) | 0.725 0.989 1.21 0.978 | (0.276) (0.511) (0.532) (0.484) | 23.8 22.7 27.8 17.9 | (13.4) (10.1) (14.3) (9.57) | 47.4 49.7 61.9 53.6 | (14.7) (18.2) (19.6) (20.3)c |
|
| Late (Month 9) | ||||||||
| 1 to less than 2 yr 1 to less than 6 yr 6 to less than 12 yr 12 to 18 yr | (4)d
(12) (11) (14) | 0.604 0.869 1.12 1.09 | (0.208) (0.479) (0.462) (0.518) | 25.6 30.4 29.2 18.1 | (4.25) (9.16) (12.6) (7.29) | 55.8 61.0 66.8 56.7 | (11.6) (10.7) (21.2) (14.0) |
|
The mycophenolate mofetil oral suspension dose of 600 mg/m2 twice daily (up to a maximum of 1 g twice daily) achieved mean MPA AUC values in pediatric patients similar to those seen in adult kidney transplant patients receiving mycophenolate mofetil capsules at a dose of 1 g twice daily in the early post-transplant period. There was wide variability in the data. As observed in adults, early post-transplant MPA AUC values were approximately 45% to 53% lower than those observed in the later post-transplant period (> 3 months). MPA AUC values were similar in the early and late post-transplant period across the 1 to 18-year age range.
Male and Female Patients
Data obtained from several studies were pooled to look at any gender-related differences in the pharmacokinetics of MPA (data were adjusted to 1 g oral dose). Mean (±SD) MPA AUC(0-12h) for males (n=79) was 32.0 (±14.5) and for females (n=41) was 36.5 (±18.8) mcg•h/mL while mean (±SD) MPA Cmax was 9.96 (±6.19) in the males and 10.6 (±5.64) mcg/mL in the females. These differences are not of clinical significance.
Drug Interaction Studies
Acyclovir
Coadministration of MMF (1 g) and acyclovir (800 mg) to 12 healthy volunteers resulted in no significant change in MPA AUC and Cmax. However, MPAG and acyclovir plasma AUCs were increased 10.6% and 21.9%, respectively.
Antacids with Magnesium and Aluminum Hydroxides
Absorption of a single dose of MMF (2 g) was decreased when administered to 10 rheumatoid arthritis patients also taking Maalox® TC (10 mL qid). The Cmax and AUC(0-24h) for MPA were 33% and 17% lower, respectively, than when MMF was administered alone under fasting conditions.
Proton Pump Inhibitors (PPIs)
Coadministration of PPIs (e.g., lansoprazole, pantoprazole) in single doses to healthy volunteers and multiple doses to transplant patients receiving mycophenolate mofetil has been reported to reduce the exposure to MPA. An approximate reduction of 30 to 70% in the Cmax and 25% to 35% in the AUC of MPA has been observed, possibly due to a decrease in MPA solubility at an increased gastric pH.
Cholestyramine
Following single-dose administration of 1.5 g MMF to 12 healthy volunteers pretreated with 4 g three times a day of cholestyramine for 4 days, MPA AUC decreased approximately 40%. This decrease is consistent with interruption of enterohepatic recirculation which may be due to binding of recirculating MPAG with cholestyramine in the intestine.
Cyclosporine
Cyclosporine (Sandimmune®) pharmacokinetics (at doses of 275 to 415 mg/day) were unaffected by single and multiple doses of 1.5 g twice daily of MMF in 10 stable kidney transplant patients. The mean (±SD) AUC(0-12h) and Cmax of cyclosporine after 14 days of multiple doses of MMF were 3290 (±822) ng•h/mL and 753 (±161) ng/mL, respectively, compared to 3245 (±1088) ng•h/mL and 700 (±246) ng/mL, respectively, 1 week before administration of MMF.
Cyclosporine A interferes with MPA enterohepatic recirculation. In kidney transplant patients, mean MPA exposure (AUC(0-12h)) was approximately 30 to 50% greater when MMF was administered without cyclosporine compared with when MMF was coadministered with cyclosporine. This interaction is due to cyclosporine inhibition of multidrug-resistance-associated protein 2 (MRP-2) transporter in the biliary tract, thereby preventing the excretion of MPAG into the bile that would lead to enterohepatic recirculation of MPA. This information should be taken into consideration when MMF is used without cyclosporine.
Drugs Affecting Glucuronidation
Concomitant administration of drugs inhibiting glucuronidation of MPA may increase MPA exposure (e.g., increase of MPA AUC(0-∞) by 35% was observed with concomitant administration of isavuconazole).
