FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
OXBRYTA is indicated for the treatment of sickle cell disease (SCD) in adults and pediatric patients 4 years of age and older.
This indication is approved under accelerated approval based on increase in hemoglobin (Hb) [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Adults and Pediatric Patients 12 Years and Older
The recommended dosage of OXBRYTA is 1,500 mg orally once daily.
2.2 Recommended Dosage for Pediatric Patients 4 Years to Less Than 12 Years
For pediatric patients 4 years to less than 12 years, select the appropriate product (OXBRYTA tablets or OXBRYTA tablets for oral suspension) based on patient's ability to swallow tablets and patient's weight.
The recommended dosage of OXBRYTA for pediatric patients 4 years to less than 12 years is shown in Table 1.
| Body Weight | Recommended Dose (once daily) |
|---|---|
|
40 kg or greater |
1,500 mg |
|
20 kg to less than 40 kg |
900 mg |
|
10 kg to less than 20 kg |
600 mg |
2.3 Recommended Dosage for Adults and Pediatric Patients 12 Years and Older with Hepatic Impairment
The recommended dosage of OXBRYTA in adults and pediatric patients 12 years and older with severe hepatic impairment (Child Pugh C) is 1,000 mg orally once daily.
No dosage adjustment of OXBRYTA is required for patients with mild or moderate hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Recommended Dosage for Pediatric Patients 4 Years to Less Than 12 Years with Hepatic Impairment
The recommended dosage of OXBRYTA in pediatric patients 4 years to less than 12 years with severe hepatic impairment (Child Pugh C) is described in Table 2.
No dosage adjustment of OXBRYTA is required for patients with mild or moderate hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
| Body Weight | Recommended Dose (once daily) |
|---|---|
|
40 kg or greater |
1,000 mg (two 500 mg tablets) |
|
20 kg to less than 40 kg |
600 mg |
|
10 kg to less than 20 kg |
300 mg |
2.5 Recommended Dosage of OXBRYTA for Adults and Pediatric Patients 12 Years and Older When Used with Concomitant Strong or Moderate CYP3A4 Inducers
CYP3A4 Inducers
Avoid concomitant use of strong or moderate CYP3A4 inducers with OXBRYTA [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
If concomitant use of strong CYP3A4 inducers is unavoidable, the recommended dosage of OXBRYTA is 2,500 mg orally once daily. If concomitant use of moderate CYP3A4 inducers is unavoidable, the recommended dosage of OXBRYTA is 2,000 mg orally once daily.
2.6 Recommended Dosage of OXBRYTA for Pediatric Patients 4 Years to Less Than 12 Years When Used with Concomitant Strong or Moderate CYP3A4 Inducers
CYP3A4 Inducers
Avoid concomitant use of strong or moderate CYP3A4 inducers with OXBRYTA [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. If concomitant use of strong or moderate CYP3A4 inducers is unavoidable, see Table 3 for dosage.
| Body Weight | Recommended Dose (once daily) | |
|---|---|---|
| Concomitant Use of Strong CYP3A4 Inducers | Concomitant Use of Moderate CYP3A4 Inducers | |
|
40 kg or greater |
2,500 mg (five 500 mg tablets) |
2,000 mg (four 500 mg tablets) |
|
20 kg to less than 40 kg |
1,500 mg |
1,200 mg |
|
10 kg to less than 20 kg |
900 mg |
900 mg |
2.7 Important Administration Instructions
Administer OXBRYTA orally, once daily with or without food. If a dose is missed, or not administered entirely, resume dosing the following day.
OXBRYTA may be given with or without hydroxyurea.
OXBRYTA 300 mg and 500 mg Tablets
Patients should swallow OXBRYTA tablets whole. Do not cut, crush, or chew the tablets.
OXBRYTA 300 mg Tablets for Oral Suspension
Patients should disperse tablets for oral suspension immediately before administration in a cup and in room temperature clear liquid (such as drinking water, clear soda, apple juice, clear electrolyte drinks, clear flavored drinks, or clear sports drinks) before swallowing.
Do not swallow whole, cut, crush, or chew the tablets for oral suspension.
| Recommended Daily Dose | Number of Tablets for Oral Suspension | Minimum Recommended Volume of Clear Drink |
|---|---|---|
|
300 mg |
1 |
5 mL (1 teaspoon) |
|
600 mg |
2 |
10 mL (2 teaspoons) |
|
900 mg |
3 |
15 mL (3 teaspoons) |
|
1,200 mg |
4 |
20 mL (4 teaspoons) |
|
1,500 mg |
5 |
25 mL (5 teaspoons) |
|
2,100 mg |
7 |
35 mL (7 teaspoons) |
|
2,400 mg |
8 |
40 mL (8 teaspoons) |
- •
- After the tablets start to disintegrate, swirl the contents of the cup until the tablets are dispersed, wait 1 to 5 minutes, swirl the contents of the cup again, and then orally administer the contents of the cup. The tablet(s) will not completely dissolve; there will still be small tablet clumps in the mixture.
