NIASPAN- niacin tablet, film coated, extended release
AbbVie Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NIASPAN® safely and effectively. See full prescribing information for NIASPAN.
NIASPAN (niacin extended-release tablets), for oral use Initial U.S. Approval: 1997 INDICATIONS AND USAGENIASPAN contains extended-release niacin (nicotinic acid), and is indicated:
Limitations of use: Addition of NIASPAN did not reduce cardiovascular morbidity or mortality among patients treated with simvastatin in a large, randomized controlled trial. (5.1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSUnscored film-coated tablets for oral administration: 500, 750 and 1000 mg niacin extended-release. (3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence >5% and greater than placebo) are flushing, diarrhea, nausea, vomiting, increased cough, and pruritus. (6.1) Flushing of the skin may be reduced in frequency or severity by pretreatment with aspirin (up to the recommended dose of 325 mg taken 30 minutes prior to NIASPAN dose). (2.2)
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 5/2022 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Therapy with lipid-altering agents should be only one component of multiple risk factor intervention in individuals at significantly increased risk for atherosclerotic vascular disease due to hyperlipidemia. Niacin therapy is indicated as an adjunct to diet when the response to a diet restricted in saturated fat and cholesterol and other nonpharmacologic measures alone has been inadequate.
- NIASPAN is indicated to reduce elevated TC, LDL-C, Apo B and TG levels, and to increase HDL-C in patients with primary hyperlipidemia and mixed dyslipidemia.
- In patients with a history of myocardial infarction and hyperlipidemia, niacin is indicated to reduce the risk of recurrent nonfatal myocardial infarction.
- In patients with a history of coronary artery disease (CAD) and hyperlipidemia, niacin, in combination with a bile acid binding resin, is indicated to slow progression or promote regression of atherosclerotic disease.
- NIASPAN in combination with a bile acid binding resin is indicated to reduce elevated TC and LDL-C levels in adult patients with primary hyperlipidemia.
- Niacin is also indicated as adjunctive therapy for treatment of adult patients with severe hypertriglyceridemia who present a risk of pancreatitis and who do not respond adequately to a determined dietary effort to control them.
Limitations of Use
Addition of NIASPAN did not reduce cardiovascular morbidity or mortality among patients treated with simvastatin in a large, randomized controlled trial (AIM-HIGH) [see Warnings and Precautions (5.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Initial Dosing
NIASPAN should be taken at bedtime, after a low-fat snack, and doses should be individualized according to patient response. Therapy with NIASPAN must be initiated at 500 mg at bedtime in order to reduce the incidence and severity of side effects which may occur during early therapy. The recommended dose escalation is shown in Table 1 below.
| Week(s) | Daily dose | NIASPAN Dosage | |
| INITIAL TITRATION | 1 to 4 | 500 mg | 1 NIASPAN 500 mg tablet at bedtime |
| SCHEDULE | 5 to 8 | 1000 mg | 1 NIASPAN 1000 mg tablet or 2 NIASPAN 500 mg tablets at bedtime |
| * | 1500 mg | 2 NIASPAN 750 mg tablets or 3 NIASPAN 500 mg tablets at bedtime |
|
| * | 2000 mg | 2 NIASPAN 1000 mg tablets or 4 NIASPAN 500 mg tablets at bedtime |
|
| * After Week 8, titrate to patient response and tolerance. If response to 1000 mg daily is inadequate, increase dose to 1500 mg daily; may subsequently increase dose to 2000 mg daily. Daily dose should not be increased more than 500 mg in a 4-week period, and doses above 2000 mg daily are not recommended. Women may respond at lower doses than men. | |||
2.2 Maintenance Dose
The daily dosage of NIASPAN should not be increased by more than 500 mg in any 4-week period. The recommended maintenance dose is 1000 mg (two 500 mg tablets or one 1000 mg tablet) to 2000 mg (two 1000 mg tablets or four 500 mg tablets) once daily at bedtime. Doses greater than 2000 mg daily are not recommended. Women may respond at lower NIASPAN doses than men [see Clinical Studies (14.2)].
Single-dose bioavailability studies have demonstrated that two of the 500 mg and one of the 1000 mg tablet strengths are interchangeable but three of the 500 mg and two of the 750 mg tablet strengths are not interchangeable.
Flushing of the skin [see Adverse Reactions (6.1)] may be reduced in frequency or severity by pretreatment with aspirin (up to the recommended dose of 325 mg taken 30 minutes prior to NIASPAN dose). Tolerance to this flushing develops rapidly over the course of several weeks. Flushing, pruritus, and gastrointestinal distress are also greatly reduced by slowly increasing the dose of niacin and avoiding administration on an empty stomach. Concomitant alcoholic, hot drinks or spicy foods may increase the side effects of flushing and pruritus and should be avoided around the time of NIASPAN ingestion.
Equivalent doses of NIASPAN should not be substituted for sustained-release (modified-release, timed-release) niacin preparations or immediate-release (crystalline) niacin [see Warnings and Precautions (5)]. Patients previously receiving other niacin products should be started with the recommended NIASPAN titration schedule (see Table 1), and the dose should subsequently be individualized based on patient response.
