FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Synagis is indicated for the prevention of serious lower respiratory tract disease caused by respiratory syncytial virus (RSV) in pediatric patients:
- with a history of premature birth (less than or equal to 35 weeks gestational age) and who are 6 months of age or younger at the beginning of RSV season,
- with bronchopulmonary dysplasia (BPD) that required medical treatment within the previous 6 months and who are 24 months of age or younger at the beginning of RSV season,
- with hemodynamically significant congenital heart disease (CHD) and who are 24 months of age or younger at the beginning of RSV season [see Clinical Studies (14)].
Limitations of Use:
The safety and efficacy of Synagis have not been established for treatment of RSV disease [see Warnings and Precautions (5.4)].
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dose of Synagis is 15 mg per kg of body weight given monthly by intramuscular injection. The first dose of Synagis should be administered prior to commencement of the RSV season and the remaining doses should be administered monthly throughout the RSV season. Children who develop an RSV infection should continue to receive monthly doses throughout the RSV season. In the northern hemisphere, the RSV season typically commences in November and lasts through April, but it may begin earlier or persist later in certain communities.
Synagis serum levels are decreased after cardio-pulmonary bypass [see Clinical Pharmacology (12.3)]. Children undergoing cardio-pulmonary bypass should receive an additional dose of Synagis as soon as possible after the cardio-pulmonary bypass procedure (even if sooner than a month from the previous dose). Thereafter, doses should be administered monthly as scheduled.
The efficacy of Synagis at doses less than 15 mg per kg, or of dosing less frequently than monthly throughout the RSV season, has not been established.
2.2 Administration Instructions
- DO NOT DILUTE THE PRODUCT.
- DO NOT SHAKE OR VIGOROUSLY AGITATE THE VIAL.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use any vials exhibiting particulate matter or discoloration.
- Using aseptic techniques, attach a sterile needle to a sterile syringe. Remove the flip top from the Synagis vial and wipe the rubber stopper with a disinfectant (e.g., 70% isopropyl alcohol). Insert the needle into the vial and withdraw into the syringe an appropriate volume of solution. Administer immediately after drawing the dose into the syringe.
- Synagis should be administered in a dose of 15 mg per kg intramuscularly using aseptic technique, preferably in the anterolateral aspect of the thigh. The gluteal muscle should not be used routinely as an injection site because of the risk of damage to the sciatic nerve. The dose (volume of injection in mL) per month = patient weight (kg) x 15 mg per kg ÷ 100 mg per mL of Synagis. Injection volumes over 1 mL should be given as a divided dose.
- Synagis is supplied as a single-dose vial and does not contain preservatives. Do not re-enter the vial after withdrawal of drug; discard unused portion. Only administer one dose per vial.
- Use sterile disposable syringes and needles. To prevent the transmission of hepatitis viruses or other infectious agents from one person to another, DO NOT reuse syringes and needles.
3 DOSAGE FORMS AND STRENGTHS
Single-dose liquid solution vials: 50 mg per 0.5 mL and 100 mg per 1 mL.
4 CONTRAINDICATIONS
Synagis is contraindicated in children who have had a previous significant hypersensitivity reaction to Synagis [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Cases of anaphylaxis and anaphylactic shock, including fatal cases, have been reported following initial exposure or re-exposure to Synagis. Other acute hypersensitivity reactions, which may be severe, have also been reported on initial exposure or re-exposure to Synagis. Signs and symptoms may include urticaria, pruritus, angioedema, dyspnea, respiratory failure, cyanosis, hypotonia, hypotension, and unresponsiveness. The relationship between these reactions and the development of antibodies to Synagis is unknown. If a significant hypersensitivity reaction occurs with Synagis, its use should be permanently discontinued. If anaphylaxis or other significant hypersensitivity reaction occurs, administer appropriate medications (e.g., epinephrine) and provide supportive care as required. If a mild hypersensitivity reaction occurs, clinical judgment should be used regarding cautious readministration of Synagis.
5.2 Coagulation Disorders
Synagis is for intramuscular use only. As with any intramuscular injection, Synagis should be given with caution to children with thrombocytopenia or any coagulation disorder.