- •
- Resuspend any residue left in the cup in more clear drink and administer. Repeat until no tablet residue is left in the cup.
Tablets for oral suspension may be substituted for tablets in adults and pediatric patients 12 years and older with difficulty swallowing the tablets. Use the number of tablets for oral suspension needed to achieve the recommended dose.
3 DOSAGE FORMS AND STRENGTHS
Tablets: 300 mg light purple to purple, oval shaped, biconvex, debossed with "G 300" on one side.
Tablets: 500 mg light yellow to yellow, oval shaped, biconvex, debossed with "GBT 500" on one side.
Tablets for oral suspension: 300 mg light yellow to yellow, round shaped, debossed with "300 D" on one side.
4 CONTRAINDICATIONS
OXBRYTA is contraindicated in patients with a history of serious drug hypersensitivity reaction to voxelotor or excipients. Clinical manifestations may include generalized rash, urticaria, mild shortness of breath, mild facial swelling, and eosinophilia [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions after administration of OXBRYTA have occurred in <1% of patients treated. Clinical manifestations may include generalized rash, urticaria, mild shortness of breath, mild facial swelling, and eosinophilia [see Adverse Reactions (6.1)].
Drug reaction with eosinophilia and systemic symptoms (DRESS) has been reported in postmarketing experience with OXBRYTA [see Adverse Reactions (6.2)]. Patients who develop a combination of skin rash, fever, peripheral eosinophilia, and internal systemic organ involvement (e.g., hepatic, renal, pulmonary) while receiving OXBRYTA should undergo medical evaluation.
Advise patients of the signs and symptoms of severe hypersensitivity reactions, including DRESS. If hypersensitivity reactions occur, discontinue OXBRYTA and administer appropriate medical therapy. Do not reinitiate OXBRYTA in patients who experience these symptoms with previous use.
5.2 Laboratory Test Interference
OXBRYTA administration may interfere with measurement of Hb subtypes (HbA, HbS, and HbF) by high-performance liquid chromatography (HPLC) [see Drug Interactions (7.3)]. If precise quantitation of Hb species is required, chromatography should be performed when the patient has not received OXBRYTA therapy in the immediately preceding 10 days.
6 ADVERSE REACTIONS
The following clinically significant adverse reaction is discussed in other sections of the labeling:
- •
- Hypersensitivity Reactions [see Contraindications (4) and Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adults and Pediatric Patients 12 Years and Older
The safety of OXBRYTA was evaluated in the HOPE trial based on data from 88 patients with SCD who received OXBRYTA 1,500 mg and 91 patients who received placebo orally once daily [see Clinical Studies (14.1)]. Seventy-four patients received OXBRYTA 1,500 mg once daily for ≥24 weeks, 65 patients for ≥48 weeks, and 63 patients completed the 72-week treatment period.
In patients who received OXBRYTA 1,500 mg once daily the median age was 24 years (range:12 to 59 years); 65% female; 66% Black or African American and 23% Arab/Middle Eastern; and 65% receiving hydroxyurea at baseline.
Serious adverse reactions occurred in 3% (3/88) of patients receiving OXBRYTA 1,500 mg, which included headache, drug hypersensitivity, and pulmonary embolism occurring in 1 patient each. Permanent discontinuation due to an adverse reaction (Grades 1-4) occurred in 5% (4/88) of patients who received OXBRYTA 1,500 mg.
Dosage modifications (dose reduction or dosing interruption) due to adverse reactions occurred in 14% (12/88) of patients who received OXBRYTA 1,500 mg. The adverse reactions requiring dosage modification included rash (4.5%), diarrhea (3.4%), headache (2.3%), nausea (2.3%), abdominal pain (1.1%), and drug hypersensitivity (1.1%).
The safety profile observed in pediatric patients 12 to <17 years treated with OXBRYTA in the HOPE trial was similar to that seen in adult patients.
The most common adverse reactions occurring in ≥10% of patients treated with OXBRYTA 1,500 mg with a difference of >3% compared to placebo are summarized in Table 4.
| Adverse Reaction* | OXBRYTA
1,500 mg (N=88) | Placebo
(N=91) |
|---|---|---|
|
||
|
Headache |
32% |
25% |
|
Diarrhea |
23% |
11% |
|
Abdominal Pain† |
23% |
16% |
|
Nausea |
19% |
10% |
|
Rash‡ |
15% |
11% |
|
Pyrexia |
15% |
8% |
Clinically relevant adverse reactions occurring in <10% of patients included:
- •
- Drug hypersensitivity
Pediatric Patients 4 to <12 Years
The safety of OXBRYTA in pediatric patients 4 to <12 years with SCD was evaluated in an open-label, Phase 2 study [see Clinical Studies (14.2)]. In this study, 45 patients 4 to <12 years of age received doses of OXBRYTA tablets for oral suspension based on weight at baseline. Thirty-five patients received OXBRYTA for 24 weeks and 26 patients for 48 weeks. The most common adverse reactions (>10%) reported in pediatric patients 4 to <12 years were pyrexia (36%), vomiting (33%), rash (20%), abdominal pain (18%), diarrhea (18%), and headache (18%).