Concomitant administration of telmisartan and mycophenolate mofetil resulted in an approximately 30% decrease in MPA concentrations. Telmisartan changes MPA's elimination by enhancing PPAR gamma (peroxisome proliferator-activated receptor gamma) expression, which in turn results in an enhanced UGT1A9 expression and glucuronidation activity.
Ganciclovir
Following single-dose administration to 12 stable kidney transplant patients, no pharmacokinetic interaction was observed between MMF (1.5 g) and intravenous ganciclovir (5 mg/kg). Mean (±SD) ganciclovir AUC and Cmax (n=10) were 54.3 (±19.0) mcg•h/mL and 11.5 (±1.8) mcg/mL, respectively, after coadministration of the two drugs, compared to 51.0 (±17.0) mcg•h/mL and 10.6 (±2.0) mcg/mL, respectively, after administration of intravenous ganciclovir alone. The mean (±SD) AUC and Cmax of MPA (n=12) after coadministration were 80.9 (±21.6) mcg•h/mL and 27.8 (±13.9) mcg/mL, respectively, compared to values of 80.3 (±16.4) mcg•h/mL and 30.9 (±11.2) mcg/mL, respectively, after administration of MMF alone.
Oral Contraceptives
A study of coadministration of mycophenolate mofetil (1 g twice daily) and combined oral contraceptives containing ethinylestradiol (0.02 mg to 0.04 mg) and levonorgestrel (0.05 mg to 0.20 mg), desogestrel (0.15 mg) or gestodene (0.05 mg to 0.10 mg) was conducted in 18 women with psoriasis over 3 consecutive menstrual cycles. Mean serum levels of LH, FSH and progesterone were not significantly affected. Mean AUC(0-24h) was similar for ethinylestradiol and 3-keto desogestrel; however, mean levonorgestrel AUC(0-24h) significantly decreased by about 15%. There was large inter-patient variability (%CV in the range of 60% to 70%) in the data, especially for ethinylestradiol.
Sevelamer
Concomitant administration of sevelamer and MMF in adult and pediatric patients decreased the mean MPA Cmax and AUC(0-12h) by 36% and 26% respectively.
Antimicrobials
Antimicrobials eliminating beta-glucuronidase-producing bacteria in the intestine (e.g. aminoglycoside, cephalosporin, fluoroquinolone, and penicillin classes of antimicrobials) may interfere with the MPAG/MPA enterohepatic recirculation thus leading to reduced systemic MPA exposure. Information concerning antibiotics is as follows:
- Trimethoprim/Sulfamethoxazole: Following single-dose administration of MMF (1.5 g) to 12 healthy male volunteers on day 8 of a 10-day course of trimethoprim 160 mg/sulfamethoxazole 800 mg administered twice daily, no effect on the bioavailability of MPA was observed. The mean (±SD) AUC and Cmax of MPA after concomitant administration were 75.2 (±19.8) mcg•h/mL and 34.0 (±6.6) mcg/mL, respectively, compared to 79.2 (±27.9) mcg•h/mL and 34.2 (±10.7) mcg/mL, respectively, after administration of MMF alone.
- Norfloxacin and Metronidazole: Following single-dose administration of MMF (1 g) to 11 healthy volunteers on day 4 of a 5-day course of a combination of norfloxacin and metronidazole, the mean MPA AUC(0-48h) was significantly reduced by 33% compared to the administration of MMF alone (p<0.05). The mean (±SD) MPA AUC(0-48h) after coadministration of MMF with norfloxacin or metronidazole separately was 48.3 (±24) mcg•h/mL and 42.7 (±23) mcg•h/mL, respectively, compared with 56.2 (±24) mcg•h/mL after administration of MMF alone.
- Ciprofloxacin and Amoxicillin Plus Clavulanic Acid: A total of 64 mycophenolate mofetil-treated kidney transplant recipients received either oral ciprofloxacin 500 mg twice daily or amoxicillin plus clavulanic acid 375 mg three times daily for 7 or at least 14 days, respectively. Approximately 50% reductions in median trough MPA concentrations (pre-dose) from baseline (mycophenolate mofetil alone) were observed in 3 days following commencement of oral ciprofloxacin or amoxicillin plus clavulanic acid. These reductions in trough MPA concentrations tended to diminish within 14 days of antimicrobial therapy and ceased within 3 days of discontinuation of antibiotics.