If NIASPAN therapy is discontinued for an extended period, reinstitution of therapy should include a titration phase (see Table 1).
NIASPAN tablets should be taken whole and should not be broken, crushed or chewed before swallowing.
2.3 Dosage in Patients with Renal or Hepatic Impairment
Use of NIASPAN in patients with renal or hepatic impairment has not been studied. NIASPAN is contraindicated in patients with significant or unexplained hepatic dysfunction. NIASPAN should be used with caution in patients with renal impairment [see Warnings and Precautions (5)].
3 DOSAGE FORMS AND STRENGTHS
- 500 mg unscored, medium-orange, film-coated, capsule-shaped tablets
- 750 mg unscored, medium-orange, film-coated, capsule-shaped tablets
- 1000 mg unscored, medium-orange, film-coated, capsule-shaped tablets
5 WARNINGS AND PRECAUTIONS
NIASPAN preparations should not be substituted for equivalent doses of immediate-release (crystalline) niacin. For patients switching from immediate-release niacin to NIASPAN, therapy with NIASPAN should be initiated with low doses (i.e., 500 mg at bedtime) and the NIASPAN dose should then be titrated to the desired therapeutic response [see Dosage and Administration (2.1)].
Caution should also be used when NIASPAN is used in patients with unstable angina or in the acute phase of an MI, particularly when such patients are also receiving vasoactive drugs such as nitrates, calcium channel blockers, or adrenergic blocking agents.
Niacin is rapidly metabolized by the liver, and excreted through the kidneys. NIASPAN is contraindicated in patients with significant or unexplained hepatic impairment [see Contraindications (4) and Warnings and Precautions (5.3)] and should be used with caution in patients with renal impairment. Patients with a past history of jaundice, hepatobiliary disease, or peptic ulcer should be observed closely during NIASPAN therapy.
5.1 Mortality and Coronary Heart Disease Morbidity
NIASPAN has not been shown to reduce cardiovascular morbidity or mortality among patients already treated with a statin.
The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial was a randomized placebo-controlled trial of 3414 patients with stable, previously diagnosed cardiovascular disease. Mean baseline lipid levels were LDL-C 74 mg/dL, HDL-C 35 mg/dL, non-HDL-C 111 mg/dL and median triglyceride level of 163-177 mg/dL. Ninety-four percent of patients were on background statin therapy prior to entering the trial. All participants received simvastatin, 40 to 80 mg per day, plus ezetimibe 10 mg per day if needed, to maintain an LDL-C level of 40-80 mg/dL, and were randomized to receive NIASPAN 1500-2000 mg/day (n=1718) or matching placebo (IR Niacin, 100-150 mg, n=1696). On-treatment lipid changes at two years for LDL-C were -12.0% for the simvastatin plus NIASPAN group and -5.5% for the simvastatin plus placebo group. HDL-C increased by 25.0% to 42 mg/dL in the simvastatin plus NIASPAN group and by 9.8% to 38 mg/dL in the simvastatin plus placebo group (P<0.001). Triglyceride levels decreased by 28.6% in the simvastatin plus NIASPAN group and by 8.1% in the simvastatin plus placebo group. The primary outcome was an ITT composite of the first study occurrence of coronary heart disease death, nonfatal myocardial infarction, ischemic stroke, hospitalization for acute coronary syndrome or symptom-driven coronary or cerebral revascularization procedures. The trial was stopped after a mean follow-up period of 3 years owing to a lack of efficacy. The primary outcome occurred in 282 patients in the simvastatin plus NIASPAN group (16.4%) and in 274 patients in the simvastatin plus placebo group (16.2%) (HR 1.02 [95% CI, 0.87-1.21], P=0.79. In an ITT analysis, there were 42 cases of first occurrence of ischemic stroke reported, 27 (1.6%) in the simvastatin plus NIASPAN group and 15 (0.9%) in the simvastatin plus placebo group, a non-statistically significant result (HR 1.79, [95%CI = 0.95-3.36], p=0.071). The on-treatment ischemic stroke events were 19 for the simvastatin plus NIASPAN group and 15 for the simvastatin plus placebo group [see Adverse Reactions (6.1)].
5.2 Skeletal Muscle
Cases of rhabdomyolysis have been associated with concomitant administration of lipid-altering doses (≥1 g/day) of niacin and statins. Elderly patients and patients with diabetes, renal failure, or uncontrolled hypothyroidism are particularly at risk. Monitor patients for any signs and symptoms of muscle pain, tenderness, or weakness, particularly during the initial months of therapy and during any periods of upward dosage titration. Periodic serum creatine phosphokinase (CPK) and potassium determinations should be considered in such situations, but there is no assurance that such monitoring will prevent the occurrence of severe myopathy.
5.3 Liver Dysfunction
Cases of severe hepatic toxicity, including fulminant hepatic necrosis, have occurred in patients who have substituted sustained-release (modified-release, timed-release) niacin products for immediate-release (crystalline) niacin at equivalent doses.
NIASPAN should be used with caution in patients who consume substantial quantities of alcohol and/or have a past history of liver disease. Active liver diseases or unexplained transaminase elevations are contraindications to the use of NIASPAN.
