FULL PRESCRIBING INFORMATION
WARNING: SERIOUS OR FATAL RESPIRATORY FAILURE
TERLIVAZ may cause serious or fatal respiratory failure. Patients with volume overload or with acute-on-chronic liver failure (ACLF) Grade 3 are at increased risk [see References (15)]. Assess oxygenation saturation (e.g., SpO2) before initiating TERLIVAZ.
Do not initiate TERLIVAZ in patients experiencing hypoxia (e.g., SpO2 <90%) until oxygenation levels improve. Monitor patients for hypoxia using continuous pulse oximetry during treatment and discontinue TERLIVAZ if SpO2 decreases below 90% [see Dosage and Administration (2.1), Contraindications (4), and Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
TERLIVAZ is indicated to improve kidney function in adults with hepatorenal syndrome with rapid reduction in kidney function.
2 DOSAGE AND ADMINISTRATION
2.1 Important Considerations Prior to Initiating and During Therapy
Obtain baseline oxygen saturation (SpO2) prior to administering the first dose of TERLIVAZ. During treatment, monitor patient oxygen saturation using continuous pulse oximetry. Do not use TERLIVAZ treatment in patients experiencing hypoxia until hypoxia resolves [see Contraindications (4), and Warnings and Precautions (5.1)].
Assess Acute-on-Chronic Liver Failure (ACLF) Grade and volume status before initiating TERLIVAZ [see Warnings and Precautions (5.1) and References (15)].
2.2 Recommended Dosage
Record last available serum creatinine (SCr) value prior to initiating treatment (baseline SCr). The recommended starting dosage is TERLIVAZ 0.85 mg every 6 hours by slow intravenous bolus injection (over 2 minutes) on Days 1 through 3. Adjust the dose on Day 4 based on changes from baseline SCr using the dosing chart (Figure 1).
Figure 1: Dosing Chart
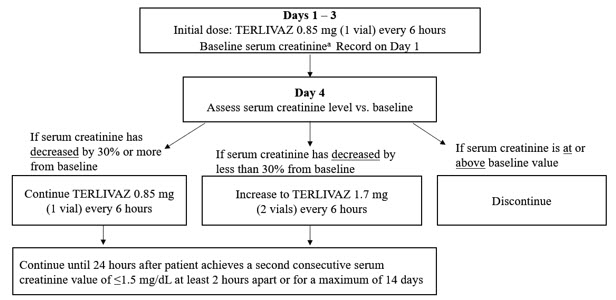
a Baseline SCr is the last available serum creatinine before initiating treatment.
2.3 Preparation and Administration
Reconstitute each vial with 5 mL of 0.9% Sodium Chloride Injection to prepare a 0.85 mg/5 mL solution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Administer TERLIVAZ through a peripheral or central line. A dedicated central line is not required. Flush the line after TERLIVAZ administration.
If not administered immediately, store TERLIVAZ at 2°C to 8°C (36°F to 46°F) for up to 48 hours. Do not freeze. The reconstituted solution does not need protection from light.
3 DOSAGE FORMS AND STRENGTHS
For injection: TERLIVAZ 0.85 mg is a white to off-white lyophilized powder in a single-dose vial for reconstitution.
4 CONTRAINDICATIONS
TERLIVAZ is contraindicated in patients experiencing hypoxia or worsening respiratory symptoms.
TERLIVAZ is contraindicated in patients with ongoing coronary, peripheral or mesenteric ischemia.
5 WARNINGS AND PRECAUTIONS
5.1 Serious or Fatal Respiratory Failure
In the primary clinical trial [see Clinical Studies (14)], serious or fatal respiratory failure occurred in 14% of patients treated with TERLIVAZ compared to 5% of patients on placebo.
Obtain baseline oxygen saturation and do not initiate TERLIVAZ in hypoxic patients [see Contraindications (4)]. Monitor patients for changes in respiratory status using continuous pulse oximetry and regular clinical assessments. Discontinue TERLIVAZ in patients experiencing hypoxia or increased respiratory symptoms.
Patients with fluid overload may be at increased risk of respiratory failure. Manage intravascular volume overload by reducing or discontinuing the administration of albumin and/or other fluids and judicious use of diuretics. Temporarily interrupt, reduce, or discontinue TERLIVAZ treatment until patient volume status improves [see Dosage and Administration (2.1)].
Avoid use in patients with ACLF Grade 3 because they are at significant risk for respiratory failure [see References (15)].
5.2 Ineligibility for Liver Transplant
TERLIVAZ-related adverse reactions (respiratory failure, ischemia) may make a patient ineligible for liver transplantation, if listed. For patients with high prioritization for liver transplantation (e.g., MELD ≥ 35), the benefits of TERLIVAZ may not outweigh its risks [see Adverse Reactions (6.1)].