- Rifampin: In a single heart-lung transplant patient, after correction for dose, a 67% decrease in MPA exposure (AUC(0-12h)) has been observed with concomitant administration of MMF and rifampin.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week oral carcinogenicity study in mice, MMF in daily doses up to 180 mg/kg was not tumorigenic. The highest dose tested was 0.4 times the recommended clinical dose (2 g/day) in renal transplant patients and 0.3 times the recommended clinical dose (3 g/day) in cardiac transplant patients when corrected for differences in body surface area (BSA). In a 104-week oral carcinogenicity study in rats, MMF in daily doses up to 15 mg/kg was not tumorigenic. The highest dose was 0.07 times the recommended clinical dose in kidney transplant patients and 0.05 times the recommended clinical dose in heart transplant patients when corrected for BSA. While these animal doses were lower than those given to patients, they were maximal in those species and were considered adequate to evaluate the potential for human risk [see Warnings and Precautions (5.2)].
The genotoxic potential of MMF was determined in five assays. MMF was genotoxic in the mouse lymphoma/thymidine kinase assay and the in vivo mouse micronucleus assay. MMF was not genotoxic in the bacterial mutation assay, the yeast mitotic gene conversion assay or the Chinese hamster ovary cell chromosomal aberration assay.
MMF had no effect on fertility of male rats at oral doses up to 20 mg/kg/day. This dose represents 0.1 times the recommended clinical dose in renal transplant patients and 0.06 times the recommended clinical dose in cardiac transplant patients when corrected for BSA. In a female fertility and reproduction study conducted in rats, oral doses of 4.5 mg/kg/day caused malformations (principally of the head and eyes) in the first generation offspring in the absence of maternal toxicity. This dose was 0.02 times the recommended clinical dose in renal transplant patients and 0.01 times the recommended clinical dose in cardiac transplant patients when corrected for BSA. No effects on fertility or reproductive parameters were evident in the dams or in the subsequent generation.
14 CLINICAL STUDIES
14.1 Kidney Transplantation
Adults
The three de novo kidney transplantation studies compared two dose levels of oral mycophenolate mofetil (1 g twice daily and 1.5 g twice daily) with azathioprine (2 studies) or placebo (1 study) to prevent acute rejection episodes. One of the two studies with azathioprine (AZA) control arm also included anti-thymocyte globulin (ATGAM®) induction therapy. The geographic location of the investigational sites of these studies are included in Table 11.
In all three de novo kidney transplantation studies, the primary efficacy endpoint was the proportion of patients in each treatment group who experienced treatment failure within the first 6 months after transplantation. Treatment failure was defined as biopsy-proven acute rejection on treatment or the occurrence of death, graft loss or early termination from the study for any reason without prior biopsy-proven rejection.
Mycophenolate mofetil, in combination with corticosteroids and cyclosporine, reduced (statistically significant at 0.05 level) the incidence of treatment failure within the first 6 months following transplantation (Table 11). Patients who prematurely discontinued treatment were followed for the occurrence of death or graft loss, and the cumulative incidence of graft loss and patient death combined are summarized in Table 12. Patients who prematurely discontinued treatment were not followed for the occurrence of acute rejection after termination.
|
*Does not include death and graft loss as reason for early termination. |
|||
| USA Studya
(N=499 patients) | Mycophenolate Mofetil
2 g/day (n=167 patients) | Mycophenolate Mofetil
3 g/day (n=166 patients) | AZA
1 to 2 mg/kg/day (n=166 patients) |
| All 3 groups received anti-thymocyte globulin induction, cyclosporine and corticosteroids | |||
| All treatment failures | 31.1% | 31.3% | 47.6% |
| Early termination without prior acute rejection | 9.6% | 12.7% | 6.0% |
| Biopsy-proven rejection episode on treatment | 19.8% | 17.5% | 38.0% |
| Europe/Canada/Australia Study (N=503 patients) | Mycophenolate Mofetil
2 g/day (n=173 patients) | Mycophenolate Mofetil
3 g/day (n=164 patients) | AZA
100 to 150 mg/day (n=166 patients) |
| No induction treatment administered; all 3 groups received cyclosporine and corticosteroids. | |||
| All treatment failures | 38.2% | 34.8% | 50.0% |
| Early termination without prior acute rejection | 13.9% | 15.2% | 10.2% |
| Biopsy-proven rejection episode on treatment | 19.7% | 15.9% | 35.5% |
| Europe Study
(N=491 patients) | Mycophenolate Mofetil
2 g/day (n=165 patients) | Mycophenolate Mofetil
3 g/day (n=160 patients) | Placebo (n=166 patients) |
| No induction treatment administered; all 3 groups received cyclosporine and corticosteroids. | |||
| All treatment failures | 30.3% | 38.8% | 56.0% |
| Early termination without prior acute rejection | 11.5% | 22.5% | 7.2% |
| Biopsy-proven rejection episode on treatment | 17.0% | 13.8% | 46.4% |
No advantage of mycophenolate mofetil at 12 months with respect to graft loss or patient death (combined) was established (Table 12). Numerically, patients receiving mycophenolate mofetil 2 g/day and 3 g/day experienced a better outcome than controls in all three studies; patients receiving mycophenolate mofetil 2 g/day experienced a better outcome than mycophenolate mofetil 3 g/day in two of the three studies. Patients in all treatment groups who terminated treatment early were found to have a poor outcome with respect to graft loss or patient death at 1 year.