Niacin preparations have been associated with abnormal liver tests. In three placebo-controlled clinical trials involving titration to final daily NIASPAN doses ranging from 500 to 3000 mg, 245 patients received NIASPAN for a mean duration of 17 weeks. No patient with normal serum transaminase levels (AST, ALT) at baseline experienced elevations to more than 3 times the upper limit of normal (ULN) during treatment with NIASPAN. In these studies, fewer than 1% (2/245) of NIASPAN patients discontinued due to transaminase elevations greater than 2 times the ULN.
Liver-related tests should be performed on all patients during therapy with NIASPAN. Serum transaminase levels, including AST and ALT (SGOT and SGPT), should be monitored before treatment begins, every 6 to 12 weeks for the first year, and periodically thereafter (e.g., at approximately 6-month intervals). Special attention should be paid to patients who develop elevated serum transaminase levels, and in these patients, measurements should be repeated promptly and then performed more frequently. If the transaminase levels show evidence of progression, particularly if they rise to 3 times ULN and are persistent, or if they are associated with symptoms of nausea, fever, and/or malaise, the drug should be discontinued.
5.4 Laboratory Abnormalities
Increase in Blood Glucose: Niacin treatment can increase fasting blood glucose. Frequent monitoring of blood glucose should be performed to ascertain that the drug is producing no adverse effects. Diabetic patients may experience a dose-related increase in glucose intolerance. Diabetic or potentially diabetic patients should be observed closely during treatment with NIASPAN, particularly during the first few months of use or dose adjustment; adjustment of diet and/or hypoglycemic therapy may be necessary.
Reduction in platelet count: NIASPAN has been associated with small but statistically significant dose-related reductions in platelet count (mean of -11% with 2000 mg). Caution should be observed when NIASPAN is administered concomitantly with anticoagulants; platelet counts should be monitored closely in such patients.
Increase in Prothrombin Time (PT): NIASPAN has been associated with small but statistically significant increases in prothrombin time (mean of approximately +4%); accordingly, patients undergoing surgery should be carefully evaluated. Caution should be observed when NIASPAN is administered concomitantly with anticoagulants; prothrombin time should be monitored closely in such patients.
Increase in Uric Acid: Elevated uric acid levels have occurred with niacin therapy, therefore use with caution in patients predisposed to gout.
Decrease in Phosphorus: In placebo-controlled trials, NIASPAN has been associated with small but statistically significant, dose-related reductions in phosphorus levels (mean of -13% with 2000 mg). Although these reductions were transient, phosphorus levels should be monitored periodically in patients at risk for hypophosphatemia.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Mortality and Coronary Heart Disease Morbidity [see Warnings and Precautions (5.1)]
- Skeletal Muscle (rhabdomyolysis) [see Warnings and Precautions (5.2)]
- Liver Dysfunction [see Warnings and Precautions (5.3)]
- Laboratory Abnormalities [see Warnings and Precautions (5.4)]
6.1 Clinical Studies Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
In the placebo-controlled clinical trials database of 402 patients (age range 21-75 years, 33% women, 89% Caucasians, 7% Blacks, 3% Hispanics, 1% Asians) with a median treatment duration of 16 weeks, 16% of patients on NIASPAN and 4% of patients on placebo discontinued due to adverse reactions. The most common adverse reactions in the group of patients treated with NIASPAN that led to treatment discontinuation and occurred at a rate greater than placebo were flushing (6% vs. 0%), rash (2% vs. 0%), diarrhea (2% vs. 0%), nausea (1% vs. 0%), and vomiting (1% vs. 0%). The most commonly reported adverse reactions (incidence >5% and greater than placebo) in the NIASPAN controlled clinical trial database of 402 patients were flushing, diarrhea, nausea, vomiting, increased cough and pruritus.
In the placebo-controlled clinical trials, flushing episodes (i.e., warmth, redness, itching and/or tingling) were the most common treatment-emergent adverse reactions (reported by as many as 88% of patients) for NIASPAN. Spontaneous reports suggest that flushing may also be accompanied by symptoms of dizziness, tachycardia, palpitations, shortness of breath, sweating, burning sensation/skin burning sensation, chills, and/or edema, which in rare cases may lead to syncope. In pivotal studies, 6% (14/245) of NIASPAN patients discontinued due to flushing. In comparisons of immediate-release (IR) niacin and NIASPAN, although the proportion of patients who flushed was similar, fewer flushing episodes were reported by patients who received NIASPAN. Following 4 weeks of maintenance therapy at daily doses of 1500 mg, the incidence of flushing over the 4-week period averaged 8.6 events per patient for IR niacin versus 1.9 following NIASPAN.
