Abrupt discontinuation of intrathecal baclofen, regardless of the cause, has resulted in sequelae that include high fever, altered mental status, exaggerated rebound spasticity, and muscle rigidity, that in rare cases has advanced to rhabdomyolysis, multiple organ-system failure and death.
Prevention of abrupt discontinuation of intrathecal baclofen requires careful attention to programming and monitoring of the infusion system, refill scheduling and procedures, and pump alarms. Patients and caregivers should be advised of the importance of keeping scheduled refill visits and should be educated on the early symptoms of baclofen withdrawal. Special attention should be given to patients at apparent risk (e.g. spinal cord injuries at T-6 or above, communication difficulties, history of withdrawal symptoms from oral or intrathecal baclofen). Consult the technical manual of the implantable infusion system for additional postimplant clinician and patient information (see WARNINGS).
DESCRIPTION
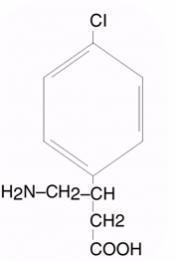
LIORESAL® INTRATHECAL (baclofen injection) is a muscle relaxant and antispastic. Its chemical name is 4-amino-3-(4-chlorophenyl) butanoic acid, and its structural formula is:
Baclofen is a white to off-white, odorless or practically odorless crystalline powder, with a molecular weight of 213.66. It is slightly soluble in water, very slightly soluble in methanol, and insoluble in chloroform.
LIORESAL INTRATHECAL is a sterile, pyrogen-free, isotonic solution free of antioxidants, preservatives or other potentially neurotoxic additives indicated only for intrathecal administration. The drug is stable in solution at 37° C and compatible with CSF. Each milliliter of LIORESAL INTRATHECAL contains baclofen U.S.P. 50 mcg, 500 mcg or 2000 mcg and sodium chloride 9 mg in Water for Injection; pH range is 5.0-7.0. Each ampule is intended for SINGLE USE ONLY. Discard any unused portion. DO NOT AUTOCLAVE.
CLINICAL PHARMACOLOGY
The precise mechanism of action of baclofen as a muscle relaxant and antispasticity agent is not fully understood. Baclofen inhibits both monosynaptic and polysynaptic reflexes at the spinal level, possibly by decreasing excitatory neurotransmitter release from primary afferent terminals, although actions at supraspinal sites may also occur and contribute to its clinical effect. Baclofen is a structural analog of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), and may exert its effects by stimulation of the GABAB receptor subtype.
LIORESAL INTRATHECAL when introduced directly into the intrathecal space permits effective CSF concentrations to be achieved with resultant plasma concentrations 100 times less than those occurring with oral administration.
In people, as well as in animals, baclofen has been shown to have general CNS depressant properties as indicated by the production of sedation with tolerance, somnolence, ataxia, and respiratory and cardiovascular depression.
Pharmacodynamics of LIORESAL INTRATHECAL:
Intrathecal Bolus:Adult Patients: The onset of action is generally one-half hour to one hour after an intrathecal bolus. Peak spasmolytic effect is seen at approximately four hours after dosing and effects may last four to eight hours. Onset, peak response, and duration of action may vary with individual patients depending on the dose and severity of symptoms.
Pediatric Patients: The onset, peak response and duration of action is similar to those seen in adult patients.
Continuous Infusion:LIORESAL INTRATHECAL’S antispastic action is first seen at 6 to 8 hours after initiation of continuous infusion. Maximum activity is observed in 24 to 48 hours.
Continuous Infusion: No additional information is available for pediatric patients.
Pharmacokinetics of LIORESAL INTRATHECAL:
The pharmacokinetics of CSF clearance of LIORESAL INTRATHECAL calculated from intrathecal bolus or continuous infusion studies approximates CSF turnover, suggesting elimination is by bulk-flow removal of CSF.
Intrathecal Bolus: After a bolus lumbar injection of 50 or 100 mcg LIORESAL INTRATHECAL in seven patients, the average CSF elimination half-life was 1.51 hours over the first four hours and the average CSF clearance was approximately 30 mL/hour.
Continuous Infusion: The mean CSF clearance for LIORESAL INTRATHECAL (baclofen injection) was approximately 30 mL/hour in a study involving ten patients on continuous intrathecal infusion. Concurrent plasma concentrations of baclofen during intrathecal administration are expected to be low (0-5 ng/mL).
Limited pharmacokinetic data suggest that a lumbar-cisternal concentration gradient of about 4:1 is established along the neuroaxis during baclofen infusion. This is based upon simultaneous CSF sampling via cisternal and lumbar tap in 5 patients receiving continuous baclofen infusion at the lumbar level at doses associated with therapeutic efficacy; the interpatient variability was great. The gradient was not altered by position.
Six pediatric patients (age 8-18 years) receiving continuous intrathecal baclofen infusion at doses of 77-400 mcg/day had plasma baclofen levels near or below 10 ng/mL.
INDICATIONS AND USAGE
LIORESAL INTRATHECAL (baclofen injection) is indicated for use in the management of severe spasticity. Patients should first respond to a screening dose of intrathecal baclofen prior to consideration for long term infusion via an implantable pump. For spasticity of spinal cord origin, chronic infusion of LIORESAL INTRATHECAL via an implantable pump should be reserved for patients unresponsive to oral baclofen therapy, or those who experience intolerable CNS side effects at effective doses. Patients with spasticity due to traumatic brain injury should wait at least one year after the injury before consideration of long term intrathecal baclofen therapy. LIORESAL INTRATHECAL is intended for use by the intrathecal route in single bolus test doses (via spinal catheter or lumbar puncture) and, for chronic use, only in implantable pumps approved by the FDA specifically for the administration of LIORESAL INTRATHECAL into the intrathecal space.
Spasticity of Spinal Cord Origin: Evidence supporting the efficacy of LIORESAL INTRATHECAL was obtained in randomized, controlled investigations that compared the effects of either a single intrathecal dose or a three day intrathecal infusion of LIORESAL INTRATHECAL to placebo in patients with severe spasticity and spasms due to either spinal cord trauma or multiple sclerosis. LIORESAL INTRATHECAL was superior to placebo on both principal outcome measures employed: change from baseline in the Ashworth rating of spasticity and the frequency of spasms.
Spasticity of Cerebral Origin: The efficacy of LIORESAL INTRATHECAL was investigated in three controlled clinical trials; two enrolled patients with cerebral palsy and one enrolled patients with spasticity due to previous brain injury. The first study, a randomized controlled cross-over trial of 51 patients with cerebral palsy, provided strong, statistically significant results; LIORESAL INTRATHECAL was superior to placebo in reducing spasticity as measured by the Ashworth Scale. A second cross-over study was conducted in 11 patients with spasticity arising from brain injury. Despite the small sample size, the study yielded a nearly significant test statistic (p=0.066) and provided directionally favorable results. The last study, however, did not provide data that could be reliably analyzed.
LIORESAL INTRATHECAL therapy may be considered an alternative to destructive neurosurgical procedures. Prior to implantation of a device for chronic intrathecal infusion of LIORESAL INTRATHECAL, patients must show a response to LIORESAL INTRATHECAL in a screening trial (see Dosage and Administration).
CONTRAINDICATIONS
Hypersensitivity to baclofen. LIORESAL INTRATHECAL is not recommended for intravenous, intramuscular, subcutaneous or epidural administration.
WARNINGS
LIORESAL INTRATHECAL is for use in single bolus intrathecal injections (via a catheter placed in the lumbar intrathecal space or injection by lumbar puncture) and in implantable pumps approved by the FDA specifically for the intrathecal administration of baclofen. Because of the possibility of potentially life-threatening CNS depression, cardiovascular collapse, and/or respiratory failure, physicians must be adequately trained and educated in chronic intrathecal infusion therapy.
The pump system should not be implanted until the patient’s response to bolus LIORESAL INTRATHECAL injection is adequately evaluated. Evaluation (consisting of a screening procedure: see Dosage and Administration) requires that LIORESAL INTRATHECAL be administered into the intrathecal space via a catheter or lumbar puncture. Because of the risks associated with the screening procedure and the adjustment of dosage following pump implantation, these phases must be conducted in a medically supervised and adequately equipped environment following the instructions outlined in the Dosage and Administration section.
Resuscitative equipment should be available.
Following surgical implantation of the pump, particularly during the initial phases of pump use, the patient should be monitored closely until it is certain that the patient’s response to the infusion is acceptable and reasonably stable.
On each occasion that the dosing rate of the pump and/or the concentration of LIORESAL INTRATHECAL (baclofen injection) in the reservoir is adjusted, close medical monitoring is required until it is certain that the patient’s response to the infusion is acceptable and reasonably stable.
It is mandatory that the patient, all patient caregivers, and the physicians responsible for the patient receive adequate information regarding the risks of this mode of treatment. All medical personnel and caregivers should be instructed in 1) the signs and symptoms of overdose, 2) procedures to be followed in the event of overdose and 3) proper home care of the pump and insertion site.
Overdose: Signs of overdose may appear suddenly or insidiously. Acute massive overdose may present as coma. Less sudden and/or less severe forms of overdose may present with signs of drowsiness, lightheadedness, dizziness, somnolence, respiratory depression, seizures, rostral progression of hypotonia and loss of consciousness progressing to coma. Should overdose appear likely, the patient should be taken immediately to a hospital for assessment and emptying of the pump reservoir. In cases reported to date, overdose has generally been related to pump malfunction, inadvertent subcutaneous injection, or dosing error. (See Drug Overdose Symptoms and Treatment.)