The overall safety profile of OXBRYTA in pediatric patients 4 to <12 years was similar to that seen in adults and pediatric patients 12 years and older.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of OXBRYTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- •
- Drug reaction with eosinophilia and systemic symptoms (DRESS), Pruritis, Angioedema (including swelling of eyelid, face edema, lip swelling, and periorbital swelling).
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Voxelotor
Strong or Moderate CYP3A4 Inducers
Coadministration of strong or moderate CYP3A4 inducers may decrease voxelotor plasma and whole blood concentrations and may lead to reduced efficacy.
Avoid coadministration of OXBRYTA with strong or moderate CYP3A4 inducers. Increase the OXBRYTA dosage when coadministration with a strong or moderate CYP3A4 inducer is unavoidable [see Dosage and Administration (2.5, 2.6) and Clinical Pharmacology (12.3)].
7.2 Effect of Voxelotor on Other Drugs
Voxelotor increased the systemic exposure of midazolam (a sensitive CYP3A4 substrate) [see Clinical Pharmacology (12.3)]. Avoid coadministration of OXBRYTA with sensitive CYP3A4 substrates with a narrow therapeutic index. If concomitant use is unavoidable, consider dose reduction of the sensitive CYP3A4 substrate(s).
7.3 Laboratory Test Interference
OXBRYTA administration may interfere with measurement of Hb subtypes (HbA, HbS, and HbF) by HPLC [see Warnings and Precautions (5.2)]. If precise quantitation of Hb species is required, chromatography should be performed when the patient has not received OXBRYTA therapy in the immediately preceding 10 days.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on OXBRYTA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, oral administration of voxelotor to pregnant rats and rabbits during organogenesis at exposures up to 2.8-times (rats) and 0.3-times (rabbits) the exposure at the maximum recommended human dose resulted in no adverse developmental effects (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is approximately 14% and up to 43%, respectively. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes.
There are adverse effects on maternal and fetal outcomes associated with SCD in pregnancy (see Clinical Considerations). OXBRYTA should only be used during pregnancy if the benefit of the drug outweighs the potential risk.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Women with SCD have an increased risk of adverse pregnancy outcomes for the mother and the fetus. Pregnant women are at greater risk for vasoocclusive crises, pre-eclampsia, eclampsia, and maternal mortality. For the fetus, there is an increased risk for intrauterine growth restriction, preterm delivery, low birth weight, and perinatal mortality.
Animal Data
In embryo-fetal development studies, voxelotor was administered orally to pregnant rats at 15, 50, and 250 mg/kg/day (gestation days 7 through 17) and rabbits at 25, 75, and 150 mg/kg/day (gestation days 7 through 19) through organogenesis. Maternal toxicity was observed at the highest dose levels in these studies equivalent to 2.8-times (rats) and 0.3-times (rabbits) the exposures in patients receiving OXBRYTA at the recommended daily dose. There was no evidence of adverse developmental outcomes in rats or rabbits.
In a pre- and postnatal development study, voxelotor was administered orally to pregnant rats at 15, 50 and 250 mg/kg/day (gestation day 6 through lactation day 20). Maternal gestational body weights were decreased at 250 mg/kg/day, which continued to the end of lactation. The findings in offspring included reduced survival and reduced body weights throughout lactation, weaning and maturation. The effects in offspring were observed at the maternal dose of 250 mg/kg/day with an exposure approximately 2.8-times the exposure in patients at the recommended dose.
8.2 Lactation
Risk Summary
There are no data on the presence of voxelotor in human milk, the effects on the breastfed child, or the effects on milk production. Voxelotor was detected in milk in lactating rats. Plasma concentrations of voxelotor in pregnant rats were higher than the concentration in milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The concentration of voxelotor in animal milk does not necessarily predict the concentration of drug in human milk. Because of the potential for serious adverse reactions in the breastfed child, including changes in the hematopoietic system, advise patients that breastfeeding is not recommended during treatment with OXBRYTA, and for at least 2 weeks after the last dose.
8.4 Pediatric Use
The safety and effectiveness of OXBRYTA for SCD have been established in pediatric patients aged 4 years and older. The safety and efficacy of OXBRYTA in pediatric patients with SCD below the age of 4 years have not been established.