| Study | Mycophenolate Mofetil 2 g/day | Mycophenolate Mofetil 3 g/day | Control (AZA or Placebo) |
| USA | 8.5% | 11.5% | 12.2% |
| Europe/Canada/Australia | 11.7% | 11.0% | 13.6% |
| Europe | 8.5% | 10.0% | 11.5% |
Pediatrics-De Novo Kidney transplantation PK Study with Long Term Follow-Up
One open-label, safety and pharmacokinetic study of mycophenolate mofetil oral suspension 600 mg/m2 twice daily (up to 1 g twice daily) in combination with cyclosporine and corticosteroids was performed at centers in the United States (9), Europe (5) and Australia (1) in 100 pediatric patients (3 months to 18 years of age) for the prevention of renal allograft rejection. Mycophenolate mofetil was well tolerated in pediatric patients [see Adverse Reactions (6.1)], and the pharmacokinetics profile was similar to that seen in adult patients dosed with 1 g twice daily mycophenolate mofetil capsules [see Clinical Pharmacology (12.3)]. The rate of biopsy-proven rejection was similar across the age groups (3 months to <6 years, 6 years to <12 years, 12 years to 18 years). The overall biopsy-proven rejection rate at 6 months was comparable to adults. The combined incidence of graft loss (5%) and patient death (2%) at 12 months post-transplant was similar to that observed in adult kidney transplant patients.
14.2 Heart Transplantation
A double-blind, randomized, comparative, parallel-group, multicenter study in primary de novo heart transplant recipients was performed at centers in the United States (20), in Canada (1), in Europe (5) and in Australia (2). The total number of patients enrolled (ITT population) was 650; 72 never received study drug and 578 received study drug (Safety Population). Patients received mycophenolate mofetil 1.5 g twice daily (n=289) or AZA 1.5 to 3 mg/kg/day (n=289), in combination with cyclosporine (Sandimmune® or Neoral®) and corticosteroids as maintenance immunosuppressive therapy. The two primary efficacy endpoints were: (1) the proportion of patients who, after transplantation, had at least one endomyocardial biopsy-proven rejection with hemodynamic compromise, or were re-transplanted or died, within the first 6 months, and (2) the proportion of patients who died or were re-transplanted during the first 12 months following transplantation. Patients who prematurely discontinued treatment were followed for the occurrence of allograft rejection for up to 6 months and for the occurrence of death for 1 year. The analyses of the endpoints showed:
- Rejection: No difference was established between mycophenolate mofetil and AZA with respect to biopsy-proven rejection with hemodynamic compromise.
- Survival: Mycophenolate mofetil was shown to be at least as effective as AZA in preventing death or re-transplantation at 1 year (see Table 13).
|
a Hemodynamic compromise occurred if any of the following criteria were met: pulmonary capillary wedge pressure ≥ 20 mm or a 25% increase; cardiac index < 2.0 L/min/m2 or a 25% decrease; ejection fraction ≤ 30%; pulmonary artery oxygen saturation ≤ 60% or a 25% decrease; presence of new S3 gallop; fractional shortening was ≤ 20% or a 25% decrease; inotropic support required to manage the clinical condition. |
||||
| All Patie | nts (ITT) | Treated | Patients | |
| AZA
N = 323 | Mycophenolate Mofetil N = 327 | AZA
N = 289 | Mycophenolate Mofetil N = 289 | |
| Biopsy-proven rejection with hemodynamic compromise at 6 monthsa | 121 (38%) | 120 (37%) | 100 (35%) | 92 (32%) |
| Death or re-transplantation at 1 year | 49 (15.2%) | 42 (12.8%) | 33 (11.4%) | 18 (6.2%) |
14.3 Liver Transplantation
A double-blind, randomized, comparative, parallel-group, multicenter study in primary hepatic transplant recipients was performed at centers in the United States (16), in Canada (2), in Europe (4) and in Australia (1). The total number of patients enrolled was 565. Per protocol, patients received mycophenolate mofetil 1 g twice daily intravenously for up to 14 days followed by mycophenolate mofetil 1.5 g twice daily orally or AZA 1 to 2 mg/kg/day intravenously followed by AZA 1 to 2 mg/kg/day orally, in combination with cyclosporine (Neoral®) and corticosteroids as maintenance immunosuppressive therapy. The actual median oral dose of AZA on study was 1.5 mg/kg/day (range of 0.3 to 3.8 mg/kg/day) initially and 1.26 mg/kg/day (range of 0.3 to 3.8 mg/kg/day) at 12 months. The two primary endpoints were: (1) the proportion of patients who experienced, in the first 6 months post-transplantation, one or more episodes of biopsy-proven and treated rejection or death or re-transplantation, and (2) the proportion of patients who experienced graft loss (death or re-transplantation) during the first 12 months post-transplantation. Patients who prematurely discontinued treatment were followed for the occurrence of allograft rejection and for the occurrence of graft loss (death or re-transplantation) for 1 year. In combination with corticosteroids and cyclosporine, mycophenolate mofetil demonstrated a lower rate of acute rejection at 6 months and a similar rate of death or retransplantation at 1 year compared to AZA (Table 14).