Extreme caution must be used when filling an FDA approved implantable pump. Such pumps should only be refilled through the reservoir refill septum. Inadvertent injection into the subcutaneous tissue can occur if the reservoir refill septum is not properly accessed. Some pumps are also equipped with a catheter access port that allows direct access to the intrathecal catheter. Direct injection into this catheter access port or inadvertent injection into the subcutaneous tissue may cause a life-threatening overdose.
Withdrawal: Abrupt withdrawal of intrathecal baclofen, regardless of the cause, has resulted in sequelae that included high fever, altered mental status, exaggerated rebound spasticity and muscle rigidity that in rare cases progressed to rhabdomyolysis, multiple organ-system failure, and death. In the first 9 years of post-marketing experience, 27 cases of withdrawal temporally related to the cessation of baclofen therapy were reported; six patients died. In most cases, symptoms of withdrawal appeared within hours to a few days following interruption of baclofen therapy. Common reasons for abrupt interruption of intrathecal baclofen therapy included malfunction of the catheter (especially disconnection), low volume in the pump reservoir, and end of pump battery life; human error may have played a causal or contributing role in some cases. Cases of intrathecal mass at the tip of the implanted catheter leading to withdrawal symptoms have also been reported, most of them involving pharmacy compounded analgesic admixtures (see PRECAUTIONS).
Prevention of abrupt discontinuation of intrathecal baclofen requires careful attention to programming and monitoring of the infusion system, refill scheduling and procedures, and pump alarms. Patients and caregivers should be advised of the importance of keeping scheduled refill visits and should be educated on the early symptoms of baclofen withdrawal.
All patients receiving intrathecal baclofen therapy are potentially at risk for withdrawal. Early symptoms of baclofen withdrawal may include return of baseline spasticity, pruritus, hypotension, and paresthesias. Priapism may develop or recur if treatment with intrathecal baclofen is interrupted. Some clinical characteristics of the advanced intrathecal baclofen withdrawal syndrome may resemble autonomic dysreflexia, infection (sepsis), malignant hyperthermia, neuroleptic-malignant syndrome, or other conditions associated with a hypermetabolic state or widespread rhabdomyolysis.
Rapid, accurate diagnosis and treatment in an emergency-room or intensive-care setting are important in order to prevent the potentially life-threatening central nervous system and systemic effects of intrathecal baclofen withdrawal. The suggested treatment for intrathecal baclofen withdrawal is the restoration of intrathecal baclofen at or near the same dosage as before therapy was interrupted. However, if restoration of intrathecal delivery is delayed, treatment with GABA-ergic agonist drugs such as oral or enteral baclofen, or oral, enteral, or intravenous benzodiazepines may prevent potentially fatal sequelae. Oral or enteral baclofen alone should not be relied upon to halt the progression of intrathecal baclofen withdrawal.
Seizures have been reported during overdose and with withdrawal from LIORESAL INTRATHECAL as well as in patients maintained on therapeutic doses of LIORESAL INTRATHECAL.
Fatalities:
Spasticity of Spinal Cord Origin: There were 16 deaths reported among the 576 U.S. patients treated with LIORESAL INTRATHECAL (baclofen injection) in pre- and post-marketing studies evaluated as of December 1992. Because these patients were treated under uncontrolled clinical settings, it is impossible to determine definitively what role, if any, LIORESAL INTRATHECAL played in their deaths.
As a group, the patients who died were relatively young (mean age was 47 with a range from 25 to 63), but the majority suffered from severe spasticity of many years duration, were nonambulatory, had various medical complications such as pneumonia, urinary tract infections, and decubiti, and/or had received multiple concomitant medications. A case-by-case review of the clinical course of the 16 patients who died failed to reveal any unique signs, symptoms, or laboratory results that would suggest that treatment with LIORESAL INTRATHECAL caused their deaths. Two patients, however, did suffer sudden and unexpected death within 2 weeks of pump implantation and one patient died unexpectedly after screening.
One patient, a 44 year-old male with MS, died in hospital on the second day following pump implantation. An autopsy demonstrated severe fibrosis of the coronary conduction system. A second patient, a 52 year-old woman with MS and a history of an inferior wall myocardial infarction, was found dead in bed 12 days after pump implantation, 2 hours after having had documented normal vital signs. An autopsy revealed pulmonary congestion and bilateral pleural effusions. It is impossible to determine whether LIORESAL INTRATHECAL contributed to these deaths. The third patient underwent three baclofen screening trials. His medical history included SCI, aspiration pneumonia, septic shock, disseminated intravascular coagulopathy, severe metabolic acidosis, hepatic toxicity, and status epilepticus. Twelve days after screening (he was not implanted), he again experienced status epilepticus with subsequent significant neurological deterioration. Based upon prior instruction, extraordinary resuscitative measures were not pursued and the patient died.
Spasticity of Cerebral Origin: There were three deaths occurring among the 211 patients treated with LIORESAL INTRATHECAL in pre-marketing studies as of March 1996. These deaths were not attributed to the therapy.
PRECAUTIONS
Children should be of sufficient body mass to accommodate the implantable pump for chronic infusion. Please consult pump manufacturer's manual for specific recommendations.
Safety and effectiveness in pediatric patients below the age of 4 have not been established.
Screening
Patients should be infection-free prior to the screening trial
with LIORESAL INTRATHECAL (baclofen injection) because the presence of a
systemic infection may interfere with an assessment of the patient’s response to
bolus LIORESAL INTRATHECAL.
Pump Implantation
Patients should be infection-free prior to pump implantation
because the presence of infection may increase the risk of surgical
complications. Moreover, a systemic infection may complicate dosing.
Pump Dose Adjustment and Titration
In most patients, it will be necessary to increase the dose
gradually over time to maintain effectiveness; a sudden requirement for
substantial dose escalation typically indicates a catheter complication (i.e.,
catheter kink or dislodgement).
Reservoir refilling must be performed by fully trained and qualified personnel following the directions provided by the pump manufacturer. Inadvertent injection into the subcutaneous tissue can occur if the reservoir refill septum is not properly accessed. Subcutaneous injection may result in symptoms of a systemic overdose or early depletion of the reservoir. Refill intervals should be carefully calculated to prevent depletion of the reservoir, as this would result in the return of severe spasticity and possibly symptoms of withdrawal.
Strict aseptic technique in filling is required to avoid bacterial contamination and serious infection. A period of observation appropriate to the clinical situation should follow each refill or manipulation of the drug reservoir.
Extreme caution must be used when filling an FDA approved implantable pump equipped with an injection port that allows direct access to the intrathecal catheter. Direct injection into the catheter through the catheter access port may cause a life-threatening overdose.
Additional considerations pertaining to dosage adjustment: It may be important to titrate the dose to maintain some degree of muscle tone and allow occasional spasms to: 1) help support circulatory function, 2) possibly prevent the formation of deep vein thrombosis, 3) optimize activities of daily living and ease of care.
Except in overdose related emergencies, the dose of LIORESAL INTRATHECAL should ordinarily be reduced slowly if the drug is discontinued for any reason.
An attempt should be made to discontinue concomitant oral antispasticity medication to avoid possible overdose or adverse drug interactions, either prior to screening or following implant and initiation of chronic LIORESAL INTRATHECAL infusion. Reduction and discontinuation of oral anti-spasmotics should be done slowly and with careful monitoring by the physician. Abrupt reduction or discontinuation of concomitant antispastics should be avoided.
Drowsiness: Drowsiness has been reported in patients on LIORESAL INTRATHECAL. Patients should be cautioned regarding the operation of automobiles or other dangerous machinery, and activities made hazardous by decreased alertness. Patients should also be cautioned that the central nervous system depressant effects of LIORESAL INTRATHECAL (baclofen injection) may be additive to those of alcohol and other CNS depressants.
Intrathecal mass: Cases of intrathecal mass at the tip of the implanted catheter have been reported, most of them involving pharmacy compounded analgesic admixtures. The most frequent symptoms associated with intrathecal mass are: 1) decreased therapeutic response (worsening spasticity, return of spasticity when previously well controlled, withdrawal symptoms, poor response to escalating doses, or frequent or large dosage increases), 2) pain, 3) neurological deficit/dysfunction. Clinicians should monitor patients on intraspinal therapy carefully for any new neurological signs or symptoms. In patients with new neurological signs or symptoms suggestive of an intrathecal mass, consider a neurosurgical consultation, since many of the symptoms of inflammatory mass are not unlike the symptoms experienced by patients with severe spasticity from their disease. In some cases, performance of an imaging procedure may be appropriate to confirm or rule-out the diagnosis of an intrathecal mass.
Precautions in special patient populations: Careful dose titration of LIORESAL INTRATHECAL is needed when spasticity is necessary to sustain upright posture and balance in locomotion or whenever spasticity is used to obtain optimal function and care.
Patients suffering from psychotic disorders, schizophrenia, or confusional states should be treated cautiously with LIORESAL INTRATHECAL and kept under careful surveillance, because exacerbations of these conditions have been observed with oral administration.
LIORESAL INTRATHECAL should be used with caution in patients with a history of autonomic dysreflexia. The presence of nociceptive stimuli or abrupt withdrawal of LIORESAL INTRATHECAL (baclofen injection) may cause an autonomic dysreflexic episode.
Because LIORESAL is primarily excreted unchanged by the kidneys, it should be given with caution in patients with impaired renal function and it may be necessary to reduce the dosage.
LABORATORY TESTS
No specific laboratory tests are deemed essential for the management of patients on LIORESAL INTRATHECAL.