Use of OXBRYTA in pediatric patients 12 to <17 years for SCD is supported by evidence from an adequate and well-controlled study in adults and pediatric patients (HOPE trial). The HOPE trial enrolled 26 pediatric patients aged 12 to <17 years, in which 12 pediatric patients received OXBRYTA 1,500 mg once daily and 14 pediatric patients received OXBRYTA 900 mg once daily [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
Use of OXBRYTA in pediatric patients 4 to <12 years for SCD is supported by evidence from an open-label, Phase 2 study. The study enrolled 45 pediatric patients aged 4 to <12 years and 11 patients aged 12 to <17 years with SCD. Patients 12 to <17 years received OXBRYTA 1,500 mg once daily. Patients 4 to <12 years were administered OXBRYTA based on body weight. OXBRYTA doses of 600 mg, 900 mg, or 1,500 mg once daily were administered to patients weighing 10 kg to <20 kg, 20 kg to <40 kg, or ≥40 kg, respectively [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.2)].
Pharmacokinetics, safety and efficacy were similar across the pediatric age groups and across pediatric and adult patients [see Dosage and Administration (2), Clinical Pharmacology (12.3) and Clinical Studies (14)].
The adverse reactions observed were similar across the pediatric age groups and across pediatric and adult patients [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Clinical studies of OXBRYTA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
Severe hepatic impairment increases voxelotor exposures [see Clinical Pharmacology (12.3)]. Reduce OXBRYTA dose [see Dosage and Administration (2.3, 2.4)].
11 DESCRIPTION
OXBRYTA contains voxelotor, a hemoglobin S polymerization inhibitor. The chemical name of voxelotor is 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde with a molecular formula of C19H19N3O3 and a molecular weight of 337.4.
The chemical structure of voxelotor is:
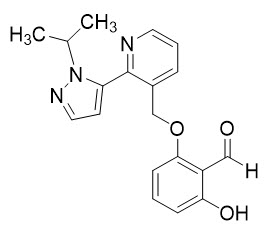
Voxelotor is a white-to-yellow-to-beige compound in crystalline Form II of its free base. It is non-hygroscopic and highly soluble in common organic solvents such as acetone and toluene and insoluble in water.
OXBRYTA film-coated tablets for oral use contain either 300 mg or 500 mg of voxelotor. Both strengths of OXBRYTA film-coated tablets contain the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. In addition, the 500 mg tablet film coating contains: polyethylene glycol 3350, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide. The 300 mg tablet film coating contains: black and red iron oxide, polyethylene glycol 3350, polyvinyl alcohol, talc, and titanium dioxide.
Each OXBRYTA tablet for oral suspension contains 300 mg of voxelotor with the following inactive ingredients: artificial grape flavor, colloidal silicon dioxide, croscarmellose sodium, iron oxide pigment, magnesium stearate, microcrystalline cellulose, and sucralose.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Voxelotor is a hemoglobin S (HbS) polymerization inhibitor that binds to HbS with a 1:1 stoichiometry and exhibits preferential partitioning to red blood cells (RBCs). By increasing the affinity of Hb for oxygen, voxelotor demonstrates dose-dependent inhibition of HbS polymerization. Nonclinical studies suggest that voxelotor may inhibit RBC sickling, improve RBC deformability, and reduce whole blood viscosity.
12.2 Pharmacodynamics
The pharmacodynamic effect of voxelotor treatment demonstrated a dose-dependent increase in Hb oxygen affinity as determined by the change in p50 (partial pressure of oxygen at which Hb oxygen saturation of 50% is achieved) that was linearly correlated with voxelotor exposure.
The pharmacodynamic effect of voxelotor treatment also demonstrated a dose-dependent reduction in clinical measures of hemolysis (indirect bilirubin and % reticulocytes).
12.3 Pharmacokinetics
Voxelotor is absorbed into plasma and is then distributed predominantly into RBCs due to its preferential binding to Hb. The major route of elimination of voxelotor is by metabolism with subsequent excretion of metabolites into urine and feces. The PK are linear and voxelotor exposures increased proportionally with either single or multiple doses (Table 5) in whole blood, plasma, and RBCs. Steady-state after repeated administration is reached within 8 days and exposures of voxelotor are consistent with accumulation predicted based on single dose data in patients with SCD.
| PK Parameter | Voxelotor 1,500 mg
Geometric Mean (%CV) |
|---|---|
|
|
|
Plasma PK |
|
|
AUC0-24h (μg∙hr/mL) |
278 (28.4) |
|
Cmax (µg/mL) |
14 (24.5) |
|
Half-life (hours) |
38.7 (30.2) |
|
Whole Blood PK |
|
|
AUC0-24h (μg∙hr/mL) |
3,830 (33.5) |
|
Cmax (µg/mL) |
180 (31) |
In healthy subjects, voxelotor exposures were comparable when administered as tablet for oral suspension dispersed in water or as oral tablet swallowed whole.