Other adverse reactions occurring in ≥5% of patients treated with NIASPAN and at an incidence greater than placebo are shown in Table 2 below.
| Placebo-Controlled Studies
NIASPAN Treatment@ |
|||||
| Recommended Daily
Maintenance Doses † |
|||||
| Placebo | 500 mg‡ | 1000 mg | 1500 mg | 2000 mg | |
| (n=157) | (n=87) | (n=110) | (n=136) | (n=95) | |
| % | % | % | % | % | |
| Gastrointestinal Disorders | |||||
| Diarrhea | 13 | 7 | 10 | 10 | 14 |
| Nausea | 7 | 5 | 6 | 4 | 11 |
| Vomiting | 4 | 0 | 2 | 4 | 9 |
| Respiratory | |||||
| Cough, Increased | 6 | 3 | 2 | < 2 | 8 |
| Skin and Subcutaneous Tissue Disorders | |||||
| Pruritus | 2 | 8 | 0 | 3 | 0 |
| Rash | 0 | 5 | 5 | 5 | 0 |
| Vascular Disorders | |||||
| Flushing& | 19 | 68 | 69 | 63 | 55 |
| Note: Percentages are calculated from the total number of patients in each column. † Adverse reactions are reported at the initial dose where they occur. @ Pooled results from placebo-controlled studies; for NIASPAN, n=245 and median treatment duration=16 weeks. Number of NIASPAN patients (n) are not additive across doses. ‡ The 500 mg/day dose is outside the recommended daily maintenance dosing range [see Dosage and Administration (2.2)]. & 10 patients discontinued before receiving 500 mg, therefore they were not included. |
|||||
In general, the incidence of adverse events was higher in women compared to men.
Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH)
In AIM-HIGH involving 3414 patients (mean age of 64 years, 15% women, 92% Caucasians, 34% with diabetes mellitus) with stable, previously diagnosed cardiovascular disease, all patients received simvastatin, 40 to 80 mg per day, plus ezetimibe 10 mg per day if needed, to maintain an LDL-C level of 40-80 mg/dL, and were randomized to receive NIASPAN 1500-2000 mg/day (n=1718) or matching placebo (IR Niacin, 100-150 mg, n=1696). The incidence of the adverse reactions of “blood glucose increased” (6.4% vs. 4.5%) and “diabetes mellitus” (3.6% vs. 2.2%) was significantly higher in the simvastatin plus NIASPAN group as compared to the simvastatin plus placebo group. There were 5 cases of rhabdomyolysis reported, 4 (0.2%) in the simvastatin plus NIASPAN group and one (<0.1%) in the simvastatin plus placebo group.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post-approval use of NIASPAN. Because the below reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders: tachycardia, palpitations, atrial fibrillation, other cardiac arrhythmias
Eye disorders: blurred vision, macular edema
Gastrointestinal disorders: peptic ulcers, eructation, flatulence
Hepatobiliary disorders: hepatitis, jaundice
Immune system disorders: hypersensitivity reactions (including anaphylaxis, angioedema, urticaria, flushing, dyspnea, tongue edema, larynx edema, face edema, peripheral edema, laryngismus, and vesiculobullous rash)
Metabolism and nutrition disorders: decreased glucose tolerance, gout
Musculoskeletal and connective tissue disorders: myalgia, myopathy
Nervous system disorders: dizziness, insomnia, asthenia, nervousness, paresthesia, migraine
Respiratory, thoracic and mediastinal disorders: dyspnea
Skin and subcutaneous tissue disorders: maculopapular rash, dry skin, sweating, burning sensation/skin burning sensation, skin discoloration, acanthosis nigricans
Vascular disorders: syncope, hypotension, postural hypotension
Clinical Laboratory Abnormalities
Chemistry: Elevations in serum transaminases, LDH, fasting glucose, uric acid, total bilirubin, amylase and creatine kinase, and reduction in phosphorus.
Hematology: Slight reductions in platelet counts and prolongation in prothrombin time.
7 DRUG INTERACTIONS
7.1 Statins
Caution should be used when prescribing niacin (≥1 gm/day) with statins as these drugs can increase risk of myopathy/rhabdomyolysis [see Warnings and Precautions (5) and Clinical Pharmacology (12.3)].
7.2 Bile Acid Sequestrants
An in vitro study results suggest that the bile acid-binding resins have high niacin binding capacity. Therefore, 4 to 6 hours, or as great an interval as possible, should elapse between the ingestion of bile acid-binding resins and the administration of NIASPAN [see Clinical Pharmacology (12.3)].
7.3 Aspirin
Concomitant aspirin may decrease the metabolic clearance of nicotinic acid. The clinical relevance of this finding is unclear.
7.4 Antihypertensive Therapy
Niacin may potentiate the effects of ganglionic blocking agents and vasoactive drugs resulting in postural hypotension.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Discontinue NIASPAN when pregnancy is recognized in patients receiving the drug for the treatment of hyperlipidemia. Assess the individual risks and benefits of continuing NIASPAN during pregnancy in patients receiving the drug for the treatment of hypertriglyceridemia. Advise patients to inform their healthcare provider of a known or suspected pregnancy.
The potential for embryofetal toxicity with the doses of niacin in NIASPAN is unknown. The available data on NIASPAN use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Animal reproduction studies have not been conducted with niacin or with NIASPAN. Treatment of hypercholesterolemia is not generally necessary during pregnancy. Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia for most patients.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
Niacin is present in human milk and the amount of niacin increases with maternal supplementation. There is no information on the effects of the doses of niacin in NIASPAN on the breastfed infant or the effects on milk production. Because of the potential for serious adverse reactions in breastfeeding infants, including hepatotoxicity, advise patients not to breastfeed during treatment with NIASPAN.