DRUG INTERACTIONS
There is inadequate systematic experience with the use of LIORESAL INTRATHECAL in combination with other medications to predict specific drug-drug interactions. Interactions attributed to the combined use of LIORESAL INTRATHECAL and epidural morphine include hypotension and dyspnea.
CARCINOGENESIS, MUTAGENESIS, AND IMPAIRMENT OF FERTILITY
No increase in tumors was seen in rats receiving baclofen orally for two years. Adequate genotoxicity assays of baclofen have not been performed.
PREGNANCY
There are no adequate and well-controlled studies in pregnant women. In animal studies, baclofen had adverse effects on embryofetal development when administered orally to pregnant rats. LIORESAL INTRATHECAL should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Baclofen given orally increased the incidence of fetal structural abnormalities (omphaloceles) in rats. Reductions in food intake and body weight gain were observed in the dams. Fetal structural abnormalities were not observed in mice or rabbits
NURSING MOTHERS
In mothers treated with oral LIORESAL (baclofen USP) in therapeutic doses, the active substance passes into the milk. It is not known whether detectable levels of drug are present in milk of nursing mothers receiving LIORESAL INTRATHECAL. As a general rule, nursing should be undertaken while a patient is receiving LIORESAL INTRATHECAL only if the potential benefit justifies the potential risks to the infant.
PEDIATRIC USE
Children should be of sufficient body mass to accommodate the implantable pump for chronic infusion. Please consult pump manufacturer's manual for specific recommendations.
Safety and effectiveness in pediatric patients below the age of 4 have not been established. Considerations based on experience with oral LIORESAL (baclofen USP)
A dose-related increase in incidence of ovarian cysts was observed in female rats treated chronically with oral LIORESAL. Ovarian cysts have been found by palpation in about 4% of the multiple sclerosis patients who were treated with oral LIORESAL for up to one year. In most cases these cysts disappeared spontaneously while patients continued to receive the drug. Ovarian cysts are estimated to occur spontaneously in approximately 1% to 5% of the normal female population.
ADVERSE REACTIONS
Spasticity of Spinal Cord Origin – Clinical Studies:
Commonly Observed in Patients with Spasticity of Spinal Origin — In pre- and post-marketing clinical trials, the most commonly observed adverse events associated with use of LIORESAL INTRATHECAL (baclofen injection) which were not seen at an equivalent incidence among placebo-treated patients were: somnolence, dizziness, nausea, hypotension, headache, convulsions and hypotonia.
Associated with Discontinuation of Treatment — 8/474 patients with spasticity of spinal cord origin receiving long term infusion of LIORESAL INTRATHECAL in pre- and post-marketing clinical studies in the U.S. discontinued treatment due to adverse events. These include: pump pocket infections (3), meningitis (2), wound dehiscence (1), gynecological fibroids (1) and pump overpressurization (1) with unknown, if any, sequela. Eleven patients who developed coma secondary to overdose had their treatment temporarily suspended, but all were subsequently re-started and were not, therefore, considered to be true discontinuations.
Fatalities — See Warnings.
Incidence in Controlled Trials — Experience with LIORESAL INTRATHECAL (baclofen injection) obtained in parallel, placebo-controlled, randomized studies provides only a limited basis for estimating the incidence of adverse events because the studies were of very brief duration (up to three days of infusion) and involved only a total of 63 patients. The following events occurred among the 31 patients receiving LIORESAL INTRATHECAL (baclofen injection) in two randomized, placebo-controlled trials: hypotension (2), dizziness (2), headache (2), dyspnea (1). No adverse events were reported among the 32 patients receiving placebo in these studies.
Events Observed during the Pre- and Post-marketing Evaluation of LIORESAL INTRATHECAL — Adverse events associated with the use of LIORESAL INTRATHECAL reflect experience gained with 576 patients followed prospectively in the United States. They received LIORESAL INTRATHECAL for periods of one day (screening) (N = 576) to over eight years (maintenance) (N = 10). The usual screening bolus dose administered prior to pump implantation in these studies was typically 50 mcg. The maintenance dose ranged from 12 mcg to 2003 mcg per day. Because of the open, uncontrolled nature of the experience, a causal linkage between events observed and the administration of LIORESAL INTRATHECAL cannot be reliably assessed in many cases and many of the adverse events reported are known to occur in association with the underlying conditions being treated. Nonetheless, many of the more commonly reported reactions—hypotonia, somnolence, dizziness, paresthesia, nausea/vomiting and headache—appear clearly drug-related.
Adverse experiences reported during all U.S. studies (both controlled and uncontrolled) are shown in the following table. Eight of 474 patients who received chronic infusion via implanted pumps had adverse experiences which led to a discontinuation of long term treatment in the pre- and post-marketing studies.
| INCIDENCE OF MOST FREQUENT (≥1%) ADVERSE EVENTS IN PATIENTS WITH SPASTICITY OF SPINAL ORIGIN IN PROSPECTIVELY MONITORED CLINICAL TRIALS | |||
|---|---|---|---|
|
a Following administration of test bolus b Two month period following implant c Beyond two months following implant N= total number of patients entering each period %=% of patients evaluated |
|||
| Percent of Patients Reporting Events | |||
| N = 576 | N = 474 | N = 430 | |
| Screeninga | Titrationb | Maintenancec | |
| Adverse Event | Percent | Percent | Percent |
| Hypotonia | 5.4 | 13.5 | 25.3 |
| Somnolence | 5.7 | 5.9 | 20.9 |
| Dizziness | 1.7 | 1.9 | 7.9 |
| Paresthesia | 2.4 | 2.1 | 6.7 |
| Nausea and Vomiting | 1.6 | 2.3 | 5.6 |
| Headache | 1.6 | 2.5 | 5.1 |
| Constipation | 0.2 | 1.5 | 5.1 |
| Convulsion | 0.5 | 1.3 | 4.7 |
| Urinary Retention | 0.7 | 1.7 | 1.9 |
| Dry Mouth | 0.2 | 0.4 | 3.3 |
| Accidental Injury | 0.0 | 0.2 | 3.5 |
| Asthenia | 0.7 | 1.3 | 1.4 |
| Confusion | 0.5 | 0.6 | 2.3 |
| Death | 0.2 | 0.4 | 3.0 |
| Pain | 0.0 | 0.6 | 3.0 |
| Speech Disorder | 0.0 | 0.2 | 3.5 |
| Hypotension | 1.0 | 0.2 | 1.9 |
| Ambylopia | 0.5 | 0.2 | 2.3 |
| Diarrhea | 0.0 | 0.8 | 2.3 |
| Hypoventilation | 0.2 | 0.8 | 2.1 |
| Coma | 0.0 | 1.5 | 0.9 |
| Impotence | 0.2 | 0.4 | 1.6 |
| Peripheral Edema | 0.0 | 0.0 | 2.3 |
| Urinary Incontinence | 0.0 | 0.8 | 1.4 |
| Insomnia | 0.0 | 0.4 | 1.6 |
| Anxiety | 0.2 | 0.4 | 0.9 |
| Depression | 0.0 | 0.0 | 1.6 |
| Dyspnea | 0.3 | 0.0 | 1.2 |
| Fever | 0.5 | 0.2 | 0.7 |
| Pneumonia | 0.2 | 0.2 | 1.2 |
| Urinary Frequency | 0.0 | 0.6 | 0.9 |
| Urticaria | 0.2 | 0.2 | 1.2 |
| Anorexia | 0.0 | 0.4 | 0.9 |
| Diplopia | 0.0 | 0.4 | 0.9 |
| Dysautonomia | 0.2 | 0.2 | 0.9 |
| Hallucinations | 0.3 | 0.4 | 0.5 |
| Hypertension | 0.2 | 0.6 | 0.5 |
In addition to the more common (1% or more) adverse events reported in the prospectively followed 576 domestic patients in pre- and post-marketing studies, experience from an additional 194 patients exposed to LIORESAL INTRATHECAL (baclofen injection) from foreign studies has been reported. The following adverse events, not described in the table, and arranged in decreasing order of frequency, and classified by body system, were reported:
Nervous System: Abnormal gait, thinking abnormal, tremor, amnesia, twitching, vasodilitation, cerebrovascular accident, nystagmus, personality disorder, psychotic depression, cerebral ischemia, emotional lability, euphoria, hypertonia, ileus, drug dependence, incoordination, paranoid reaction and ptosis.
Digestive System: Flatulence, dysphagia, dyspepsia and gastroenteritis.
Cardiovascular: Postural hypotension, bradycardia, palpitations, syncope, arrhythmia ventricular, deep thrombophlebitis, pallor and tachycardia.
Respiratory: Respiratory disorder, aspiration pneumonia, hyperventilation, pulmonary embolus and rhinitis.
Urogenital: Hematuria and kidney failure.
Skin and Appendages: Alopecia and sweating.
Metabolic and Nutritional Disorders: Weight loss, albuminuria, dehydration and hyperglycemia.
Special Senses: Abnormal vision, abnormality of accommodation, photophobia, taste loss and tinnitus.
Body as a Whole: Suicide, lack of drug effect, abdominal pain, hypothermia, neck rigidity, chest pain, chills, face edema, flu syndrome and overdose.
Hemic and Lymphatic System: Anemia.
Spasticity of Cerebral Origin – Clinical Studies:
Commonly Observed — In pre-marketing clinical trials, the most commonly observed adverse events associated with use of LIORESAL INTRATHECAL (baclofen injection) which were not seen at an equivalent incidence among placebo-treated patients included: agitation, constipation, somnolence, leukocytosis, chills, urinary retention and hypotonia.