Absorption
The median plasma and whole blood Tmax of voxelotor after oral administration is 2 hours. The mean peak concentrations in whole blood and RBCs are observed between 6 and 18 hours after oral administration.
Effect of Food
A high-fat, high-calorie meal increased voxelotor AUC by 42% and Cmax by 45% in whole blood relative to AUC and Cmax in the fasted state. Similarly, AUC increased by 42% and Cmax increased by 95% in plasma.
Distribution
Voxelotor apparent volume of distribution of the central compartment and peripheral compartment are 333 L and 72.3 L in plasma, respectively. Protein binding is 99.8% in vitro. The blood-to-plasma ratio is approximately 17:1 in patients with SCD.
Elimination
The geometric mean (%CV) terminal elimination half-life of voxelotor in patients with SCD is 38.7 hours (30.2%) with concentrations in plasma, whole blood, and RBCs declining in parallel. The apparent oral clearance of voxelotor was estimated as 6.1 L/h in plasma in patients with SCD.
Metabolism
In vitro and in vivo studies indicate that voxelotor is extensively metabolized through Phase I (oxidation and reduction), Phase II (glucuronidation) and combinations of Phase I and II metabolism. Metabolism of voxelotor is mediated by CYP3A4, CYP3A5, CYP2B6, CYP2C19, CYP2C9, UGT1A1, and UGT1A9.
Excretion
Following the administration of radiolabeled voxelotor, approximately 62.6% of the dose and its metabolites are excreted into feces (33.3% unchanged) and 35.5% in urine (0.08% unchanged).
Specific Populations
No clinically significant differences in the pharmacokinetics of voxelotor were observed based on age (12 to 59 years), sex, body weight (28 to 135 kg), or mild to severe renal impairment (creatinine clearance [CLcr] 15-89 mL/min).
Pediatric Patients
The pharmacokinetic exposures of voxelotor in whole blood and plasma were similar between pediatric patients 4 to <17 years and adults following the recommended dosage [see Dosage and Administration (2)].
Patients with Renal Impairment
There was no clinically significant effect of renal function on the excretion of voxelotor. Following a single 900 mg dose of voxelotor, whole blood exposures in subjects with severe renal impairment (eGFR <30 mL/min/1.73 m2) were 25% lower compared to healthy controls.
The unbound plasma concentrations were comparable. OXBRYTA has not been evaluated in patients with end stage renal disease requiring dialysis.
Patients with Hepatic Impairment
The voxelotor AUC in whole blood were 14% and 15% higher in subjects with mild and moderate hepatic impairment (Child Pugh A and B) and 90% higher in subjects with severe hepatic impairment (Child Pugh C) compared to subjects with normal hepatic function.
Patients with HbSC Genotype
Voxelotor steady state whole blood AUC and Cmax were 50% and 45% higher in HbSC genotype patients (n=11) compared to HbSS genotype (n=220) patients and voxelotor steady state plasma AUC and Cmax were 23% and 15% higher in HbSC genotype patients compared to HbSS genotype patients.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Strong CYP3A4 Inhibitors on Voxelotor: concomitant use of OXBRYTA with itraconazole increased voxelotor AUC in healthy subjects by 11%.
Effect of Strong or Moderate CYP3A4 Inducers on Voxelotor: concomitant use of OXBRYTA with rifampin (a strong CYP3A4 inducer) is predicted to decrease voxelotor AUC in patients by up to 40%, and efavirenz (a moderate CYP3A4 inducer) is predicted to decrease voxelotor AUC in patients by up to 24%.
Effect of Acid Reducing Agents on Voxelotor: coadministration of omeprazole (proton pump inhibitor) with OXBRYTA did not alter voxelotor exposure.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Voxelotor was not carcinogenic in a 26-week study in RasH2 transgenic mice at oral doses of 30, 150, or 500 mg/kg/day.
Voxelotor was not genotoxic in the reverse mutation bacterial (Ames) test, rat Comet assay, or rat micronucleus assay.
In a fertility and early embryonic development study, voxelotor was administered orally to rats at 15, 50, and 250 mg/kg/day. Males were dosed 28 days prior to mating through cohabitation and females were dosed 14 days prior to mating through gestation Day 7. Voxelotor had no effect on fertility or reproductive function. Sperm motility was decreased and changes in sperm morphology occurred at 250 mg/kg/day (approximately 5-times the human exposure at 1,500 mg/day).