| AZA
N = 287 | Mycophenolate Mofetil N = 278 |
|
| Biopsy-proven, treated rejection at 6 months (includes death or re-transplantation) | 137 (47.7%) | 107 (38.5%) |
| Death or re-transplantation at 1 year | 42 (14.6%) | 41 (14.7%) |
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Handling and Disposal
Mycophenolate mofetil (MMF) has demonstrated teratogenic effects in humans [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)]. Wearing disposable gloves is recommended during reconstitution and when wiping the outer surface of the bottle/cap and the table after reconstitution. Avoid inhalation or direct contact with skin or mucous membranes of the powder contained in Mycophenolate Mofetil for Injection, USP (during or after preparation) [see Dosage and Administration (2.6)]. Follow applicable special handling and disposal procedures1.
17 PATIENT COUNSELING INFORMATION
Information for Patients
See FDA-approved patient labeling (Medication Guide and Instructions for Use).
17.1 Embryofetal Toxicity
Pregnancy loss and malformations
- Inform females of reproductive potential and pregnant women that use of mycophenolate mofetil during pregnancy is associated with an increased risk of first trimester pregnancy loss and an increased risk of congenital malformations. Advise that they must use an acceptable form of contraception [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
- Encourage pregnant women to enroll in the Pregnancy Exposure Registry. This registry monitors pregnancy outcomes in women exposed to mycophenolate [see Use in Specific Populations (8.1)].
Contraception
- Discuss pregnancy testing, pregnancy prevention and planning with females of reproductive potential [see Use in Specific Populations (8.3)].
- Females of reproductive potential must use an acceptable form of birth control during the entire mycophenolate mofetil therapy and for 6 weeks after stopping mycophenolate mofetil, unless the patient chooses abstinence. Mycophenolate mofetil may reduce effectiveness of oral contraceptives. Use of additional barrier contraceptive methods is recommended [see Use in Specific Populations (8.3)].
- For patients who are considering pregnancy, discuss appropriate alternative immunosuppressants with less potential for embryofetal toxicity. Risks and benefits of mycophenolate mofetil should be discussed with the patient.
- Advise sexually active male patients and/or their partners to use effective contraception during the treatment of the male patient and for at least 90 days after cessation of treatment. This recommendation is based on findings of animal studies [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
17.2 Development of Lymphoma and Other Malignancies
- Inform patients that they are at increased risk of developing lymphomas and other malignancies, particularly of the skin, due to immunosuppression [see Warnings and Precautions (5.2)].
- Advise patients to limit exposure to sunlight and ultraviolet (UV) light by wearing protective clothing and use of broad-spectrum sunscreen with high protection factor.
17.3 Increased Risk of Serious Infections
Inform patients that they are at increased risk of developing a variety of infections due to immunosuppression. Instruct them to contact their physician if they develop any of the signs and symptoms of infection explained in the Medication Guide.
17.4 Blood Dyscrasias
Inform patients that they are at increased risk for developing blood adverse effects such as anemia or low white blood cells. Advise patients to immediately contact their healthcare provider if they experience any evidence of infection, unexpected bruising, or bleeding, or any other manifestation of bone marrow suppression [see Warnings and Precautions (5.4)].
17.5 Gastrointestinal Tract Complications
Inform patients that mycophenolate mofetil can cause gastrointestinal tract complications including bleeding, intestinal perforations, and gastric or duodenal ulcers. Advise the patient to contact their healthcare provider if they have symptoms of gastrointestinal bleeding, or sudden onset or persistent abdominal pain [see Warnings and Precautions (5.5)].