5.3 Ischemic Events
TERLIVAZ may cause cardiac, cerebrovascular, peripheral, or mesenteric ischemia. Avoid use of TERLIVAZ in patients with a history of severe cardiovascular conditions, cerebrovascular and ischemic disease. Discontinue TERLIVAZ in patients who experience signs or symptoms suggestive of ischemic adverse reactions [see Dosage and Administration (2.1) and Adverse Reactions (6.1)].
5.4 Embryo-Fetal Toxicity
TERLIVAZ may cause fetal harm when administered to a pregnant woman based on the mechanism of action and data from published literature. Terlipressin induces uterine contractions and endometrial ischemia in both humans and animals. If this drug is used during pregnancy, the patient should be apprised of the potential risk to the fetus [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Serious or Fatal Respiratory Failure [see Warnings and Precautions (5.1)]
- Ischemic Events [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of TERLIVAZ cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of TERLIVAZ was evaluated in the CONFIRM trial [see Clinical Studies (14)]. The average daily dose of TERLIVAZ was 3.1 mg (range 0.8 to 5.8 mg), with a mean duration of exposure to TERLIVAZ of 6.2 days (range 1 to 15 days).
Treatment discontinuation due to adverse events occurred in 12.0% (24/200) of patients receiving TERLIVAZ and 5.1% (5/99) of patients receiving placebo. The most common adverse reactions that led to TERLIVAZ discontinuation were respiratory failure, abdominal pain, and intestinal ischemia/obstruction.
Table 1 lists adverse reactions that occurred more commonly on TERLIVAZ than on placebo, and in at least 4% of patients treated with TERLIVAZ in the CONFIRM trial. The most commonly observed adverse reactions in TERLIVAZ-treated patients (≥10%) were abdominal pain, nausea, respiratory failure, diarrhea, and dyspnea.
| Patients, n (%) | ||
|---|---|---|
| TERLIVAZ (N=200) | Placebo (N=99) |
|
|
||
| Abdominal pain | 39 (19.5) | 6 (6.1) |
| Nausea | 32 (16.0) | 10 (10.1) |
| Respiratory failure | 31 (15.5) | 7 (7.1) |
| Diarrhea | 26 (13.0) | 7 (7.1) |
| Dyspnea | 25 (12.5) | 5 (5.1) |
| Fluid overload | 17 (8.5) | 3 (3.0) |
| Pleural effusion | 11 (5.5) | 0 (0.0) |
| Sepsis | 11 (5.5) | 1 (1.0) |
| Bradycardia | 10 (5.0) | 0 (0.0) |
| Ischemia-related events* | 9 (4.5) | 0 (0.0) |
6.2 Postmarketing Experience
Adverse reactions reported from the worldwide postmarketing experience with terlipressin include headache, hyponatremia, skin necrosis and gangrene. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to terlipressin exposure.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from the published literature and on its mechanism of action, TERLIVAZ may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. In small, published studies, administration of a single intravenous dose of terlipressin to pregnant women during the first trimester induced uterine contractions and endometrial ischemia. The limited published data are not sufficient to determine a drug-associated risk for major birth defects or miscarriage. If TERLIVAZ is used during pregnancy, the patient should be informed of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In published reproductive toxicity animal studies, administration of terlipressin to pregnant guinea pigs at doses lower than the maximum recommended human dose of 4 mg/day caused a marked decrease in blood flow to the uterus and placenta. In rabbits, terlipressin is both embryotoxic and teratogenic (increased resorptions, increased implantation loss, fetal anomalies and fetal deformities).
8.2 Lactation
Risk Summary
There are no data on the presence of terlipressin in human or animal milk, the effects on the breastfed infant, or the effect on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TERLIVAZ and any potential adverse effects on the breastfed child from TERLIVAZ or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of TERLIVAZ have not been established in pediatric patients.
8.5 Geriatric Use
Of the total number of patients in clinical studies treated with TERLIVAZ, 55 (16%) were ≥65 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects; other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
No dose adjustment is required in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Manifestations of TERLIVAZ overdose are expected to be similar to the adverse reactions described with therapeutic doses. In case of overdose, initiate close monitoring of vital signs, electrolytes, and potential ischemic events and initiate appropriate symptomatic treatment.