14 CLINICAL STUDIES
14.1 Adults and Pediatric Patients 12 Years and Older
The efficacy and safety of OXBRYTA in SCD was evaluated in HOPE, a Phase 3 randomized, double-blind, placebo-controlled, multicenter trial [NCT 03036813]. In this study, 274 patients were randomized to daily oral administration of OXBRYTA 1,500 mg (N=90), OXBRYTA 900 mg (N=92), or placebo (N=92). Patients were included if they had from 1 to 10 vasoocclusive crisis (VOC) events within 12 months prior to enrollment and baseline hemoglobin (Hb) ≥5.5 to ≤10.5 g/dL. Eligible patients on stable doses of hydroxyurea for at least 90 days were allowed to continue hydroxyurea therapy throughout the study. Randomization was stratified by patients already receiving hydroxyurea (yes, no), geographic region (North America, Europe, Other), and age (12 to <17 years, 18 to 65 years). The trial excluded patients who received red blood cell (RBC) transfusions within 60 days and erythropoietin within 28 days of enrollment, had renal insufficiency, uncontrolled liver disease, were pregnant, or lactating.
The majority of patients had HbSS or HbS/beta0-thalassemia genotype (90%) and were receiving background hydroxyurea therapy (65%). The median age was 24 years (range: 12 to 64 years); 46 (17%) patients were 12 to <17 years. Median baseline Hb was 8.5 g/dL (5.9 to 10.8 g/dL). One hundred and fifteen (42%) had 1 VOC event and 159 (58%) had 2 to 10 events within 12 months prior to enrollment. In the OXBRYTA 1,500 mg group, 63 (70%) patients completed the study through Week 72.
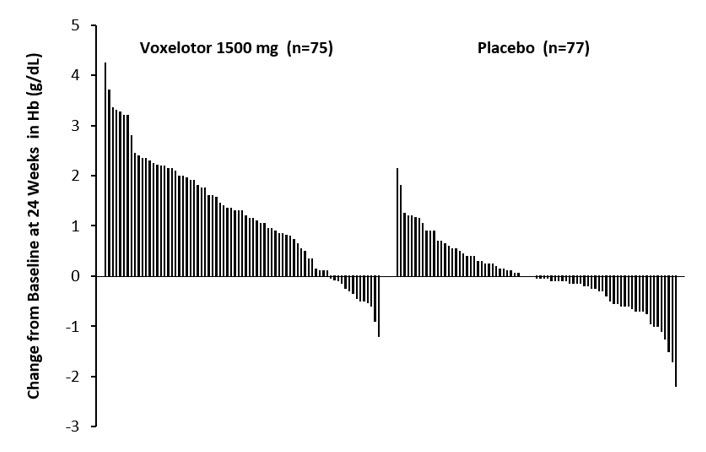
Efficacy was based on Hb response rate defined as a Hb increase of >1 g/dL from baseline to Week 24 in patients treated with OXBRYTA 1,500 mg versus placebo. The response rate for OXBRYTA 1,500 mg was 51.1% (46/90) compared to 6.5% (6/92) in the placebo group (p < 0.001). No outlier subgroups were observed. The distribution of Hb change from baseline for individual patients completing 24 weeks of treatment with OXBRYTA 1,500 mg or placebo is depicted in Figure 1.
|
|
Figure 1: Subject-level Change from Baseline in Hemoglobin at Week 24 in Patients Who Completed 24 Weeks of Treatment* |
 |
Additional efficacy evaluation included change in Hb and percent change in indirect bilirubin and percent reticulocyte count from baseline to Week 24 (Table 6).
| OXBRYTA 1,500 mg QD
(N=90) | Placebo
(N=92) | P Value | |
|---|---|---|---|
| QD = once daily; SE = standard error | |||
|
Hemoglobin |
1.1 g/dL |
-0.1 g/dL |
< 0.001 |
|
Indirect Bilirubin |
-29.1% |
-2.8% |
< 0.001 |
|
Percent Reticulocyte Count |
-18.0% |
6.8% |
< 0.001 |
14.2 Pediatric Patients 4 to <12 Years
The efficacy and safety of OXBRYTA in patients 4 to <12 years with SCD was evaluated in an open-label, multi-center, Phase 2 trial [NCT 02850406]. In this study, 45 patients 4 to <12 years and 11 patients 12 to <17 years received OXBRYTA. Patients 4 to <12 years received tablets for oral suspension based on body weight at baseline. OXBRYTA doses of 600 mg, 900 mg, or 1,500 mg once daily were administered to patients weighing 10 kg to <20 kg, 20 kg to <40 kg, or ≥40 kg, respectively. Patients 12 to <17 years received OXBRYTA 1,500 mg once daily.
Patients were included if their baseline hemoglobin (Hb) was ≤10.5 g/dL. Eligible patients on stable doses of hydroxyurea for at least 90 days were allowed to continue hydroxyurea therapy throughout the study. The trial excluded patients who had a VOC event within 14 days prior to enrollment, received red blood cell (RBC) transfusions within 30 days of enrollment, and had renal insufficiency or uncontrolled liver disease.