17.6 Immunizations
Inform patients that mycophenolate mofetil can interfere with the usual response to immunizations. Before seeking vaccines on their own, advise patients to discuss first with their physician [see Warnings and Precautions (5.7)].
17.7 Administration Instructions
- Advise patients to take a missed dose as soon as they remember, except if it is closer than 2 hours to the next scheduled dose; in this case they should continue to take mycophenolate mofetil at the usual times.
17.8 Blood Donation
Advise patients not to donate blood during therapy and for at least 6 weeks following discontinuation of mycophenolate mofetil.
17.9 Semen Donation
Advise males of childbearing potential not to donate semen during therapy and for 90 days following discontinuation of mycophenolate mofetil.
17.10 Potential to Impair Driving and Use of Machinery
Advise patients that mycophenolate mofetil can affect the ability to drive or operate machines. Patients should avoid driving or operating machines if they experience somnolence, confusion, dizziness, tremor or hypotension during treatment with mycophenolate mofetil.
Novaplus is a registered trademark of Vizient, Inc.

Manufactured by: Akorn, Inc.
Lake Forest, IL 60045
NMM00N Rev. 02/20
MEDICATION GUIDE
Mycophenolate Mofetil
(my-koh-FEH-noh-layt MOH-feh-til)
for Injection
Read the Medication Guide that comes with mycophenolate mofetil before you start taking it and each time you refill your prescription. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment.
What is the most important information I should know about mycophenolate mofetil? Mycophenolate mofetil can cause serious side effects, including:
Increased risk of loss of a pregnancy (miscarriage) and higher risk of birth defects. Females who take mycophenolate mofetil during pregnancy have a higher risk of miscarriage during the first 3 months (first trimester), and a higher risk that their baby will be born with birth defects.
-
If you are a female who can become pregnant, your doctor must talk with you about acceptable birth control methods (contraceptive counseling) to use while taking mycophenolate mofetil. You should have 1 pregnancy test immediately before starting mycophenolate mofetil and another pregnancy test 8 to 10 days later. Pregnancy tests should be repeated during routine follow-up visits with your doctor. Talk to your doctor about the results of all of your pregnancy tests.
You must use acceptable birth control during your entire mycophenolate mofetil treatment and for 6 weeks after stopping mycophenolate mofetil, unless at any time you choose to avoid sexual intercourse (abstinence) with a man completely. Mycophenolate mofetil decreases blood levels of the hormones in birth control pills that you take by mouth. Birth control pills may not work as well while you take mycophenolate mofetil, and you could become pregnant. If you take birth control pills while using mycophenolate mofetil you must also use another form of birth control. Talk to your doctor about other birth control methods that you can use while taking mycophenolate mofetil. - If you are a sexually active male whose female partner can become pregnant while you are taking mycophenolate mofetil, use effective contraception during treatment and for at least 90 days after stopping mycophenolate mofetil.
- If you plan to become pregnant, talk with your doctor. Your doctor will decide if other medicines to prevent rejection may be right for you.
-
If you become pregnant while taking mycophenolate mofetil, do not stop taking mycophenolate mofetil. Call your doctor right away. You and your doctor may decide that other medicines to prevent rejection may be right for you. You and your doctor should report your pregnancy to the Mycophenolate Pregnancy Registry either:
- By phone at 1-800-617-8191 or
- By visiting the REMS website at: www.mycophenolateREMS.com
The purpose of this registry is to gather information about the health of you and your baby.
Increased risk of getting certain cancers. People who take mycophenolate mofetil have a higher risk of getting lymphoma, and other cancers, especially skin cancer. Tell your doctor if you have:
- unexplained fever, prolonged tiredness, weight loss or lymph node swelling
- a brown or black skin lesion with uneven borders, or one part of the lesion does not look like the other
- a change in the size and color of a mole
- a new skin lesion or bump
- any other changes to your health
Increased risk of getting serious infections. Mycophenolate mofetil weakens the body's immune system and affects your ability to fight infections. Serious infections can happen with mycophenolate mofetil and can lead to hospitalizations and death. These serious infections can include:
-
Viral infections. Certain viruses can live in your body and cause active infections when your immune system is weak. Viral infections that can happen with mycophenolate mofetil include:
- Shingles, other herpes infections, and cytomegalovirus (CMV). CMV can cause serious tissue and blood infections.
- BK virus. BK virus can affect how your kidney works and cause your transplanted kidney to fail.
- Hepatitis B and C viruses. Hepatitis viruses can affect how your liver works. Talk to your doctor about how hepatitis viruses may affect you.