8.4 Pediatric Use
Safety and effectiveness of niacin therapy in pediatric patients (≤16 years) have not been established.
8.5 Geriatric Use
Of 979 patients in clinical studies of NIASPAN, 21% of the patients were age 65 and over. No overall differences in safety and effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No studies have been performed in this population. NIASPAN should be used with caution in patients with renal impairment [see Warnings and Precautions (5)].
8.7 Hepatic Impairment
No studies have been performed in this population. NIASPAN should be used with caution in patients with a past history of liver disease and/or who consume substantial quantities of alcohol. Active liver disease, unexplained transaminase elevations and significant or unexplained hepatic dysfunction are contraindications to the use of NIASPAN [see Contraindications (4) and Warnings and Precautions (5.3)].
11 DESCRIPTION
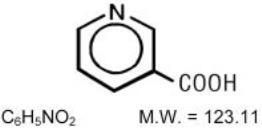
NIASPAN (niacin tablet, film-coated extended-release), contains niacin, which at therapeutic doses is an antihyperlipidemic agent. Niacin (nicotinic acid, or 3-pyridinecarboxylic acid) is a white, crystalline powder, very soluble in water, with the following structural formula:
NIASPAN is an unscored, medium-orange, film-coated tablet for oral administration and is available in three tablet strengths containing 500, 750, and 1000 mg niacin. NIASPAN tablets also contain the inactive ingredients hypromellose, povidone, stearic acid, and polyethylene glycol, and the following coloring agents: FD&C yellow #6/sunset yellow FCF Aluminum Lake, synthetic red and yellow iron oxides, and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which niacin alters lipid profiles has not been well defined. It may involve several actions including partial inhibition of release of free fatty acids from adipose tissue, and increased lipoprotein lipase activity, which may increase the rate of chylomicron triglyceride removal from plasma. Niacin decreases the rate of hepatic synthesis of VLDL and LDL, and does not appear to affect fecal excretion of fats, sterols, or bile acids.
12.3 Pharmacokinetics
Absorption
Due to extensive and saturable first-pass metabolism, niacin concentrations in the general circulation are dose dependent and highly variable. Time to reach the maximum niacin plasma concentrations was about 5 hours following NIASPAN. To reduce the risk of gastrointestinal (GI) upset, administration of NIASPAN with a low-fat meal or snack is recommended.
Single-dose bioavailability studies have demonstrated that the 500 mg and 1000 mg tablet strengths are dosage form equivalent but the 500 mg and 750 mg tablet strengths are not dosage form equivalent.
Metabolism
The pharmacokinetic profile of niacin is complicated due to extensive first-pass metabolism that is dose-rate specific and, at the doses used to treat dyslipidemia, saturable. In humans, one pathway is through a simple conjugation step with glycine to form nicotinuric acid (NUA). NUA is then excreted in the urine, although there may be a small amount of reversible metabolism back to niacin. The other pathway results in the formation of nicotinamide adenine dinucleotide (NAD). It is unclear whether nicotinamide is formed as a precursor to, or following the synthesis of, NAD. Nicotinamide is further metabolized to at least N-methylnicotinamide (MNA) and nicotinamide-N-oxide (NNO). MNA is further metabolized to two other compounds, N-methyl-2-pyridone-5-carboxamide (2PY) and N-methyl-4-pyridone-5-carboxamide (4PY). The formation of 2PY appears to predominate over 4PY in humans. At the doses used to treat hyperlipidemia, these metabolic pathways are saturable, which explains the nonlinear relationship between niacin dose and plasma concentrations following multiple-dose NIASPAN administration.
Nicotinamide does not have hypolipidemic activity; the activity of the other metabolites is unknown.
Elimination
Following single and multiple doses, approximately 60 to 76% of the niacin dose administered as NIASPAN was recovered in urine as niacin and metabolites; up to 12% was recovered as unchanged niacin after multiple dosing. The ratio of metabolites recovered in the urine was dependent on the dose administered.
Pediatric Use
No pharmacokinetic studies have been performed in this population (≤16 years) [see Use in Specific Populations (8.4)].
Geriatric Use
No pharmacokinetic studies have been performed in this population (>65 years) [see Use in Specific Populations (8.5)].
Renal Impairment
No pharmacokinetic studies have been performed in this population. NIASPAN should be used with caution in patients with renal disease [see Warnings and Precautions (5)].
Hepatic Impairment
No pharmacokinetic studies have been performed in this population. Active liver disease, unexplained transaminase elevations and significant or unexplained hepatic dysfunction are contraindications to the use of NIASPAN [see Contraindications (4) and Warnings and Precautions (5.3)].
Gender
Steady-state plasma concentrations of niacin and metabolites after administration of NIASPAN are generally higher in women than in men, with the magnitude of the difference varying with dose and metabolite. This gender differences observed in plasma levels of niacin and its metabolites may be due to gender-specific differences in metabolic rate or volume of distribution. Recovery of niacin and metabolites in urine, however, is generally similar for men and women, indicating that absorption is similar for both genders [see Gender (8.8)].