5.3 RSV Diagnostic Test Interference
Palivizumab may interfere with immunological-based RSV diagnostic tests such as some antigen detection-based assays. In addition, palivizumab inhibits virus replication in cell culture, and therefore may also interfere with viral culture assays. Palivizumab does not interfere with reverse transcriptase-polymerase chain reaction based assays. Assay interference could lead to false-negative RSV diagnostic test results. Therefore, diagnostic test results, when obtained, should be used in conjunction with clinical findings to guide medical decisions [see Microbiology (12.4)].
6 ADVERSE REACTIONS
The most serious adverse reactions occurring with Synagis are anaphylaxis and other acute hypersensitivity reactions [see Warnings and Precautions (5.1)].
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to Synagis (n=1639) compared with placebo (n=1143) in children 3 days to 24.1 months of age at high risk of RSV-related hospitalization in two clinical trials. Trial 1 was conducted during a single RSV season and studied a total of 1502 children less than or equal to 24 months of age with BPD or infants with premature birth (less than or equal to 35 weeks gestation) who were less than or equal to 6 months of age at study entry. Trial 2 was conducted over four consecutive seasons among a total of 1287 children less than or equal to 24 months of age with hemodynamically significant congenital heart disease.
In Trials 1 and 2 combined, fever and rash were each reported more frequently among Synagis than placebo recipients, 27% versus 25%, and 12% versus 10%, respectively. Adverse reactions observed in the 153-patient crossover study comparing the liquid and lyophilized formulations were comparable for the two formulations, and were similar to those observed with Synagis in Trials 1 and 2.
Immunogenicity
In Trial 1, the incidence of anti-palivizumab antibody following the fourth injection was 1.1% in the placebo group and 0.7% in the Synagis group. In children receiving Synagis for a second season, one of the fifty-six children had transient, low titer reactivity. This reactivity was not associated with adverse events or alteration in serum concentrations. Immunogenicity was not assessed in Trial 2.
A trial of high-risk preterm children less than or equal to 24 months of age was conducted to evaluate the immunogenicity of the lyophilized formulation of Synagis (used in Trials 1 and 2 above) and the liquid formulation of Synagis. Three hundred seventy-nine children contributed to the 4 to 6 months post-final dose analysis. The rate of anti-palivizumab antibodies at this time point was low in both formulation groups (anti-palivizumab antibodies were not detected in any subject in the liquid formulation group and were detected in one subject in the lyophilized group (0.5%), with an overall rate of 0.3% for both treatment groups combined).
These data reflect the percentage of children whose test results were considered positive for antibodies to palivizumab in an enzyme-linked immunosorbent assay (ELISA) and are highly dependent on the sensitivity and specificity of the assay.
The ELISA has substantial limitations in detecting anti-palivizumab antibodies in the presence of palivizumab. Immunogenicity samples tested with the ELISA assay likely contained palivizumab at levels that may interfere with the detection of anti-palivizumab antibodies.
An electrochemical luminescence (ECL) based immunogenicity assay, with a higher tolerance for palivizumab presence compared to the ELISA, was used to evaluate the presence of anti-palivizumab antibodies in subject samples from two additional clinical trials. The rates of anti-palivizumab antibody positive results in these trials were 1.1% and 1.5%.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of Synagis. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: severe thrombocytopenia (platelet count less than 50,000 per microliter)
General Disorders and Administration Site Conditions: injection site reactions
Limited information from post-marketing reports suggests that, within a single RSV season, adverse events after a sixth or greater dose of Synagis are similar in character and frequency to those after the initial five doses.
7 DRUG INTERACTIONS
No formal drug-drug interaction studies were conducted. In Trial 1, the proportions of children in the placebo and Synagis groups who received routine childhood vaccines, influenza vaccine, bronchodilators, or corticosteroids were similar and no incremental increase in adverse reactions was observed among children receiving these agents.
8 USE IN SPECIFIC POPULATIONS
8.4 Pediatric Use
The safety and effectiveness of Synagis in children older than 24 months of age at the start of dosing have not been established [see Clinical Studies (14)].