11 DESCRIPTION
TERLIVAZ contains terlipressin, a vasopressin receptor agonist. Terlipressin is a 12-amino acid peptide with the chemical name N-[N-(N-glycylglycyl)glycyl]-8-L-lysinevasopressin. The structure of terlipressin acetate is shown below:
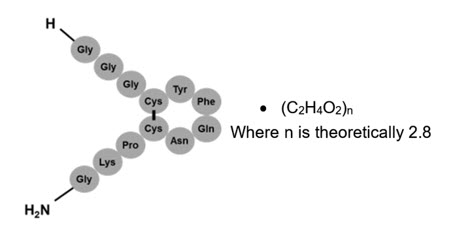
Molecular formula: C52H74N16O15S2 ∙ (C2H4O2)n; (n=number of acetate molecules; theoretical n=2.8)
Average molecular weight: 1227.38 (as free base)
TERLIVAZ is supplied as a sterile, preservative-free, lyophilized, white-to off-white powder for intravenous administration. Each vial contains 0.85 mg terlipressin, equivalent to 1 mg terlipressin acetate, and 10.0 mg mannitol. Glacial acetic acid and/or sodium hydroxide may be added to adjust pH at the time of manufacture.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Terlipressin is a synthetic vasopressin analogue with twice the selectivity for vasopressin V1 receptors versus V2 receptors. Terlipressin acts as both a prodrug for lysine-vasopressin, as well as having pharmacologic activity on its own. Terlipressin is thought to increase renal blood flow in patients with hepatorenal syndrome by reducing portal hypertension and blood circulation in portal vessels and increasing effective arterial volume and mean arterial pressure (MAP).
12.2 Pharmacodynamics
After administration of a single 0.85 mg dose of terlipressin in patients with hepatorenal syndrome type 1 (HRS-1), an increase in the diastolic, systolic, and mean arterial pressure (MAP), and decrease in heart rate were evident within 5 minutes after dosing and were maintained for at least 6 hours after dosing. The maximum change in blood pressure and heart rate occurred at 1.2 to 2 hours post dose. For MAP, the estimated maximum effect was an increase of 16.2 mmHg. The estimated maximum effect for heart rate was a decrease of 10.6 beats/minute.
Cardiac Electrophysiology
The effect of terlipressin on QTc interval was evaluated in 41 patients with HRS-1. Patients received an initial dose of 1 mg terlipressin acetate every 6 hours for a period of up to 14 days. No clinically meaningful changes from baseline were detected in the trial based on the Fridericia correction method. Increases of the mean QTc interval of <10 ms were reported.
12.3 Pharmacokinetics
The pharmacokinetic parameters of terlipressin and its major active metabolite, lysine-vasopressin, were derived from population pharmacokinetic modeling with sparse PK samples from 69 patients with HRS-1.
Following a 1 mg IV injection of terlipressin acetate, the median Cmax, AUC24h and Cave of terlipressin at steady state was 70.5 ng/mL, 123 ng×hr/mL and 14.2 ng/mL, respectively. The median Cmax, AUC24h and Cave of lysine-vasopressin were 1.2 ng/mL, 11.2 ng×hr/mL and 0.5 ng/mL, respectively.
Terlipressin and lysine-vasopressin exhibit linear pharmacokinetics in healthy subjects. Plasma concentrations of terlipressin demonstrate proportional increases with the dose administered.
Distribution
The volume of distribution (Vd) of terlipressin was 6.3 L and 1370 L for lysine-vasopressin.
Elimination
The clearance of terlipressin was 27.4 L/hr and 318 L/hr for lysine-vasopressin. There were no dose-dependent changes in the elimination rate constant of terlipressin in healthy subjects. Clearance of terlipressin in HRS-1 patients increased with body weight, while body weight had no effect on the clearance of lysine-vasopressin.
The terminal half-life of terlipressin was 0.9 hours and 3.0 hours for lysine-vasopressin.
Metabolism
Terlipressin is metabolized by cleavage of the N-terminal glycyl residues of terlipressin by various tissue peptidases, resulting in release of the pharmacologically active metabolite lysine-vasopressin. Once formed, lysine-vasopressin is metabolized by body tissue via various peptidase-mediated routes. Terlipressin is not metabolized in blood or plasma. Due to the ubiquitous nature of peptidases in body tissue, it is unlikely that the metabolism of terlipressin will be affected by disease state or other drugs.
Excretion
Less than 1% of terlipressin and <0.1% of lysine-vasopressin is excreted in urine in healthy subjects.
Specific Populations
Gender, age, creatinine clearance, Child-Pugh score, serum alkaline phosphatase, serum alanine aminotransferase (ALT), serum aspartate aminotransferase (AST), and total bilirubin do not appear to have any clinically significant effect on clearance of either terlipressin or lysine-vasopressin.
Drug Interactions
In vitro studies in human liver microsomes demonstrated that there was little or no evidence that terlipressin was a direct-, time-, or metabolism-dependent inhibitor and inducer of any of the CYP enzymes evaluated. In addition, there was little or no evidence that terlipressin is an inhibitor and substrate of human ABC and SLC transporters. No significant drug-drug interactions are anticipated with TERLIVAZ.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with terlipressin.
Terlipressin was not mutagenic or clastogenic in the following tests: in vitro bacterial reverse mutation assay, in vivo mouse micronucleus assay, and in vitro mammalian cell (CHO) chromosome aberration assay.