Drug interactions
Fluvastatin
Niacin did not affect fluvastatin pharmacokinetics [see Drug Interactions (7.1)].
Lovastatin
When NIASPAN 2000 mg and lovastatin 40 mg were co-administered, NIASPAN increased lovastatin Cmax and AUC by 2% and 14%, respectively, and decreased lovastatin acid Cmax and AUC by 22% and 2%, respectively. Lovastatin reduced NIASPAN bioavailability by 2-3% [see Drug Interactions (7.1)].
Simvastatin
When NIASPAN 2000 mg and simvastatin 40 mg were co-administered, NIASPAN increased simvastatin Cmax and AUC by 1% and 9%, respectively, and simvastatin acid Cmax and AUC by 2% and 18%, respectively. Simvastatin reduced NIASPAN bioavailability by 2% [see Drug Interactions (7.1)].
Bile Acid Sequestrants
An in vitro study was carried out investigating the niacin-binding capacity of colestipol and cholestyramine. About 98% of available niacin was bound to colestipol, with 10 to 30% binding to cholestyramine [see Drug Interactions (7.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis and Mutagenesis and Impairment of Fertility
Niacin administered to mice for a lifetime as a 1% solution in drinking water was not carcinogenic. The mice in this study received approximately 6 to 8 times a human dose of 3000 mg/day as determined on a mg/m2 basis. Niacin was negative for mutagenicity in the Ames test. No studies on impairment of fertility have been performed. No studies have been conducted with NIASPAN regarding carcinogenesis, mutagenesis, or impairment of fertility.
14 CLINICAL STUDIES
14.1 Niacin Clinical Studies
Niacin’s ability to reduce mortality and the risk of definite, nonfatal myocardial infarction (MI) has been assessed in long-term studies. The Coronary Drug Project, completed in 1975, was designed to assess the safety and efficacy of niacin and other lipid-altering drugs in men 30 to 64 years old with a history of MI. Over an observation period of 5 years, niacin treatment was associated with a statistically significant reduction in nonfatal, recurrent MI. The incidence of definite, nonfatal MI was 8.9% for the 1119 patients randomized to nicotinic acid versus 12.2% for the 2789 patients who received placebo (p<0.004). Total mortality was similar in the two groups at 5 years (24.4% with nicotinic acid versus 25.4% with placebo; p=N.S.). At the time of a 15-year follow-up, there were 11% (69) fewer deaths in the niacin group compared to the placebo cohort (52.0% versus 58.2%; p=0.0004). However, mortality at 15 years was not an original endpoint of the Coronary Drug Project. In addition, patients had not received niacin for approximately 9 years, and confounding variables such as concomitant medication use and medical or surgical treatments were not controlled.
The Cholesterol-Lowering Atherosclerosis Study (CLAS) was a randomized, placebo-controlled, angiographic trial testing combined colestipol and niacin therapy in 162 non-smoking males with previous coronary bypass surgery. The primary, per-subject cardiac endpoint was global coronary artery change score. After 2 years, 61% of patients in the placebo cohort showed disease progression by global change score (n=82), compared with only 38.8% of drug-treated subjects (n=80), when both native arteries and grafts were considered (p<0.005); disease regression also occurred more frequently in the drug-treated group (16.2% versus 2.4%; p=0.002). In a follow-up to this trial in a subgroup of 103 patients treated for 4 years, again, significantly fewer patients in the drug-treated group demonstrated progression than in the placebo cohort (48% versus 85%, respectively; p<0.0001).
The Familial Atherosclerosis Treatment Study (FATS) in 146 men ages 62 and younger with Apo B levels ≥125 mg/dL, established coronary artery disease, and family histories of vascular disease, assessed change in severity of disease in the proximal coronary arteries by quantitative arteriography. Patients were given dietary counseling and randomized to treatment with either conventional therapy with double placebo (or placebo plus colestipol if the LDL-C was elevated); lovastatin plus colestipol; or niacin plus colestipol. In the conventional therapy group, 46% of patients had disease progression (and no regression) in at least one of nine proximal coronary segments; regression was the only change in 11%. In contrast, progression (as the only change) was seen in only 25% in the niacin plus colestipol group, while regression was observed in 39%. Though not an original endpoint of the trial, clinical events (death, MI, or revascularization for worsening angina) occurred in 10 of 52 patients who received conventional therapy, compared with 2 of 48 who received niacin plus colestipol.