10 OVERDOSAGE
Overdoses with doses up to 85 mg per kg have been reported in clinical studies and post-marketing experience with Synagis, and in some cases, adverse reactions were reported. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions and appropriate symptomatic treatment instituted.
11 DESCRIPTION
Palivizumab is a humanized monoclonal antibody (IgG1κ) produced by recombinant DNA technology, directed to an epitope in the A antigenic site of the F protein of RSV. Palivizumab is a composite of human (95%) and murine (5%) antibody sequences. The human heavy chain sequence was derived from the constant domains of human IgG1 and the variable framework regions of the VH genes Cor and Cess. The human light chain sequence was derived from the constant domain of Cκ and the variable framework regions of the VL gene K104 with Jκ -4. The murine sequences were derived from a murine monoclonal antibody, Mab 1129, in a process that involved the grafting of the murine complementarity determining regions into the human antibody frameworks. Palivizumab is composed of two heavy chains and two light chains and has a molecular weight of approximately 148,000 Daltons.
Synagis is supplied as a sterile, preservative-free liquid solution at 100 mg per mL to be administered by intramuscular injection. Thimerosal or other mercury-containing salts are not used in the production of Synagis. The solution has a pH of 6.0 and should appear clear or slightly opalescent.
Each 100 mg single-dose vial of Synagis liquid solution contains 100 mg of palivizumab and also contains chloride (0.5 mg), glycine (0.1 mg), and histidine (3.9 mg), in a volume of 1 mL.
Each 50 mg single-dose vial of Synagis liquid solution contains 50 mg of palivizumab and also contains chloride (0.2 mg), glycine (0.06 mg), and histidine (1.9 mg), in a volume of 0.5 mL.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Palivizumab is a recombinant humanized monoclonal antibody with anti-RSV activity [see Microbiology (12.4)].
12.3 Pharmacokinetics
In children less than or equal to 24 months of age without congenital heart disease (CHD), the mean half-life of palivizumab was 20 days and monthly intramuscular doses of 15 mg per kg achieved mean ± SD 30 day trough serum drug concentrations of 37 ± 21 mcg per mL after the first injection, 57 ± 41 mcg per mL after the second injection, 68 ± 51 mcg per mL after the third injection, and 72 ± 50 mcg per mL after the fourth injection. Trough concentrations following the first and fourth Synagis dose were similar in children with CHD and in non-cardiac patients. In children given Synagis for a second season, the mean ± SD serum concentrations following the first and fourth injections were 61 ± 17 mcg per mL and 86 ± 31 mcg per mL, respectively.
In 139 children less than or equal to 24 months of age with hemodynamically significant CHD who received Synagis and underwent cardio-pulmonary bypass for open-heart surgery, the mean ± SD serum palivizumab concentration was 98 ± 52 mcg per mL before bypass and declined to 41 ± 33 mcg per mL after bypass, a reduction of 58% [see Dosage and Administration (2.1)]. The clinical significance of this reduction is unknown.
Specific studies were not conducted to evaluate the effects of demographic parameters on palivizumab systemic exposure. However, no effects of gender, age, body weight, or race on palivizumab serum trough concentrations were observed in a clinical study with 639 children with CHD (less than or equal to 24 months of age) receiving five monthly intramuscular injections of 15 mg per kg of Synagis.
The pharmacokinetics and safety of Synagis liquid solution and Synagis lyophilized formulation administered via intramuscular injection at 15 mg per kg were studied in a cross-over trial of 153 infants less than or equal to 6 months of age with a history of prematurity. The results of this trial indicated that the trough serum concentrations of palivizumab were comparable between the liquid solution and the lyophilized formulation, which was the formulation used in the clinical studies.
A population pharmacokinetic analysis was performed across 22 studies in 1800 patients (1684 pediatric and 116 adult patients) to characterize palivizumab pharmacokinetics and inter-subject variability in serum concentrations. Palivizumab pharmacokinetics was described by a two-compartment linear model with an elimination half-life of 24.5 days in pediatric patients. Clearance of palivizumab in a typical pediatric patient (body weight 4.5 kg) less than or equal to 24 months of age without CHD was estimated to be 11 mL per day with a bioavailability of 70% following intramuscular administration. The inter-patient variability in drug clearance was 48.7% (CV%). Covariate analysis did not identify any factors that could account for the inter-patient variability in order to predict serum concentrations a priori in an individual patient.