Associated with Discontinuation of Treatment — Nine of 211 patients receiving LIORESAL INTRATHECAL in pre-marketing clinical studies in the U.S. discontinued long term infusion due to adverse events associated with intrathecal therapy.
The nine adverse events leading to discontinuation were: infection (3), CSF leaks (2), meningitis (2), drainage (1), and unmanageable trunk control (1).
Fatalities — Three deaths, none of which were attributed to LIORESAL INTRATHECAL, were reported in patients in clinical trials involving patients with spasticity of cerebral origin. See Warnings on other deaths reported in spinal spasticity patients.
Incidence in Controlled Trials — Experience with LIORESAL INTRATHECAL (baclofen injection) obtained in parallel, placebo-controlled, randomized studies provides only a limited basis for estimating the incidence of adverse events because the studies involved a total of 62 patients exposed to a single 50 mcg intrathecal bolus. The following events occurred among the 62 patients receiving LIORESAL INTRATHECAL in two randomized, placebo-controlled trials involving cerebral palsy and head injury patients, respectively: agitation, constipation, somnolence, leukocytosis, nausea, vomiting, nystagmus, chills, urinary retention, and hypotonia.
Events Observed during the Pre-marketing Evaluation of LIORESAL INTRATHECAL — Adverse events associated with the use of LIORESAL INTRATHECAL reflect experience gained with a total of 211 U.S. patients with spasticity of cerebral origin, of whom 112 were pediatric patients (under age 16 at enrollment). They received LIORESAL INTRATHECAL for periods of one day (screening) (N=211) to 84 months (maintenance) (N=1). The usual screening bolus dose administered prior to pump implantation in these studies was 50-75 mcg. The maintenance dose ranged from 22 mcg to 1400 mcg per day. Doses used in this patient population for long term infusion are generally lower than those required for patients with spasticity of spinal cord origin.
Because of the open, uncontrolled nature of the experience, a causal linkage between events observed and the administration of LIORESAL INTRATHECAL cannot be reliably assessed in many cases. Nonetheless, many of the more commonly reported reactions—somnolence, dizziness, headache, nausea, hypotension, hypotonia and coma—appear clearly drug-related.
The most frequent (≥1%) adverse events reported during all clinical trials are shown in the following table. Nine patients discontinued long term treatment due to adverse events.
| INCIDENCE OF MOST FREQUENT (≥ 1%) ADVERSE EVENTS IN PATIENTS WITH SPASTICITY OF CEREBRAL ORIGIN IN PROSPECTIVELY MONITORED CLINICAL TRIALS | |||
|---|---|---|---|
|
a Following administration of test bolus b Two month period following implant c Beyond two months following implant N= Total number of patients entering each period. 211 patients received drug; (1 of 212) received placebo only. |
|||
| Percent of Patients Reporting Events | |||
| N = 211 | N = 153 | N = 150 | |
| Screeninga | Titrationb | Maintenancec | |
| Adverse Event | Percent | Percent | Percent |
| Hypotonia | 2.4 | 14.4 | 34.7 |
| Somnolence | 7.6 | 10.5 | 18.7 |
| Headache | 6.6 | 7.8 | 10.7 |
| Nausea and Vomiting | 6.6 | 10.5 | 4.0 |
| Vomiting | 6.2 | 8.5 | 4.0 |
| Urinary Retention | 0.9 | 6.5 | 8.0 |
| Convulsion | 0.9 | 3.3 | 10.0 |
| Dizziness | 2.4 | 2.6 | 8.0 |
| Nausea | 1.4 | 3.3 | 7.3 |
| Hypoventilation | 1.4 | 1.3 | 4.0 |
| Hypertonia | 0.0 | 0.7 | 6.0 |
| Paresthesia | 1.9 | 0.7 | 3.3 |
| Hypotension | 1.9 | 0.7 | 2.0 |
| Increased Salivation | 0.0 | 2.6 | 2.7 |
| Back Pain | 0.9 | 0.7 | 2.0 |
| Constipation | 0.5 | 1.3 | 2.0 |
| Pain | 0.0 | 0.0 | 4.0 |
| Pruritus | 0.0 | 0.0 | 4.0 |
| Diarrhea | 0.5 | 0.7 | 2.0 |
| Peripheral Edema | 0.0 | 0.0 | 3.3 |
| Thinking Abnormal | 0.5 | 1.3 | 0.7 |
| Agitation | 0.5 | 0.0 | 1.3 |
| Asthenia | 0.0 | 0.0 | 2.0 |
| Chills | 0.5 | 0.0 | 1.3 |
| Coma | 0.5 | 0.0 | 1.3 |
| Dry Mouth | 0.5 | 0.0 | 1.3 |
| Pneumonia | 0.0 | 0.0 | 2.0 |
| Speech Disorder | 0.5 | 0.7 | 0.7 |
| Tremor | 0.5 | 0.0 | 1.3 |
| Urinary Incontinence | 0.0 | 0.0 | 2.0 |
| Urination Impaired | 0.0 | 0.0 | 2.0 |
The more common (1% or more) adverse events reported in the prospectively followed 211 patients exposed to LIORESAL INTRATHECAL (baclofen injection) have been reported. In the total cohort, the following adverse events, not described in the table, and arranged in decreasing order of frequency, and classified by body system, were reported:
Nervous System: Akathisia, ataxia, confusion, depression, opisthotonos, amnesia, anxiety, hallucinations, hysteria, insomnia, nystagmus, personality disorder, reflexes decreased, and vasodilitation.
Digestive System: Dysphagia, fecal incontinence, gastrointestinal hemorrhage and tongue disorder.
Cardiovascular: Bradycardia.
Respiratory: Apnea, dyspnea and hyperventilation.
Urogenital: Abnormal ejaculation, kidney calculus, oliguria and vaginitis.
Skin and Appendages: Rash, sweating, alopecia, contact dermatitis and skin ulcer.
Special Senses: Abnormality of accommodation.
Body as a Whole: Death, fever, abdominal pain, carcinoma, malaise and hypothermia.
Hemic and Lymphatic System: Leukocytosis and petechial rash.
Postmarketing Experience:
The following adverse events have been reported during post-approval use of LIORESAL INTRATHECAL. Because these events are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency.
Musculoskeletal: The onset of scoliosis or worsening of a pre-existing scoliosis has been reported.
Urogenital: Sexual dysfunction in men and women, including decreased libido and orgasm dysfunction, have been reported. Erectile dysfunction in men has also been reported. Priapism has been reported following baclofen withdrawal.
OVERDOSAGE
Special attention must be given to recognizing the signs and symptoms of overdosage, especially during the initial screening and dose-titration phase of treatment, but also during reintroduction of LIORESAL INTRATHECAL after a period of interruption in therapy.
Symptoms of LIORESAL INTRATHECAL Overdose: Drowsiness, lightheadedness, dizziness, somnolence, respiratory depression, hypothermia, seizures, rostral progression of hypotonia and loss of consciousness progressing to coma of up to 72 hr. duration. In most cases reported, coma was reversible without sequelae after drug was discontinued. Symptoms of LIORESAL INTRATHECAL overdose were reported in a sensitive adult patient after receiving a 25 mcg intrathecal bolus.
Treatment Suggestions for Overdose:
There is no specific antidote for treating overdoses of LIORESAL INTRATHECAL (baclofen injection); however, the following steps should ordinarily be undertaken:
- Residual LIORESAL INTRATHECAL solution should be removed from the pump as soon as possible.
- Patients with respiratory depression should be intubated if necessary, until the drug is eliminated.
If lumbar puncture is not contraindicated, consideration should be given to withdrawing 30-40 mL of CSF to reduce CSF baclofen concentration.
DOSAGE AND ADMINISTRATION
Refer to the manufacturer's manual for the implantable pump approved for intrathecal infusion for specific instructions and precautions for programming the pump and/or refilling the reservoir. There are various pumps with varying reservoir volumes and there are various refill kits available. It is important to be familiar with all of these products in order to select the appropriate refill kit for the particular pump in use.
Screening Phase: Prior to pump implantation and initiation of chronic infusion of LIORESAL INTRATHECAL (baclofen injection), patients must demonstrate a positive clinical response to a LIORESAL INTRATHECAL bolus dose administered intrathecally in a screening trial. The screening trial employs LIORESAL INTRATHECAL at a concentration of 50 mcg/mL. A 1 mL ampule (50 mcg/mL) is available for use in the screening trial. The screening procedure is as follows. An initial bolus containing 50 micrograms in a volume of 1 milliliter is administered into the intrathecal space by barbotage over a period of not less than one minute. The patient is observed over the ensuing 4 to 8 hours. A positive response consists of a significant decrease in muscle tone and/or frequency and/or severity of spasms. If the initial response is less than desired, a second bolus injection may be administered 24 hours after the first. The second screening bolus dose consists of 75 micrograms in 1.5 milliliters. Again, the patient should be observed for an interval of 4 to 8 hours. If the response is still inadequate, a final bolus screening dose of 100 micrograms in 2 milliliters may be administered 24 hours later.
Pediatric Patients: The starting screening dose for pediatric patients is the same as in adult patients, i.e., 50 mcg. However, for very small patients, a screening dose of 25 mcg may be tried first. Patients who do not respond to a 100 mcg intrathecal bolus should not be considered candidates for an implanted pump for chronic infusion.
Post-Implant Dose Titration Period: To determine the initial total daily dose of LIORESAL INTRATHECAL following implant, the screening dose that gave a positive effect should be doubled and administered over a 24-hour period, unless the efficacy of the bolus dose was maintained for more than 8 hours, in which case the starting daily dose should be the screening dose delivered over a 24-hour period. No dose increases should be given in the first 24 hours (i.e., until the steady state is achieved).