-
A brain infection called Progressive Multifocal Leukoencephalopathy (PML). In some patients, mycophenolate mofetil may cause an infection of the brain that may cause death. You are at risk for this brain infection because you have a weakened immune system. Call your doctor right away if you have any of the following symptoms:
- weakness on one side of the body
- you do not care about things you usually care about (apathy)
- you are confused or have problems thinking
- you cannot control your muscles
- Fungal infections. Yeasts and other types of fungal infections can happen with mycophenolate mofetil and can cause serious tissue and blood infections (See “What are the possible side effects of mycophenolate mofetil?”).
Call your doctor right away if you have any of the following signs and symptoms of infection:
- temperature of 100.5°F or greater
- cold symptoms, such as a runny nose or sore throat
- flu symptoms, such as an upset stomach, stomach pain, vomiting or diarrhea
- earache or headache
- pain during urination
- white patches in the mouth or throat
- unexpected bruising or bleeding
- cuts, scrapes or incisions that are red, warm and oozing pus
See “What are the possible side effects of mycophenolate mofetil?” for information about other serious side effects.
What is mycophenolate mofetil?
- Mycophenolate mofetil is a prescription medicine to prevent rejection (antirejection medicine) in people who have received a kidney, heart or liver transplant. Rejection is when the body's immune system perceives the new organ as a “foreign” threat and attacks it.
- Mycophenolate mofetil is used with other medicines containing cyclosporine and corticosteroids.
Who should not take mycophenolate mofetil?
Do not take mycophenolate mofetil if you are allergic to mycophenolate mofetil or any of the ingredients in mycophenolate mofetil. See the end of this Medication Guide for a complete list of ingredients in mycophenolate mofetil.
What should I tell my doctor before taking mycophenolate mofetil?
Tell your doctor about all of your medical conditions, including if you:
- have any digestive problems, such as ulcers.
- have Lesch-Nyhan syndrome, Kelley-Seegmiller syndrome, or another rare inherited deficiency hypoxanthine-guanine phosphoribosyl-transferase (HGPRT). You should not take mycophenolate mofetil if you have one of these disorders.
- plan to receive any vaccines. People taking mycophenolate mofetil should not receive live vaccines. Some vaccines may not work as well during treatment with mycophenolate mofetil.
- are pregnant or plan to become pregnant. See “What is the most important information I should know about mycophenolate mofetil?”
- are breastfeeding or plan to breastfeed. It is not known if mycophenolate mofetil passes into breast milk. You and your doctor will decide if you will take mycophenolate mofetil or breastfeed.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Some medicines may affect the way mycophenolate mofetil works, and mycophenolate mofetil may affect how some medicines work.
Especially tell your doctor if you take:
- birth control pills (oral contraceptives). See “What is the most important information I should know about mycophenolate mofetil?”
- sevelamer (Renagel®, Renvela™). These products should be taken at least 2 hours after taking mycophenolate mofetil.
- acyclovir (Zovirax®), valacyclovir (Valtrex®), ganciclovir (CYTOVENE®-IV, Vitrasert®), valganciclovir (VALCYTE®).
- rifampin (Rifater®, Rifamate®, Rimactane®, Rifadin®).
- antacids that contain magnesium and aluminum (mycophenolate mofetil and the antacid should not be taken at the same time).
- proton pump inhibitors (PPIs) (Prevacid®, Protonix®).
- sulfamethoxazole/trimethoprim (BACTRIM™, BACTRIM DS™).
- norfloxacin (Noroxin®) and metronidazole (Flagyl®, Flagyl® ER, Flagyl® IV, Metro IV, Helidac®, Pylera™).
- ciprofloxacin (Cipro®, Cipro® XR, Ciloxan®, Proquin® XR) and amoxicillin plus clavulanic acid (Augmentin®, Augmentin XR™).
- azathioprine (Azasan®, Imuran®).
- cholestyramine (Questran Light®, Questran®, Locholest Light, Locholest, Prevalite®).
Know the medicines you take. Keep a list of them to show to your doctor or nurse and pharmacist when you get a new medicine. Do not take any new medicine without talking with your doctor.
How should I take mycophenolate mofetil?
- Take mycophenolate mofetil exactly as prescribed.
- Do not stop taking mycophenolate mofetil or change the dose unless your doctor tells you to.
- If you miss a dose of mycophenolate mofetil, or you are not sure when you took your last dose, take your prescribed dose of mycophenolate mofetil as soon as you remember. If your next dose is less than 2 hours away, skip the missed dose and take your next dose at your normal scheduled time. Do not take 2 doses at the same time. Call your doctor if you are not sure what to do.
- If you take too much mycophenolate mofetil, call your doctor or the poison control center right away.
What should I avoid while taking mycophenolate mofetil?