12.4 Microbiology
Mechanism of Action
Palivizumab, a recombinant humanized monoclonal antibody which provides passive immunity against RSV, acts by binding the RSV envelope fusion protein (RSV F) on the surface of the virus and blocking a critical step in the membrane fusion process. Palivizumab also prevents cell-to-cell fusion of RSV-infected cells.
Antiviral Activity
The antiviral activity of palivizumab was assessed in a microneutralization assay in which increasing concentrations of antibody were incubated with RSV prior to addition of the human epithelial cells HEp-2. After incubation for 4-5 days, RSV antigen was measured in an ELISA assay. The neutralization titer (50% effective concentration [EC50]) is expressed as the antibody concentration required to reduce detection of RSV antigen by 50% compared with untreated virus-infected cells. Palivizumab exhibited median EC50 values of 0.65 mcg per mL (mean 0.75 ± 0.53 mcg per mL; n=69, range 0.07-2.89 mcg per mL) and 0.28 mcg per mL (mean 0.35 ± 0.23 mcg per mL; n=35, range 0.03-0.88 mcg per mL) against clinical RSV A and RSV B isolates, respectively. The majority of clinical RSV isolates tested (n=96) were collected from subjects across the United States (CA, CO, CT, IL, MA, NC, NY, PA, RI, TN, TX, VA), with the remainder from Japan (n=1), Australia (n=5) and Israel (n=2). These isolates encoded the most common RSV F sequence polymorphisms found among clinical isolates worldwide.
Palivizumab serum concentrations of greater than or equal to 40 mcg per mL have been shown to reduce pulmonary RSV replication in the cotton rat model of RSV infection by 100-fold.
Resistance
Palivizumab binds a highly conserved region on the extracellular domain of mature RSV F, referred to as antigenic site II or site A, which encompasses amino acids 262 to 275. All RSV mutants that exhibit resistance to palivizumab have been shown to contain amino acid changes in this region on the F protein.
F protein sequence variations within antigenic site A: Amino acid substitutions in antigenic site A selected either in cell culture, in animal models, or in human subjects that resulted in palivizumab resistance were N262D, N268I, K272E/M/N/Q/T, and S275F/L. RSV variants expressing the K272N substitution in F protein showed a 5164 ± 1731-fold decrease in susceptibility (i.e., fold increase in EC50 value) when compared to the wild-type RSV, while variants containing the N262D, S275F/L, or K272E/M/Q/T substitutions showed a greater than 25,000-fold decrease in susceptibility to palivizumab. The N268I substitution conferred partial resistance to palivizumab; however, fold changes in susceptibility were not quantified for this mutant. Studies carried out to investigate the mechanism of virus escape from palivizumab showed a correlation between antibody binding and virus neutralization. RSV with substitutions in antigenic site A that were resistant to neutralization by palivizumab did not bind to palivizumab.
At least one of the palivizumab resistance-associated substitutions, N262D, K272E/Q, or S275F/L was identified in 8 of 126 clinical RSV (59 RSV A and 67 RSV B) isolates from subjects who failed immunoprophylaxis, resulting in a combined resistance-associated mutation frequency of 6.3%. A review of clinical findings revealed no association between antigenic A site sequence changes and RSV disease severity among children receiving palivizumab immunoprophylaxis who develop RSV lower respiratory tract disease.
Analysis of 254 clinical RSV isolates (145 RSV A and 109 RSV B) collected from immunoprophylaxis-naïve subjects revealed palivizumab resistance-associated substitutions in 2 (1 with N262D and 1 with S275F), resulting in a resistance-associated mutation frequency of 0.79%.