Adult Patients with Spasticity of Spinal Cord Origin: After the first 24 hours, for adult patients, the daily dosage should be increased slowly by 10-30% increments and only once every 24 hours, until the desired clinical effect is achieved.
Adult Patients with Spasticity of Cerebral Origin: After the first 24 hours, the daily dose should be increased slowly by 5-15% only once every 24 hours, until the desired clinical effect is achieved.
Pediatric Patients: After the first 24 hours, the daily dose should be increased slowly by 5-15% only once every 24 hours, until the desired clinical effect is achieved. If there is not a substantive clinical response to increases in the daily dose, check for proper pump function and catheter patency. Patients must be monitored closely in a fully equipped and staffed environment during the screening phase and dose-titration period immediately following implant. Resuscitative equipment should be immediately available for use in case of life-threatening or intolerable side effects.
Maintenance Therapy:
Spasticity of Spinal Cord Origin Patients: The clinical goal is to maintain muscle tone as close to normal as possible, and to minimize the frequency and severity of spasms to the extent possible, without inducing intolerable side effects. Very often, the maintenance dose needs to be adjusted during the first few months of therapy while patients adjust to changes in life style due to the alleviation of spasticity. During periodic refills of the pump, the daily dose may be increased by 10-40%, but no more than 40%, to maintain adequate symptom control. The daily dose may be reduced by 10-20% if patients experience side effects. Most patients require gradual increases in dose over time to maintain optimal response during chronic therapy. A sudden large requirement for dose escalation suggests a catheter complication (i.e., catheter kink or dislodgement).
Maintenance dosage for long term continuous infusion of LIORESAL INTRATHECAL (baclofen injection) has ranged from 12 mcg/day to 2003 mcg/day, with most patients adequately maintained on 300 micrograms to 800 micrograms per day. There is limited experience with daily doses greater than 1000 mcg/day. Determination of the optimal LIORESAL INTRATHECAL dose requires individual titration. The lowest dose with an optimal response should be used.
Spasticity of Cerebral Origin Patients: The clinical goal is to maintain muscle tone as close to normal as possible and to minimize the frequency and severity of spasms to the extent possible, without inducing intolerable side effects, or to titrate the dose to the desired degree of muscle tone for optimal functions. Very often the maintenance dose needs to be adjusted during the first few months of therapy while patients adjust to changes in life style due to the alleviation of spasticity. During periodic refills of the pump, the daily dose may be increased by 5-20%, but no more than 20%, to maintain adequate symptom control. The daily dose may be reduced by 10-20% if patients experience side effects. Many patients require gradual increases in dose over time to maintain optimal response during chronic therapy. A sudden large requirement for dose escalation suggests a catheter complication (i.e., catheter kink or dislodgement).
Maintenance dosage for long term continuous infusion of LIORESAL INTRATHECAL (baclofen injection) has ranged from 22 mcg/day to 1400 mcg/day, with most patients adequately maintained on 90 micrograms to 703 micrograms per day. In clinical trials, only 3 of 150 patients required daily doses greater than 1000 mcg/day.
Pediatric Patients: Use same dosing recommendations for patients with spasticity of cerebral origin. Pediatric patients under 12 years seemed to require a lower daily dose in clinical trials. Average daily dose for patients under 12 years was 274 mcg/day, with a range of 24 to 1199 mcg/day. Dosage requirement for pediatric patients over 12 years does not seem to be different from that of adult patients. Determination of the optimal LIORESAL INTRATHECAL dose requires individual titration. The lowest dose with an optimal response should be used.
Potential need for dose adjustments in chronic use : During long term treatment, approximately 5% (28/627) of patients become refractory to increasing doses. There is not sufficient experience to make firm recommendations for tolerance treatment; however, this “tolerance” has been treated on occasion, in hospital, by a “drug holiday” consisting of the gradual reduction of LIORESAL INTRATHECAL over a 2 to 4 week period and switching to alternative methods of spasticity management. After the “drug holiday,” LIORESAL INTRATHECAL may be restarted at the initial continuous infusion dose.
Stability
Parenteral drug products should be inspected for particulate matter and discoloration prior to administration, whenever solution and container permit.
Delivery Specifications
The specific concentration that should be used depends upon the total daily dose required as well as the delivery rate of the pump. LIORESAL INTRATHECAL may require dilution when used with certain implantable pumps. Please consult manufacturer's manual for specific recommendations.
Preparation Instruction:
Screening
Use the 1 mL screening ampule only (50 mcg/mL) for bolus injection into the subarachnoid space. For a 50 mcg bolus dose, use 1 mL of the screening ampule. Use 1.5 mL of 50 mcg/mL baclofen injection for a 75 mcg bolus dose. For the maximum screening dose of 100 mcg, use 2 mL of 50 mcg/mL baclofen injection (2 screening ampules).
Maintenance
For patients who require concentrations other than 500 mcg/mL or 2000 mcg/mL, LIORESAL INTRATHECAL must be diluted.
LIORESAL INTRATHECAL must be diluted with sterile preservative free Sodium Chloride for Injection, U.S.P.
Delivery Regimen:
LIORESAL INTRATHECAL is most often administered in a continuous infusion mode immediately following implant. For those patients implanted with programmable pumps who have achieved relatively satisfactory control on continuous infusion, further benefit may be attained using more complex schedules of LIORESAL INTRATHECAL delivery. For example, patients who have increased spasms at night may require a 20% increase in their hourly infusion rate. Changes in flow rate should be programmed to start two hours before the time of desired clinical effect.
HOW SUPPLIED
LIORESAL INTRATHECAL (baclofen injection) is packaged in single use ampules containing
0.05 mg/1 mL (50 mcg/mL), 10 mg/20 mL (500 mcg/mL), 10 mg/5 mL (2000 mcg/mL), or
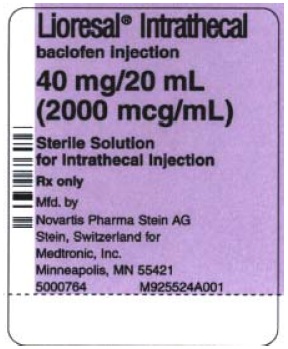
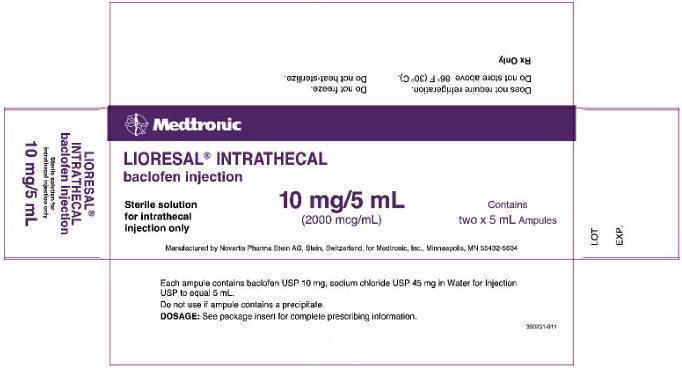
40 mg/20 mL (2000 mcg/mL) supplied as follows:
Screening dose (Model 8563s): one ampule containing 0.05 mg/1 mL (50 mcg/mL) (NDC 58281-562-01).
LIORESAL INTRATHECAL (baclofen injection) Refill Kits. Each refill kit includes the indicated amount of LIORESAL INTRATHECAL, a drug preparation kit, a pump refill kit with accessories that are compatible with Medtronic SynchroMed® Infusion Systems, and associated instructions.
Model 8561: one ampule containing 10 mg/20 mL (500 mcg/mL) (NDC 58281-560-01).
Model 8562: two ampules, each contains 10 mg/5 mL (2000 mcg/mL) (NDC 58281-561-02).
Model 8564: one ampule containing 40 mg/20 mL (2000 mcg/mL) (NDC 58281-563-01).
Model 8565: two ampules, each contains 10 mg/20 mL (500 mcg/mL) (NDC 58281-560-02).
Model 8566: two ampules, each contains 40 mg/20 mL (2000 mcg/mL) (NDC 58281-563-02).
Storage:
Does not require refrigeration.
Do not store above 86°F (30°C).
Do not freeze.
Do not heat sterilize.
Manufactured by Novartis Pharma Stein AG, Stein, Switzerland, for
Medtronic, Inc., Minneapolis, Minnesota 55432-5604 USA.
Lioresal® is a registered trademark of Saol
SynchroMed® is a registered trademark of Medtronic, Inc.
Medtronic, Inc.
710 Medtronic Parkway NE
Minneapolis, MN 55432-5604
USA
www.medtronic.com
Tel. 763-505-5000
Toll-free 1-800-328-0810
Fax 763-505-1000
Rev. 09/2016
Refill Kit 856X for use with Medtronic Implantable Programmable Infusion Pumps
Instructions for Use
Rx only
Explanation of symbols on product or package labeling
Refer to the appropriate product for symbols that apply.
![]() Open here
Open here
Medtronic®, lsoMed®, and SynchroMed® are trademarks of Medtronic, registered in the U.S.
MiniMed® is a registered trademark of Medtronic MiniMed, Inc.
Lioresal® is a registered trademark of Saol
Table of contents
Instructions for use
Sterilization
Preliminary procedures
Emptying the SynchroMed pump
Refilling the SynchroMed pump
Programming the SynchroMed pump
After the refill procedure
Reservoir rinse procedure
Performing a reservoir rinse
Emergency procedures
Lioresal Intrathecal (baclofen injection) overdose
Lioresal Intrathecal (baclofen injection) underdose/withdrawal
Emergency procedure to empty pump reservoir
Limited Warranty
Medtronic® Neuromodulation MODEL 856X REFILL KIT LIMITED WARRANTY
| Refer to the Indications, Drug Stability, and Emergency Procedures reference manual for indications and related information.