14.2 NIASPAN Clinical Studies
Placebo-Controlled Clinical Studies in Patients with Primary Hyperlipidemia and Mixed Dyslipidemia: In two randomized, double-blind, parallel, multi-center, placebo-controlled trials, NIASPAN dosed at 1000, 1500 or 2000 mg daily at bedtime with a low-fat snack for 16 weeks (including 4 weeks of dose escalation) favorably altered lipid profiles compared to placebo (Table 3). Women appeared to have a greater response than men at each NIASPAN dose level (see Gender Effect, below).
| Mean Percent Change from Baseline to Week 16* | ||||||
| Treatment | n | TC | LDL-C | HDL-C | TG | Apo B |
| NIASPAN 1000 mg at bedtime | 41 | -3 | -5 | +18 | -21 | -6 |
| NIASPAN 2000 mg at bedtime | 41 | -10 | -14 | +22 | -28 | -16 |
| Placebo | 40 | 0 | -1 | +4 | 0 | +1 |
| NIASPAN 1500 mg at bedtime | 76 | -8 | -12 | +20 | -13 | -12 |
| Placebo | 73 | +2 | +1 | +2 | +12 | +1 |
| n = number of patients at baseline; * Mean percent change from baseline for all NIASPAN doses was significantly different (p<0.05) from placebo. |
||||||
In a double-blind, multi-center, forced dose-escalation study, monthly 500 mg increases in NIASPAN dose resulted in incremental reductions of approximately 5% in LDL-C and Apo B levels in the daily dose range of 500 mg through 2000 mg (Table 4). Women again tended to have a greater response to NIASPAN than men (see Gender Effect, below).
| Mean Percent Change from Baseline* | ||||||
| Treatment | n | TC | LDL-C | HDL-C | TG | Apo B |
| Placebo‡ | 44 | -2 | -1 | +5 | -6 | -2 |
| NIASPAN | 87 | |||||
| 500 mg at bedtime | -2 | -3 | +10 | -5 | -2 | |
| 1000 mg at bedtime | -5 | -9 | +15 | -11 | -7 | |
| 1500 mg at bedtime | -11 | -14 | +22 | -28 | -15 | |
| 2000 mg at bedtime | -12 | -17 | +26 | -35 | -16 | |
| n = number of patients enrolled; ‡ Placebo data shown are after 24 weeks of placebo treatment. * For all NIASPAN doses except 500 mg, mean percent change from baseline was significantly different (p<0.05) from placebo for all lipid parameters shown. |
||||||
Pooled results for major lipids from these three placebo-controlled studies are shown below (Table 5).
| Mean Baseline and Median Percent Change from Baseline
(25th, 75th Percentiles) |
||||
| NIASPAN
Dose | n | LDL-C | HDL-C | TG |
| 1000 mg at bedtime | 104 | |||
| Baseline (mg/dL) | 218 | 45 | 172 | |
| Percent Change | -7 (-15, 0) | +14 (+7, +23) | -16 (-34, +3) | |
| 1500 mg at bedtime | 120 | |||
| Baseline (mg/dL) | 212 | 46 | 171 | |
| Percent Change | -13 (-21, -4) | +19 (+9, +31) | -25 (-45, -2) | |
| 2000 mg at bedtime | 85 | |||
| Baseline (mg/dL) | 220 | 44 | 160 | |
| Percent Change | -16 (-26, -7) | +22 (+15, +34) | -38 (-52, -14) | |
| * Represents pooled analyses of results; minimum duration on therapy at each dose was 4 weeks. | ||||
Gender Effect: Combined data from the three placebo-controlled NIASPAN studies in patients with primary hyperlipidemia and mixed dyslipidemia suggest that, at each NIASPAN dose level studied, changes in lipid concentrations are greater for women than for men (Table 6).
| Mean Percent Change from Baseline | |||||||||
| NIASPAN | n | LDL-C | HDL-C | TG | Apo B | ||||
| Dose | (M/F) | M | F | M | F | M | F | M | F |
| 500 mg at bedtime | 50/37 | -2 | -5 | +11 | +8 | -3 | -9 | -1 | -5 |
| 1000 mg at bedtime | 76/52 | -6* | -11* | +14 | +20 | -10 | -20 | -5* | -10* |
| 1500 mg at bedtime | 104/59 | -12 | -16 | +19 | +24 | -17 | -28 | -13 | -15 |
| 2000 mg at bedtime | 75/53 | -15 | -18 | +23 | +26 | -30 | -36 | -16 | -16 |
| n = number of male/female patients enrolled. * Percent change significantly different between genders (p<0.05). |
|||||||||
Other Patient Populations: In a double-blind, multi-center, 19-week study the lipid-altering effects of NIASPAN (forced titration to 2000 mg at bedtime) were compared to baseline in patients whose primary lipid abnormality was a low level of HDL-C (HDL-C ≤40 mg/dL, TG ≤400 mg/dL, and LDL-C ≤160, or <130 mg/dL in the presence of CHD). Results are shown below (Table 7).
| Mean Baseline and Mean Percent Change from Baseline* | ||||||
| n | TC | LDL-C | HDL-C | TG | Apo B† | |
| Baseline (mg/dL) | 88 | 190 | 120 | 31 | 194 | 106 |
| Week 19 (% Change) | 71 | -3 | 0 | +26 | -30 | -9 |
| n = number of patients * Mean percent change from baseline was significantly different (p<0.05) for all lipid parameters shown except LDL-C. † n=72 at baseline and 69 at week 19. |
||||||
At NIASPAN 2000 mg/day, median changes from baseline (25th, 75th percentiles) for LDL-C, HDL-C, and TG were -3% (-14, +12%), +27% (+13, +38%), and -33% (-50, -19%), respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
NIASPAN tablets are supplied as unscored, medium-orange, film-coated, capsule-shaped (containing 500 or 750 mg of niacin) or oval shaped (containing 1000 mg of niacin) tablets, in an extended-release formulation. Tablets are supplied in bottles of 90 as shown below.