- Avoid becoming pregnant. See “What is the most important information I should know about mycophenolate mofetil?”
- Limit the amount of time you spend in sunlight. Avoid using tanning beds or sunlamps. People who take mycophenolate mofetil have a higher risk of getting skin cancer (See “What is the most important information I should know about mycophenolate mofetil?”). Wear protective clothing when you are in the sun and use a broad-spectrum sunscreen with a high protection factor. This is especially important if your skin is very fair or if you have a family history of skin cancer.
- You should not donate blood while taking mycophenolate mofetil and for at least 6 weeks after stopping mycophenolate mofetil.
- You should not donate sperm while taking mycophenolate mofetil and for 90 days after stopping mycophenolate mofetil.
- Mycophenolate mofetil may influence your ability to drive and use machines (See “What are the possible side effects of mycophenolate mofetil?”. If you experience drowsiness, confusion, dizziness, tremor, or low blood pressure during treatment with mycophenolate mofetil, you should be cautious about driving or using heavy machines.
What are the possible side effects of mycophenolate mofetil? Mycophenolate mofetil can cause serious side effects, including:
- See “What is the most important information I should know about mycophenolate mofetil?”
-
Low blood cell counts. People taking high doses of mycophenolate mofetil each day may have a decrease in blood counts, including:
- white blood cells, especially neutrophils. Neutrophils fight against bacterial infections. You have a higher chance of getting an infection when your white blood cell count is low. This is most common from 1 month to 6 months after your transplant.
- red blood cells. Red blood cells carry oxygen to your body tissues. You have a higher chance of getting severe anemia when your red blood cell count is low.
- platelets. Platelets help with blood clotting.
Your doctor will do blood tests before you start taking mycophenolate mofetil and during treatment with mycophenolate mofetil to check your blood cell counts. Tell your doctor right away if you have any signs of infection (See “What is the most important information I should know about mycophenolate mofetil?”), including any unexpected bruising or bleeding. Also, tell your doctor if you have unusual tiredness, lack of energy, dizziness or fainting.
- Stomach problems. Stomach problems including intestinal bleeding, a tear in your intestinal wall (perforation) or stomach ulcers can happen in people who take mycophenolate mofetil. Bleeding can be severe and you may have to be hospitalized for treatment. Call your doctor right away if you have sudden or severe stomach-area pain or stomach-area pain that does not go away, or if you have diarrhea.
The most common side effects of mycophenolate mofetil include:
- Diarrhea
- blood problems including low white and red blood cell counts
- infections
- blood pressure problems
- fast heart beat
- swelling of the lower legs, ankles and feet
- changes in laboratory blood levels, including high levels of blood sugar (hyperglycemia)
- stomach problems including diarrhea, constipation, nausea and vomiting
- rash
- nervous system problems such as headache, dizziness and tremor
Side effects that can happen more often in children than in adults taking mycophenolate mofetil include:
|
|
These are not all of the possible side effects of mycophenolate mofetil. Tell your doctor about any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Akorn, Inc. at 1-800-932-5676.
How should I store mycophenolate mofetil?
Keep mycophenolate mofetil and all medicines out of the reach of children.
General Information about the safe and effective use of mycophenolate mofetil.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use mycophenolate mofetil for a condition for which it was not prescribed. Do not give mycophenolate mofetil to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about mycophenolate mofetil. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about mycophenolate mofetil that is written for health professionals.
What are the ingredients in mycophenolate mofetil for injection?
Active Ingredient: mycophenolate mofetil
Inactive Ingredients: polysorbate 80 and citric acid. Sodium hydroxide may have been used in the manufacture of mycophenolate mofetil for injection to adjust the pH.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
All product names, trademarks and registered trademarks are the property of their respective owners.
Novaplus is a registered trademark of Vizient, Inc.

Manufactured by: Akorn, Inc.
Lake Forest, IL 60045
NMM00N Rev. 02/20
For additional copies of this Medication Guide, please call Akorn at 1-800-932-5676 or visit www.akorn.com.
Principal Display Panel Text for Container Label:
NDC 17478-957-40
Mycophenolate
Mofetil for
Injection, USP
500 mg
For Intravenous
Infusion Only
novaplusTM+ Rx ONLY
Principal Display Panel Text for Carton Label:
NDC 17478-957-40
Mycophenolate Mofetil
for Injection, USP
500 mg
For Intravenous
Infusion Only
Attention Pharmacist: Dispense
the accompanying Medication Guide to each
patient. For additional Medication Guides
call 1-800-932-5676 or visit www.akorn.com.
4 Vials
novaplusTM+ Rx ONLY