Refer to the appropriate information for prescribers booklet for contraindications, warnings, precautions, adverse events summary, individualization of treatment, patient selection, use in specific populations, and component disposal. Refer to the Lioresal Intrathecal (baclofen injection) drug labeling for indications, contraindications, warnings, precautions, dosage and administration information, and screening procedures. |
Introduction
These instructions include only the procedure for refilling the pump reservoir with Lioresal Intrathecal (baclofen injection). Refer to the appropriate pump technical manual for implanting instructions.
Package contents
The Model 856X Refill Kit contains the following sterile components that are not made with natural rubber latex:
- 20-mL syringe
- Extension set with a clamp
- Template
- 0.22-micron filter
- Two 22-gauge noncoring needles
- Fenestrated drape
- Accessories (povidone iodine swabs, gauze pads, alcohol pads, gloves)
Indications
The Model 856X Refill Kit is intended for use in refilling Medtronic implantable programmable infusion pumps with the exception of Medtronic MiniMed infusion pumps.
Contraindications
Medtronic refill kits are contraindicated for all catheter access port procedures.
Warnings
Withdrawal - Abrupt discontinuation of intrathecal baclofen, regardless of the cause, has resulted in sequelae that include high fever, altered mental status, exaggerated rebound spasticity, and muscle rigidity, that in rare cases has advanced to rhabdomyolysis, multiple organ-system failure and death.
Prevention of abrupt discontinuation of intrathecal baclofen requires careful attention to programming and monitoring of the infusion system, refill scheduling and procedures, and pump alarms. Patients and caregivers should be advised of the importance of keeping scheduled refill visits and should be educated on the early symptoms of baclofen withdrawal. Special attention should be given to patients at apparent risk (e.g. spinal cord injuries at T-6 or above, communication difficulties, history of withdrawal symptoms from oral or intrathecal baclofen). Consult the technical manual of the implantable infusion system for additional postimplant clinician and patient information.
User instructions - Comply with all product instructions for initial preparation and filling, implantation, programming, refilling, and accessing the catheter access port (if present) of the pump. Failure to comply with all instructions can lead to technical errors or improper use of implanted infusion pumps and result in additional surgical procedures, a return of underlying symptoms, drug withdrawal symptoms, or a clinically significant or fatal drug underdose or overdose.
Implantation and system management - Implantation and ongoing system management must be performed by individuals trained in the operation and handling of the infusion system and must be in compliance with procedures described in the appropriate technical instructions. Inadequate training or failure to follow instructions can require surgical revision or replacement, and result in a clinically significant or fatal drug underdose or overdose.
Calculating catheter volume - Use the catheter length recorded at implant or catheter revision when calculating catheter volume. The actual implanted catheter length and catheter model number are required to accurately calculate catheter volume. A universal value does not exist that can be used as a substitute for this knowledge. An inaccurate catheter volume calculation can result in a clinically significant or fatal drug underdose or overdose.
Contrast medium (pumps with a catheter access port) - Do not inject any contrast medium into the pump reservoir. Injecting contrast medium into the pump reservoir can impair pump operation.
Refill - Patients must return to the clinic for refills at the prescribed times. Failure to return to the clinic for refills at the prescribed times can result in the actual flow rate of the pump being less than expected, resulting in a loss of or change in therapy, which may lead to a return of underlying symptoms, drug withdrawal symptoms, or a clinically significant or fatal drug underdose. Failure to return at the prescribed times can also damage the pump, requiring surgical replacement.
Refill kit components – Use the appropriate Medtronic refill kit during all refill procedures for Medtronic implantable infusion pumps. Using components other than Medtronic components or a kit other than the appropriate refill kit can damage Medtronic components, requiring surgical revision or replacement, and allow drug leakage into surrounding tissue, resulting in loss of or change in therapy, which may lead to a return of underlying symptoms, drug withdrawal symptoms, or a clinically significant or fatal drug underdose or overdose.
Injection error during a pump refill procedure - Be certain you are accessing the reservoir fill port when injecting fluids into an implanted pump. ALWAYS:
- identify the pump model and reservoir volume.
- identify the location of the reservoir fill port.
- Avoid pocket fill, i.e. improper injection of medication into the subcutaneous tissue (see below)
- Avoid inadvertent injection into the catheter access port (see below)
- use the instructions, noncoring needles, appropriate template edge, and other accessories provided in the appropriate kit.
- verify the location of the reservoir fill port during needle insertion according to the instructions provided AND using other medical procedures as appropriate.
- refer to the drug labeling for indications, contraindications, warnings, precautions, adverse events, and dosage and administration information.
Pocket fill is the improper injection of refill medication into the subcutaneous tissue, which includes the pump pocket. The injection of drug into the subcutaneous tissue can lead to an acute systemic overdose, which can be fatal. After a pocket fill, the pump reservoir will become empty sooner than anticipated, and this may cause underdose symptoms and/or baclofen withdrawal syndrome, which can be fatal. If it is suspected or known that all or part of the drug was injected into the pocket during the refill procedure, monitor the patient closely for signs and symptoms of overdose in an appropriate facility for a sufficient amount of time or until the symptoms have resolved. Refer to the refill kit manual or the Indications, Drug Stability, and Emergency Procedures for SynchroMed and IsoMed Implantable Infusion Systems Reference Manual for emergency procedures associated with drug underdose and overdose. Refer to the drug product information label for specific drug underdose and overdose symptoms and methods of management.
Inadvertent injection into the catheter access port may result in a clinically significant or fatal drug overdose. If it is suspected or known that all or part of the drug was injected into the catheter access port during the refill procedure, monitor the patient closely for signs and symptoms of overdose in an appropriate facility for a sufficient amount of time or until the symptoms have resolved. Refer to the catheter access port kit manual or the Indications, Drug Stability, and Emergency Procedures for SynchroMed and IsoMed Implantable Infusion Systems Reference Manual for emergency procedures associated with drug overdose. Refer to the drug labeling for specific drug overdose symptoms and methods of management.
Changing drug or decreasing drug concentrations - Rinse the reservoir twice between solutions when changing drug or decreasing drug concentrations in the pump reservoir. A significant amount of drug may be present in the pump reservoir after emptying the pump. This residual volume cannot be removed by emptying the pump. Rinsing the reservoir between solutions minimizes the amount of drug in this residual volume but does not eliminate it. Failure to account for residual drug in the pump reservoir can result in a concentration that is different than intended and a clinically significant or fatal drug underdose or overdose. Program a bridge bolus after rinsing the reservoir twice. The bridge bolus advances the remaining old drug (the drug left in the pump tubing, catheter access port, and catheter after emptying and refilling the pump) to the catheter tip at the prior flow rate.
Refer to "Performing a reservoir rinse" on page 18 of this manual. Refer to the programming guide for bridge bolus procedures.
Connections - Firmly secure all connections. Failure to secure connections can allow drug to leak onto the surrounding skin and may result in inadequate therapy or infection.
Reservoir fill port injections - Do not use excessive force when accessing the reservoir fill port. Excessive force can result in damage to the needle or pump requiring surgical revision or replacement, and leakage into surrounding tissue, resulting in loss of or change in therapy, which may lead to a return of underlying symptoms, drug withdrawal symptoms, or a clinically significant or fatal drug underdose or overdose.
Intrathecal therapy - For intrathecal therapy, use ONLY a preservative-free sterile solution indicated for intrathecal use. Nonindicated fluids containing preservatives or endotoxins can be neurotoxic in intrathecal applications. Using nonindicated fluids can result in adverse events including, but not limited to, extreme pain, cramps, seizures, and death.
Drug information - Refer to the drug labeling for indications, contraindications, warnings, precautions, dosage and administration information, and screening procedures. Refer to the drug labeling for specific drug underdose or overdose symptoms and methods of management. Failure to refer to the drug labeling can result in inappropriate patient selection and management, inadequate therapy, intolerable side effects, or a clinically significant or fatal drug underdose or overdose. Consider the possibility of a drug error if the patient experiences unusual side effects. Failure to do so can result in misdiagnosis of patient symptoms.
Mixing drugs - The effects that drug mixtures have on pump operation are unknown. Drugs can precipitate when mixed. These precipitates can inhibit pump flow or block the catheter, resulting in loss of therapy or a clinically significant or fatal drug underdose.
Drug interaction and side effects - Inform patients of the appropriate warnings and precautions regarding drug interactions, potential side effects, and signs and symptoms that require medical attention, including prodromal signs and symptoms of inflammatory mass. Failure to recognize the signs and symptoms and to seek appropriate medical intervention can result in serious patient injury or death.
Drug overdose symptoms and management - Refer to the emergency procedures included at the end of this manual and the drug labeling for specific drug overdose symptoms and methods of management.
Drug underdose/overdose - Inform patients and caregivers of the signs and symptoms of a drug underdose and overdose. Inform patients and caregivers:
- to be aware and report any unusual signs or symptoms at anytime during or after a refill or catheter access port procedure.
- to be alert for any burning sensations in the area of the pump pocket during their refill or catheter access port procedure.
- to especially watch for signs of underdose and overdose.
- to stay alert for signs or symptoms that may indicate changes to their prescribed drug concentration or programmed dose.
- to seek emergency assistance as necessary. Refer to the refill kit or catheter access port kit manual or the Indications, Drug Stability and Emergency Procedures for SynchroMed and IsoMed Implantable Infusion Systems Reference Manual for emergency procedures associated with drug underdose and overdose.