- 500 mg tablets: NDC# 0074-3265-90 (printed with 500)
- 750 mg tablets: NDC# 0074-3274-90 (printed with 750)
- 1000 mg tablets: NDC# 0074-3275-90 (printed with 1000)
Storage: Store at room temperature 20° to 25°C (68° to 77°F).
17 PATIENT COUNSELING INFORMATION
17.1 Patient Counseling
Patients should be advised to adhere to their National Cholesterol Education Program (NCEP) recommended diet, a regular exercise program, and periodic testing of a fasting lipid panel.
Patients should be advised to inform other healthcare professionals prescribing a new medication that they are taking NIASPAN.
The patient should be informed of the following:
Dosing Time
NIASPAN tablets should be taken at bedtime, after a low-fat snack. Administration on an empty stomach is not recommended.
Tablet Integrity
NIASPAN tablets should not be broken, crushed or chewed, but should be swallowed whole.
Dosing Interruption
If dosing is interrupted for any length of time, their physician should be contacted prior to restarting therapy; re-titration is recommended.
Muscle Pain
Notify their physician of any unexplained muscle pain, tenderness, or weakness promptly. They should discuss all medication, both prescription and over the counter, with their physician.
Flushing
Flushing (warmth, redness, itching and/or tingling of the skin) is a common side effect of niacin therapy that may subside after several weeks of consistent NIASPAN use. Flushing may vary in severity and is more likely to occur with initiation of therapy, or during dose increases. By dosing at bedtime, flushing will most likely occur during sleep. However, if awakened by flushing at night, the patient should get up slowly, especially if feeling dizzy, feeling faint, or taking blood pressure medications. Advise patients of the symptoms of flushing and how they differ from the symptoms of a myocardial infarction.
Use of Aspirin Medication
Taking aspirin (up to the recommended dose of 325 mg) approximately 30 minutes before dosing can minimize flushing.
Diet
Avoid ingestion of alcohol, hot beverages and spicy foods around the time of taking NIASPAN to minimize flushing.
Supplements
Notify their physician if they are taking vitamins or other nutritional supplements containing niacin or nicotinamide.
Dizziness
Notify their physician if symptoms of dizziness occur.
Diabetics
If diabetic, to notify their physician of changes in blood glucose.
Pregnancy
Advise patients to inform their healthcare provider of a known or suspected pregnancy to discuss if NIASPAN should be discontinued [see Use in Specific Populations (8.1)].
Lactation
Advise patients not to breastfeed during treatment with NIASPAN.
© 1997-2022 AbbVie. All rights reserved.
NIASPAN® is a registered trademark of AbbVie Respiratory LLC.
Manufactured by:
AbbVie LTD, Barceloneta, PR 00617
For AbbVie Inc.
North Chicago, IL 60064, USA
May, 2022
| PATIENT INFORMATION
NIASPAN® (ny-a-span) (niacin extended-release tablets) for oral use |
| Read this information carefully before you start taking NIASPAN and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. |
| What is NIASPAN?
NIASPAN is a prescription medicine used with diet and exercise to increase the good cholesterol (HDL) and lower the bad cholesterol (LDL) and fats (triglycerides) in your blood.
|
| Who should not take NIASPAN?
Do not take NIASPAN if you have:
|
| What should I tell my doctor before taking NIASPAN?
Before you take NIASPAN, tell your doctor about all your medical problems including, if you:
Especially tell your doctor if you take:
|
How should I take NIASPAN?
|
| What are the possible side effects of NIASPAN?
NIASPAN may cause serious side effects, including:
The most common side effects of NIASPAN include:
If you wake up at night because of flushing, get up slowly, especially if you:
Call your doctor right away if you have any symptoms of a heart attack. Tell your doctor if you have any side effect that bothers you or does not go away. These are not all the possible side effects of NIASPAN. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
How should I store NIASPAN?
|
| General information about the safe and effective use of NIASPAN.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use NIASPAN for a condition for which it was not prescribed. Do not give NIASPAN to other people, even if they have the same symptoms that you have. It may harm them. This Patient Information leaflet summarizes the most important information about NIASPAN. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about NIASPAN that is written for health professionals. |
| What are the ingredients in NIASPAN?
Active ingredient: niacin Inactive Ingredients: hypromellose, povidone, stearic acid, and polyethylene glycol, and the following coloring agents: FD&C yellow #6/sunset yellow FCF Aluminum Lake, synthetic red and yellow iron oxides, and titanium dioxide Manufactured by AbbVie LTD, Barceloneta, PR 00617 For AbbVie Inc. North Chicago, IL 60064, USA For more information, go to www.NIASPAN.com or call AbbVie Inc. Medical Information at 1-800-633-9110. |
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 5/2022







| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| NIASPAN
niacin tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - AbbVie Inc. (078458370) |