All patients had HbSS or HbS/beta0-thalassemia genotype (100%) and most were receiving background hydroxyurea therapy (80%). The median age was 8 years (range: 4 to 15 years); 45 (80%) patients were 4 to <12 years. In this age group, mean baseline Hb was 8.6 g/dL (range: 6.1 to 10.5 g/dL).
Efficacy was based on Hb response rate, which is defined as a Hb increase of >1 g/dL from baseline to Week 24. Hb response rate for OXBRYTA in patients aged 4 to <12 years who took at least one dose of OXBRYTA was 36% (16/45) (95% CI: 21.6%, 49.5%).
16 HOW SUPPLIED/STORAGE AND HANDLING
The 300 mg tablet is film-coated, light purple to purple, oval shaped, biconvex, debossed with "G 300" on one side, available in:
- •
- Bottles of 60 tablets with one desiccant canister, a polyester coil and child-resistant closure: NDC 72786-102-02
- •
- Bottles of 90 tablets with one desiccant canister, a polyester coil and child-resistant closure: NDC 72786-102-03
The 500 mg tablet is film-coated, light yellow to yellow, oval shaped, biconvex, debossed with "GBT 500" on one side, and available in:
- •
- Bottles of 90 tablets with one desiccant canister, a polyester coil and child-resistant closure: NDC 72786-101-01
The 300 mg tablet for oral suspension is light yellow to yellow, round shaped, debossed with "300 D" on one side, and available in:
- •
- Bottles of 60 tablets for oral suspension with a polyester coil and child-resistant closure: NDC 72786-111-02
- •
- Bottles of 90 tablets for oral suspension with a polyester coil and child-resistant closure: NDC 72786-111-03
Do not eat the desiccant canister or the polyester coil.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Hypersensitivity Reactions
Advise patients that serious hypersensitivity reactions including DRESS may occur, and to notify their healthcare providers if they develop generalized rash, urticaria, shortness of breath, facial swelling, swelling around their eyes, lips, or tongue, fever, fatigue, muscle and/or joint pain, swollen glands, and eosinophilia [see Warnings and Precautions (5.1)]. Inform patients that some severe hypersensitivity reactions may affect their internal organs (such as liver, kidneys, lungs) and their blood cells. Advise patients to stop OXBRYTA if a severe hypersensitivity reaction is suspected.
Breastfeeding
Advise women not to breastfeed while they are on OXBRYTA therapy [see Use in Specific Populations (8.2)].
Dosage and Administration
To avoid a dosing error from using the wrong formulation of OXBRYTA, strongly advise patients and caregivers to visually inspect the tablets to verify the correct formulation each time the prescription is filled [see Dosage and Administration (2) and How Supplied/Storage and Handling (16)].
Advise patients to:
- •
- Store OXBRYTA at 20°C to 30°C (68°F to 86°F).
- •
- Continue taking OXBRYTA every day for as long as their physician tells them.
- •
- Do not take St John's wort while taking OXBRYTA.
- •
- Swallow OXBRYTA tablets whole. Do not cut, crush, or chew the tablets.
- •
- Do not swallow whole, cut, crush, or chew OXBRYTA tablets for oral suspension. Disperse OXBRYTA tablets for oral suspension in room temperature clear drink (such as drinking water, clear soda, apple juice, clear electrolyte drinks, clear flavored drinks, or clear sports drinks) before administration. The amount of liquid needed to disperse the tablets for oral suspension will depend on the dose (number of tablets prescribed) [see Dosage and Administration (2.7)].
- •
- Take OXBRYTA with or without food.
- •
- If a dose is missed or not consumed entirely, resume dosing the following day [see Dosage and Administration (2.7)].
This product's label may have been updated. For most recent prescribing information, please visit www.pfizer.com. For medical information about OXBRYTA, please visit www.pfizermedinfo.com or call 1-800-438-1985.

Distributed by
Global Blood Therapeutics, Inc
A subsidiary of Pfizer Inc.