Failure to recognize these signs and symptoms and to seek appropriate medical intervention can result in serious patient injury or death.
Patient travel - Patients should notify their clinicians of any travel plans. Clinicians need this information to coordinate patient care and pump refills and help prevent a loss of or change in therapy, which may lead to a return of underlying symptoms, drug withdrawal symptoms, or a clinically significant or fatal drug underdose.
Precautions
Compatibility, all components - Follow these guidelines when selecting system components:
- Medtronic components: For proper therapy, use only components that are compatible with the appropriate indication.
- Non-Medtronic components: No claims of safety, efficacy, or compatibility are made with regard to the use of non-Medtronic components with Medtronic components. Refer to the non-Medtronic documentation for information.
Component packaging - Before shipment the components in the sterile package were sterilized by the process indicated on the package label. Do not use or implant a component if the following circumstances have occurred:
- The storage package or sterile seal has been pierced or altered because component sterility cannot be guaranteed and infection may occur.
- The component shows signs of damage because the component may not function properly.
- The use-by date has expired because component sterility cannot be guaranteed and infection may occur; also, device battery longevity may be reduced and may require early replacement.
Storage temperature: kits and accessories - Do not store or transport the kit device components or accessories above 57 °C (135 °F) or below –34 °C (–30 °F). Temperatures outside this range can damage device components.
Aseptic technique - Use strict aseptic technique when accessing the reservoir fill port or the catheter access port of an implanted pump. Failure to use aseptic technique can contaminate fluids or tissues and result in local or systemic infection.
Infection - Use extreme caution when accessing the reservoir fill port or catheter access port of the implanted pump if local or systemic infection is suspected. Avoid contaminating the system or further spreading the infection. Local or systemic infection may require pump revision or removal.
Therapy discontinuance - If therapy is discontinued for an extended period, fill the pump reservoir with preservative-free saline. Program the pump to infuse at the minimum flow rate. Refill the pump as needed to ensure the pump always contains fluid in the reservoir and fluid pathway. Stopping the pump for extended periods or allowing the pump reservoir to empty completely can damage the system and require surgical replacement.
Single use only - Do not reuse any component. Components are intended for single use only. Reusing components can result in inadequate therapy and an increased risk of infection.
Reservoir valve activation - Do not prematurely activate the pump reservoir valve. Activation of the pump reservoir valve seals the pump reservoir valve closed. Unusual resistance or the inability to inject the entire fill volume may indicate activation of the pump reservoir valve. If the valve closes, a portion of the reservoir contents must be delivered or removed before filling can be completed, and procedural delays can occur. To prevent activation of the pump reservoir valve during emptying and filling procedures:
- completely aspirate all contents of the pump reservoir before filling;
- do not allow air into the pump reservoir through an open needle in the septum or an unclamped extension; and
- do not exceed the maximum reservoir volume indicated in the pump labeling.
Adverse events
The adverse events associated with the use of this device may include, but may not be limited to, the following:
- Meningitis
- Infection
- Reservoir contamination
- Overpressurization of the reservoir
- Injection into pocket or subcutaneous tissue
- Activation of reservoir valve
Instructions for use
Become thoroughly familiar with all product literature before using this refill kit.
Sterilization
All components of the kit are sterile. Do not resterilize. Should sterility of the kit be in question, discard and use a new kit.
Preliminary procedures
- Gather the following sterile equipment:
- Syringe(s) containing prescribed fluid
From the refill kit: - 20-mL empty syringe
- Extension set with a clamp
- Template
- 0.22-micron filter
- 22-gauge noncoring needle
- Fenestrated drape
- Cleansing agent
- Alcohol pads
- Sterile gloves (not made with natural rubber latex)
Locally supplied: - Adhesive bandage, optional
- Syringe(s) containing prescribed fluid
- Refer to the drug labeling for indications, contraindications, warnings, precautions, dosage and administration information, and screening procedures.
- Prepare the programmer for use. Refer to the appropriate programming guide for instructions.
- Confirm the:
- pump model
- reservoir volume
- location of the pump
Note: The model and reservoir volume can be confirmed by the programmer. Alternatively, a radiopaque identifier in the pump shows the pump model and identifies Medtronic as the pump manufacturer on a standard x-ray (Figure 1). A three-letter code designates the pump model.

Figure 1. A radiopaque identifier on a SynchroMed II pump.
- Confirm that the volume of the prescribed fluid does not exceed the reservoir volume of the pump.
Emptying the SynchroMed pump
- Prepare the injection site by cleansing the area.
- Open the kit. Put on sterile gloves.
- Place the drape, exposing the pump site.
- Using sterile procedures, assemble the needle, extension set, and empty syringe as follows:
- Connect the empty syringe to the extension set.
- Connect the needle to the extension set.
- Palpate the pump and identify the location of the catheter access port and the edges of pump.
Factors that may make it difficult to locate the pump include, but are not limited to:- deep implant
- patient position (eg, a seated patient)
- scar tissue at the pump implant site
- seroma
- the pump is tilted in the pocket
- obesity
- pump movement within the pocket
- weight gain after implant
- weight loss after implant
If you have difficulty identifying the pump features, you may seek assistance from another clinician. If deemed necessary by the clinician, x-ray and fluoroscopy can be used to assist in locating or determining the orientation of the pump.
- Place the template on the skin over the pump, and align the refill template correctly based on the model of the pump that is being refilled (Figure 2). Align the edges of the template with the edges of the pump. Use the center circle of the template to insert the needle into the reservoir fill port.
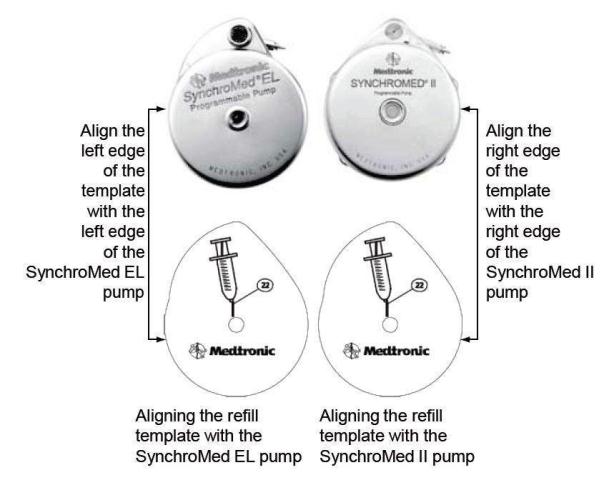
Figure 2. Aligning the refill template according to the pump model.
- Close the clamp.
- Gently insert the 22-gauge needle perpendicular to the surface of the pump through the center of the template and into the center of the reservoir fill port until the needle touches the bottom of the reservoir fill port (Figures: 3 – 4).
Note: The pump may be tilted within the pocket and therefore the needle angle may not be perpendicular to the patient's body.
During proper needle insertion, you will feel the needle:- pass through the patient's skin and subcutaneous tissue,
- hit the silicone septum,
(Scar tissue, if present, can feel similar to the septum.) - pass through the septum, and
- hit the metal bottom of the reservoir fill port.
(The top of the pump is metal and hitting the top of the pump can feel similar to hitting the bottom of the reservoir fill port.)
If excessive resistance is encountered during needle insertion, reassess placement. Do not force the needle. The feel of abnormal resistance during the procedure may be an indication that the needle is not in the center of the reservoir fill port.
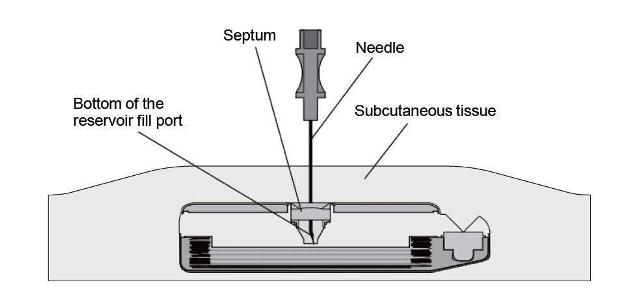
Figure 3. View inside of a SynchroMed programmable pump while the needle is fully and properly inserted.
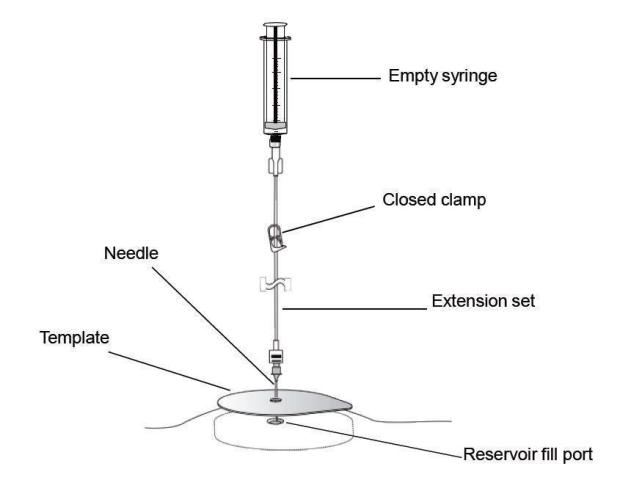
Figure 4. Close the clamp and insert the needle into the reservoir fill port.
Note: At any point during the procedure, if in doubt about the needle location, reassess its position. Factors that may contribute to difficulty inserting the needle into the reservoir fill port include, but are not limited to:
- the pump is flipped in the pocket
- deep implant
- patient position (eg, a seated patient)
- patient movement (eg, spasticity, difficulty hold still)
- localized muscle spasms at the pump implant site
- scar tissue at the pump implant site
- seroma
- the pump is tilted in the pocket
- obesity
- pump movement within the pocket
- weight gain after implant
- weight loss after implant
- Open the clamp and slowly withdraw the fluid from the reservoir into the empty syringe.