F protein sequence variations outside antigenic site A: In addition to the sequence variations in antigenic site A known to confer palivizumab resistance, F protein substitutions T100A, G139S, N165D/V406I; T326A, V450A in RSV A, and T74I, A147V, I206L, S285G, V450I, T455I in RSV B were identified in viruses isolated from failures of immunoprophylaxis. These substitutions were not identified in RSV F sequences derived from 254 clinical isolates from immunoprophylaxis-naïve subjects and thus are considered treatment-associated and non-polymorphic. Recombinant RSV B encoding the S285G substitution exhibited palivizumab sensitivity (EC50 value = 0.39 ± 0.02 mcg per mL) similar to recombinant wild-type RSV B (EC50 value = 0.17 ± 0.02 mcg per mL).
Palivizumab susceptibility of RSV encoding common F protein sequence polymorphisms located proximal to antigenic site A was evaluated. Recombinant RSV A encoding N276S (EC50 value = 0.72 ± 0.07 mcg per mL), and recombinant RSV B with S276N (EC50 value = 0.42 ± 0.04 mcg per mL), exhibited sensitivities comparable to the corresponding recombinant wild-type RSV A (EC50 value = 0.63 ± 0.22 mcg per mL) and RSV B (EC50 value = 0.23 ± 0.07 mcg per mL). Likewise, RSV B clinical isolates containing the polymorphic variation V278A were at least as sensitive to neutralization by palivizumab (EC50 range 0.08-0.45 mcg per mL) as laboratory strains of wild-type RSV B (EC50 value = 0.54 ± 0.08 mcg per mL). No known polymorphic or non-polymorphic sequence variations outside the antigenic site A on RSV F have been demonstrated to render RSV resistant to neutralization by palivizumab.
Interference of RSV Diagnostic Assays by Palivizumab
Interference with immunologically-based RSV diagnostic assays by palivizumab has been observed in laboratory studies. Rapid chromatographic/enzyme immunoassays (CIA/EIA), immunofluorescence assays (IFA), and direct immunofluorescence assays (DFA) using monoclonal antibodies targeting RSV F protein may be inhibited. Therefore, caution should be used in interpreting negative immunological assay results when clinical observations are consistent with RSV infection. A reverse transcriptase-polymerase chain reaction (RT-PCR) assay, which is not inhibited by palivizumab, may prove useful for laboratory confirmation of RSV infection [see Warnings and Precautions (5.3)].
14 CLINICAL STUDIES
The safety and efficacy of Synagis were assessed in two randomized, double-blind, placebo-controlled trials of prophylaxis against RSV infection in children at high risk of an RSV-related hospitalization. Trial 1 was conducted during a single RSV season and studied a total of 1502 children less than or equal to 24 months of age with BPD or infants with premature birth (less than or equal to 35 weeks gestation) who were less than or equal to 6 months of age at study entry. Trial 2 was conducted over four consecutive seasons among a total of 1287 children less than or equal to 24 months of age with hemodynamically significant congenital heart disease. In both trials participants received 15 mg per kg Synagis or an equivalent volume of placebo via intramuscular injection monthly for five injections and were followed for 150 days from randomization. In Trial 1, 99% of all subjects completed the study and 93% completed all five injections. In Trial 2, 96% of all subjects completed the study and 92% completed all five injections. The incidence of RSV hospitalization is shown in Table 1. The results were shown to be statistically significant using Fisher's exact test.
| Trial | Placebo | Synagis | Difference Between Groups | Relative Reduction | |
| Trial 1
Impact-RSV | N | 500 | 1002 | ||
| Hospitalization | 53 (10.6%) | 48 (4.8%) | 5.8% | 55% | |
| Trial 2
CHD | N | 648 | 639 | ||
| Hospitalization | 63 (9.7%) | 34 (5.3%) | 4.4% | 45% |
In Trial 1, the reduction of RSV hospitalization was observed both in children with BPD (34/266 [12.8%] placebo versus 39/496 [7.9%] Synagis) and in premature infants without BPD (19/234 [8.1%] placebo versus 9/506 [1.8%] Synagis). In Trial 2, reductions were observed in acyanotic (36/305 [11.8%] placebo versus 15/300 [5.0%] Synagis) and cyanotic children (27/343 [7.9%] placebo versus 19/339 [5.6%] Synagis).