No studies with terlipressin have been conducted in animals to evaluate its effect on fertility.
14 CLINICAL STUDIES
The efficacy of TERLIVAZ was assessed in a multicenter, double-blind, randomized, placebo-controlled study (CONFIRM) (NCT02770716). Patients with cirrhosis, ascites, and a diagnosis of HRS-1 with a rapidly progressive worsening in renal function to a serum creatinine (SCr) ≥2.25 mg/dL and meeting a trajectory for SCr to double over two weeks, and without sustained improvement in renal function (<20% decrease in SCr and SCr ≥2.25 mg/dL) 48 hours after both diuretic withdrawal and the beginning of plasma volume expansion with albumin were eligible to participate. All patients underwent fluid challenge with intravenous albumin (1 g/kg on the first day (maximum 100 g) and 20 g/day to 40 g/day thereafter as clinically indicated). Patients with a baseline serum creatinine level >7.0 mg/dL, shock, sepsis, and/or uncontrolled bacterial infection were excluded from the study. Use of vasopressors was prohibited during the treatment period.
A total of 300 patients were enrolled; the median age was 55 years (range: 23 to 82), 60% were male, and 90% were White. At baseline, 40% had alcoholic hepatitis and 19% had ACLF Grade 3; the mean serum creatinine was 3.5 mg/dL, and the mean MELD score was 33.
Patients were randomized 2:1 to treatment with TERLIVAZ (N=199) or placebo (N=101). Patients received 1 mg terlipressin acetate (equivalent to TERLIVAZ 0.85 mg) or placebo every 6 hours administered as an IV bolus injection over 2 minutes for a maximum of 14 days. On Day 4 of therapy, if SCr decreased by less than 30% from the baseline value, the dose was increased to 2 mg terlipressin acetate (equivalent to TERLIVAZ 1.7 mg) every 6 hours. If SCr was at or above the baseline value on Day 4, then treatment was discontinued. Both treatment groups received albumin therapy during the study (median dose 50 g/day). Concomitant diuretics were used in 26% of patients treated with TERLIVAZ and 13% of patients treated with placebo. Median treatment duration was 5 days for TERLIVAZ-treated patients and 4 days for placebo-treated patients.
The primary efficacy endpoint was the incidence of Verified HRS Reversal, defined as the percentage of patients with 2 consecutive SCr values of ≤1.5 mg/dL, obtained at least 2 hours apart while on treatment by Day 14 or discharge. To be included in the primary efficacy endpoint analysis, patients had to be alive and without intervening renal replacement therapy (e.g., dialysis) at least 10 days after achieving Verified HRS Reversal.
A greater proportion of patients achieved Verified HRS Reversal in the TERLIVAZ arm compared to the placebo arm (Table 2).
| TERLIVAZ N = 199 | Placebo N = 101 | P value | |
|---|---|---|---|
| CI = confidence interval | |||
| Verified HRS Reversal*, n (%) | 58 (29.1) | 16 (15.8) | 0.012 |
| 95% CI | (0.2, 0.4) | (0.1, 0.2) | |
| Durability of HRS Reversal†,‡, n (%) | 63 (31.7) | 16 (15.8) | 0.003 |
| 95% CI | (0.3, 0.4) | (0.1, 0.2) | |
| Incidence of HRS Reversal† in the Systemic Inflammatory Response Syndrome (SIRS) Subgroup, n (%) | N=84
28 (33.3) | N=48
3 (6.3) | <0.001 |
| 95% CI | (0.2, 0.4) | (0.0, 0.1) | |
| Incidence of Verified HRS Reversal without HRS Recurrence by Day 30, n (%) | 48 (24.1) | 16 (15.8) | 0.092 |
| 95% CI | (0.2, 0.3) | (0.1, 0.2) | |
15 REFERENCES
Jalan R, et al; Development and validation of a prognostic score to predict mortality in patients with acute-on-chronic liver failure. J Hepatol. 2014 Nov;61(5):1038-47.
16 HOW SUPPLIED/STORAGE AND HANDLING
TERLIVAZ is supplied as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials containing 0.85 mg of terlipressin. Each vial is supplied in a carton (NDC 43825-200-01).
17 PATIENT COUNSELING INFORMATION
Embryo-Fetal Toxicity
Inform female patients of reproductive potential that TERLIVAZ may cause fetal harm and to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Distributed by:
Mallinckrodt Hospital Products Inc.
Bridgewater, NJ 08807, USA
Mallinckrodt, the "M" brand mark and the Mallinckrodt Pharmaceuticals logo are trademarks of a Mallinckrodt company. Other brands are trademarks of a Mallinckrodt company or their respective owners. ©2023 Mallinckrodt.
For a list of patents, see https://www.mallinckrodt.com/patents/
Part # PCR-750-17846