South San Francisco, CA 94080
LAB-1554-1.0
|
This Patient Information has been approved by the U.S. Food and Drug Administration |
Revised: 08/2023 |
|||||
|
PATIENT INFORMATION |
||||||
|
OXBRYTA® (ox brye ta)
|
|
OXBRYTA® (ox brye ta)
| ||||
|
What is OXBRYTA?
|
||||||
|
Do not take OXBRYTA if you or your child have had an allergic reaction to voxelotor or any of the ingredients in OXBRYTA. See the end of this leaflet for a list of the ingredients in OXBRYTA. |
||||||
|
Before taking OXBRYTA, tell your healthcare provider about all of your medical conditions, including if you or your child:
Tell your healthcare provider about all the medicines you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how OXBRYTA works. OXBRYTA may also affect how other medicines work and may affect the results of certain blood tests. Keep a list of all your medicines and show it to your healthcare provider. |
||||||
|
How should I take OXBRYTA?
|
||||||
|
What should I avoid while taking OXBRYTA?
|
||||||
|
What are the possible side effects of OXBRYTA?
|
||||||
|
|
|||||
|
The most common side effects of OXBRYTA include: |
||||||
|
|
|||||
|
The most common side effects of OXBRYTA in children ages 4 to less than 12 years of age include: |
||||||
|
|
|||||
|
These are not all the possible side effects of OXBRYTA. |
||||||
|
How should I store OXBRYTA?
Keep OXBRYTA and all medicines out of the reach of children. |
||||||
|
General information about the safe and effective use of OXBRYTA.
|
||||||
|
What are the ingredients of OXBRYTA?
Distributed by Global Blood Therapeutics, Inc A subsidiary of Pfizer Inc. South San Francisco, CA 94080 For more information, go to www.pfizer.com or call 1-800-438-1985. |
||||||

|
This Instructions for Use has been approved by the U.S. Food and Drug Administration. |
Revised: 08/2023 |
|||
|
INSTRUCTIONS FOR USE
|
||||
|
This Instructions for Use contains information on how to take OXBRYTA tablets for oral suspension. |
||||
|
Important Information You Need to Know Before Taking OXBRYTA Tablets for Oral Suspension:
|
||||
|
Gather supplies |
||||
|
You will need the following items to prepare the dose of OXBRYTA tablets for oral suspension (not included with OXBRYTA tablets for oral suspension):
You will also need:
|  |
|||
|
Preparing a dose of OXBRYTA tablets for oral suspension |
||||
|
Step 1. |
Wash and dry your hands well before preparing the dose. |
|||
|
Step 2. |
Pour room temperature clear drink into the cup. The table below shows the amount of clear drink needed for your prescribed dose. You may add more clear drink if needed to mix the tablets. | 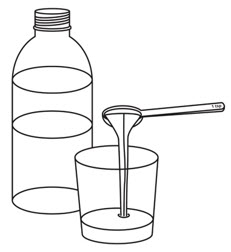 |
||
|
Number of OXBRYTA Tablets for Oral Suspension |
Amount of Clear Drink | |||
|
1 |
1 teaspoon (5 mL) | |||
|
2 |
2 teaspoons (10 mL) | |||
|
3 |
3 teaspoons (15 mL) | |||
|
4 |
4 teaspoons (20 mL) | |||
|
5 |
5 teaspoons (25 mL) | |||
|
7 |
7 teaspoons (35 mL) | |||
|
8 |
8 teaspoons (40 mL) | |||
|
|
||||
|
Step 3. |
Add the prescribed number of OXBRYTA tablets for oral suspension into the cup. | 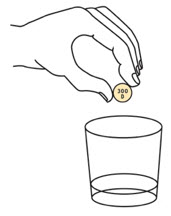 |
||
|
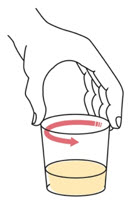
Step 4. |
Swirl the cup until the tablet(s) break apart (disperse) in the drink. Be careful not to spill the mixture.
|  |
||
|
Step 5. |
Wait for 1 to 5 minutes. |  |
||
|
Giving the dose |
||||
|
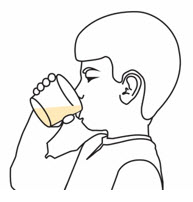
Step 6. |
Swirl the cup again. Take or give all of the prepared medicine right away.
|  |
||
|
Step 7. |
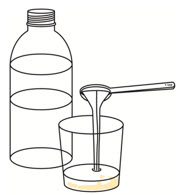
Add 1 or 2 teaspoons of room temperature clear drink to the cup to make sure the full dose is taken. Swirl the cup until the remaining medicine is mixed and take or give it right away.
|  |
||
|
Step 8. |
Wash the teaspoon and cup with warm soap and water. |
|||
|
Storing OXBRYTA
Keep OXBRYTA and all medicines out of the reach of children. |
||||
|
Disposing of OXBRYTA
 Distributed by Global Blood Therapeutics, Inc A subsidiary of Pfizer Inc. South San Francisco, CA 94080 For more information, go to www.pfizer.com or call 1-800-438-1985. |
||||
PRINCIPAL DISPLAY PANEL - 500 mg Tablet Bottle Label
NDC 72786-101-01
Pfizer
Oxbryta®
(voxelotor)
tablets
500 mg
Swallow tablets whole.
Do not cut, crush, or chew the tablets.
90 tablets
Rx only