The clinical studies do not suggest that RSV infection was less severe among children hospitalized with RSV infection who received Synagis for RSV prophylaxis compared to those who received placebo.
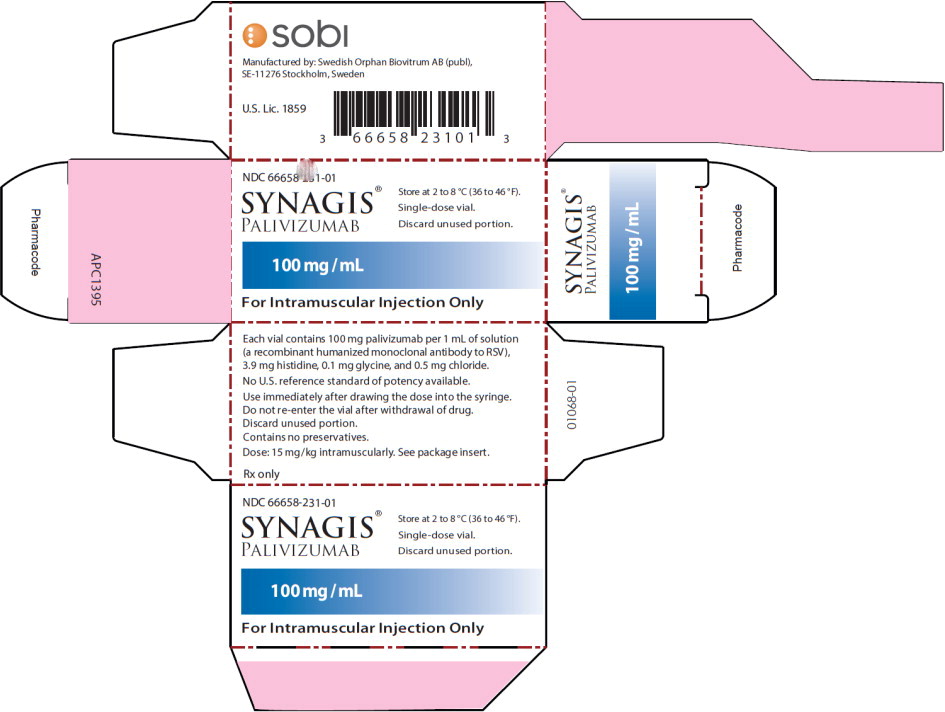
16 HOW SUPPLIED/STORAGE AND HANDLING
Synagis is supplied in single-dose vials as a preservative-free, sterile liquid solution at 100 mg per mL for intramuscular injection.
50 mg vial NDC 66658-230-01
The 50 mg vial contains 50 mg Synagis in 0.5 mL.
100 mg vial NDC 66658-231-01
The 100 mg vial contains 100 mg Synagis in 1 mL.
The rubber stopper used for sealing vials of Synagis is not made with natural rubber latex.
17 PATIENT COUNSELING INFORMATION
Advise the patient's caregiver to read the FDA-approved patient labeling (Patient Information)
Hypersensitivity Reactions
Inform the patient's caregiver of the signs and symptoms of potential hypersensitivity reactions, and advise the caregiver to seek medical attention immediately if the child experiences a severe hypersensitivity reaction to Synagis [see Contraindications (4) and Warnings and Precautions (5.1)].
Administration
Advise the patient's caregiver that Synagis should be administered by a healthcare provider once a month during the RSV season by intramuscular injection and the importance of compliance with the full course of therapy [see Dosage and Administration (2)].
Synagis® is a registered trademark of Arexis AB c/o Swedish Orphan Biovitrum AB (publ).
sobi
Manufactured by:
Swedish Orphan Biovitrum AB (publ)
Stockholm, Sweden
Distributed by:
Sobi Inc.
77 4th Avenue, 3rd FloorWaltham, MA 02451-7559
U.S License No. 1859
Issued: 11/2020
|
This Patient Information has been approved by the U.S. Food and Drug Administration |
Revised: 11/2021 |
|
| PATIENT INFORMATION
SYNAGIS® (Si-na-jis) (palivizumab) injection |
||
| What is SYNAGIS?
SYNAGIS is a prescription medication that is used to help prevent a serious lung disease caused by Respiratory Syncytial Virus (RSV) in children:
|
||
| SYNAGIS contains man-made, disease-fighting proteins called antibodies. It is not known if SYNAGIS is safe and effective to treat the symptoms of RSV in a child who already has RSV. Synagis is used to help prevent RSV disease. It is not known if SYNAGIS is safe and effective in children who are older than 24 months of age at the start of dosing. |
||
|
Who should not receive SYNAGIS?
|
||
|
|
|
Before your child receives SYNAGIS, tell your healthcare provider about all of your child's medical conditions, including if your child:
|
||
| Tell your child's healthcare provider about all the medicines your child takes, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | ||
How is SYNAGIS given?
|
||
| What are the possible side effects of SYNAGIS?
SYNAGIS may cause serious side effects including:
|
||
| The most common side effects of SYNAGIS include fever and rash. These are not all the possible side effects of SYNAGIS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects at 1866-773-5274. |
||
| General information about the safe and effective use of SYNAGIS.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. You can ask your pharmacist or healthcare provider for information about SYNAGIS that is written for health professionals. |
||
| What are the ingredients in SYNAGIS?
Active ingredient: palivizumab Inactive ingredients: chloride, glycine, and histidine |
||
| Manufactured by: Swedish Orphan Biovitrum AB (publ) , Stockholm, Sweden Synagis® is a registered trademark of Arexis AB c/o Swedish Orphan Biovitrum AB (publ). | sobi | |
| For more information, go to www.synagis.com or call 1866-773-5274. | ||
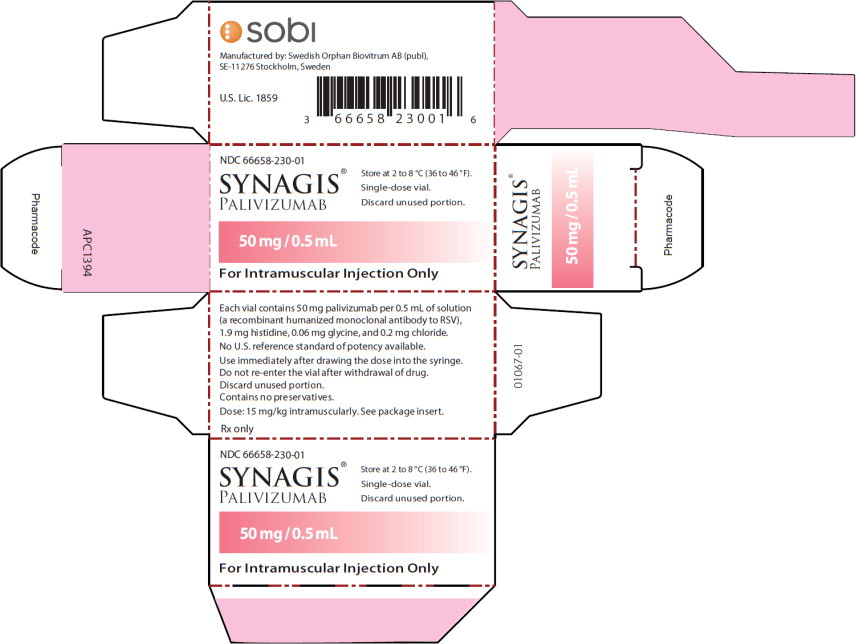
Principal Display Panel - 50 mg/0.5 mL Carton Label
NDC 66658-230-01
SYNAGIS®
PALIVIZUMAB
50 mg/0.5 mL
For Intramuscular Injection Only
Store at 2 to 8°C (36 to 46°F).
Single-dose vial.
Discard unused portion.
Principal Display Panel - 50 mg/0.5 mL Vial Label
NDC 66658-230-01
50 mg/0.5 mL
SYNAGIS®
PALIVIZUMAB
For Intramuscular
Injection Only
US Lic. 1859