Note: If the withdrawn fluid has an unexpected appearance (eg, evidence of blood), this may indicate that the needle is not properly inserted into the pump. - If the syringe maximum capacity is reached before the reservoir is completely empty, more than one syringe will be needed to empty the pump.
- Close the clamp.
- Remove the full syringe.
- Attach an empty syringe.
- Verify that the needle is in the pump reservoir fill port.
- Repeat step 9, then continue to step 11.
- Completely empty the pump. When the pump is empty, the bubbles will stop forming, and negative pressure in the syringe can be felt.
- Close the clamp and remove the syringe from the extension set.
Note: Keep the needle in the reservoir fill port and the clamp closed for the pump refill procedure that follows. - Note the amount withdrawn from the pump for entry in the patient's record.
- Compare the amount withdrawn from the pump to the expected volume.
See the pump programmer for the expected volume. The amount withdrawn should approximately equal the expected volume. - Discard the fluid and syringe as appropriate for the fluid content in accordance with institutional policies and applicable regulations.
Refilling the SynchroMed pump
|
|
Warning: Pocket fill is the improper injection into the subcutaneous tissue, which includes the pump pocket. Pocket fill can result in a loss of or change in symptom control, drug withdrawal symptoms, or a clinically significant or fatal drug underdose or overdose. Observe the patient after the pump refill procedure for any signs or symptoms that could indicate a pocket fill or any other drug- related adverse event due to the refill procedure. Inadvertent injection into the catheter access port may result in a clinically significant or fatal drug overdose. Observe the patient after the pump refill procedure for any signs or symptoms that could indicate a drug-related adverse event due to the pump refill procedure. |
|
| Warning: If it is suspected or known that all or part of the drug was inadvertently injected into the pocket or the catheter access port during the refill procedure, monitor the patient closely for signs and symptoms of overdose in an appropriate facility for a sufficient amount of time or until the symptoms have resolved. Refer to the kit manual or the Indications, Drug Stability, and Emergency Procedures for SynchroMed and IsoMed Infusion Systems Reference Manual for emergency procedures associated with drug underdose and overdose. Refer to the drug product information label for specific drug underdose and overdose symptoms and methods of management. |
|
| Warning: Swelling at the injection site may indicate that the needle tip is not properly located within the pump reservoir, and the result could be pocket fill. Pocket fill can result in a loss of or change in symptom control, drug withdrawal symptoms, or a clinically significant or fatal drug underdose or overdose. Absence of swelling does not in all cases demonstrate that the needle tip is properly located. If swelling is present, stop injecting and observe the patient for any signs or symptoms that could indicate a pocket fill or any other drug-related adverse event. |
- If changing drug or drug concentrations, refer to "Performing a reservoir rinse" on page 18. Otherwise, proceed to the next step.
- Confirm that the refill volume of the prescribed fluid does not exceed the reservoir volume of the pump.
- Purge the air from the syringe containing the prescribed fluid.
- Attach the filter to the syringe with the prescribed fluid.
- Purge all air from the filter.
- Attach the syringe with the prescribed fluid and filter to the extension set (Figure 5).
- Before and during injection, verify that the needle remains fully inserted to the bottom of the reservoir fill port. Do not apply tension to the extension tubing because the needle may be pulled out from the reservoir.
- Open the clamp and as the clamp is opened, observe the following indications that the needle continues to be properly positioned:
- The bubbles in the extension set are immediately drawn into the pump.
- The plunger may move slightly when the drug is initially drawn into the pump.
- Slowly depress the plunger on the syringe to inject the prescribed fluid into the pump reservoir. While injecting the prescribed fluid, verify that the needle remains properly located within the reservoir (Figure 5).
- Periodically withdraw and observe a portion of the drug to confirm that the drug has the expected appearance.
- After confirming that the needle remains in the reservoir, resume injecting fluid.
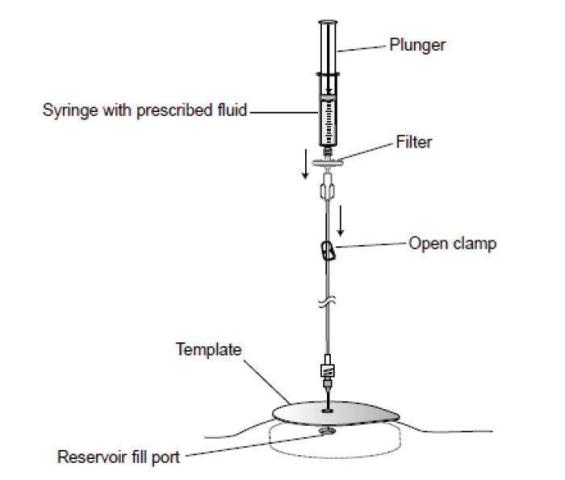
Figure 5. Open the clamp and inject into the pump reservoir.
Caution: If you encounter unusual resistance before the maximum reservoir volume is injected or you are unable to inject fluid, the reservoir valve may have been activated. Activation of the pump reservoir valve seals the pump reservoir valve closed. If the valve closes, a portion of the reservoir contents must be delivered or removed before filling can be completed, and procedural delays can occur.
To prevent activation of the pump reservoir valve during emptying and filling procedures:
- completely aspirate all contents of the pump reservoir before filling;
- do not allow air into the pump reservoir through an open needle in the septum or an unclamped extension; and
- do not exceed the maximum reservoir volume indicated in the pump labeling.
- If you have activated the reservoir valve, complete steps a – g below. Otherwise, proceed to step 11.
- Discontinue injection.
- Close the clamp.
- Remove the syringe with prescribed fluid and attached filter.
- Attach an empty 20-mL syringe to the extension set.
- Open the clamp, and aspirate until all fluid/air is removed.
- Close the clamp and remove the syringe containing the aspirate from the extension set and discard the syringe.
- Repeat steps 2 – 10.
Note: For pumps with a reservoir valve, the amount of time before the valve will release is dependent on the duration and the amount of pressure applied after the valve is first activated. The more pressure exerted, the longer it may take to release the valve.
- If more than one syringe of prescribed fluid is needed to fill the pump reservoir, complete steps a – d below. Otherwise, proceed to step 12.
- Close the clamp.
- Keep the filter from the extension set in place, and remove the first syringe from the filter.
Caution: Do not remove the filter from the extension set. Removal of the filter could compromise the sterile barrier, which could result in an infection for the patient. - Purge the air from the second syringe, and attach the second syringe to the filter (Figure 5).
- With the second syringe in a vertical position, open the clamp and slowly depress the plunger on the syringe to inject the prescribed fluid into the pump reservoir.
- When filling is complete, close the clamp and carefully remove the needle from the reservoir fill port.
Note: If you are unsure whether drug was injected correctly into the pump, completely aspirate the pump to verify that all of the injected drug can be removed. - Remove the cleansing agent from the patient’s skin using an alcohol pad.
- Apply an adhesive bandage, if desired.
- Discard all components of the kit.
Programming the SynchroMed pump
- If the drug concentration or drug has been changed, program a bridge bolus. Refer to the programming guide for the pump software.
- If any prescription information has changed, enter the changed information into the clinician programmer: for example drug name, drug concentration, infusion information, or volume of prescribed fluid in the pump reservoir.
- Update the pump.
Reservoir rinse procedure
Performing a reservoir rinse
To prevent drug overdose or underdose when changing concentrations or changing solutions in the pump reservoir, always rinse the reservoir twice between solutions to remove the drug that remains in the reservoir after emptying the pump. This remaining volume is known as the residual volume.
The procedure for performing a reservoir rinse is outlined below. Use the components of the appropriate refill kit to perform the rinse and follow the applicable empty and refill procedures for that kit.
- Empty the pump completely.
- Fill the pump with 10 mL of sterile preservative-free Sodium Chloride Injection, USP.
- Empty the pump completely.
- Repeat steps 2 and 3.
- Fill the pump to capacity with the prescribed fluid.
- Program a bridge bolus. Refer to the programming guide for the pump software.
Technical support
A toll-free technical support service is available 24 hours a day for clinicians managing patients with Medtronic implantable infusion pumps. Telephone Customer Service at: 1-800-707-0933.
Emergency procedures
Lioresal Intrathecal (baclofen injection) overdose
Consult the patient's medical record or with the patient's physician to confirm the drug or drug concentration within the pump reservoir.
Symptoms
Drowsiness, lightheadedness, dizziness, somnolence, respiratory depression, hypothermia, seizures, rostral progression of hypotonia, and loss of consciousness progressing to coma.
There is no specific antidote for treating overdoses of intrathecal baclofen injection.
Actions
See Table 1.
|
||
| Maintain airway/breathing/circulation. Intubation and respiratory support may be necessary. | ||
|
| ||
| Empty pump reservoir to stop drug flow. Record amount withdrawn. | ||
|
| ||
| FOR INTRATHECAL OVERDOSE: | FOR SUBCUTANEOUS OVERDOSE: (eg, pocket fill) | |
|
|
|
|
| If not contraindicated, withdraw 30 – 40 mL CSF by lumbar puncture or through the catheter access port to reduce the concentration of baclofen in the CSF. Use only a 24-gauge or smaller, 1.5- or 2.0-inch (3.8- or 5.1-cm), needle for withdrawal from the catheter access port.* | Proceed immediately to the next step. | |
|
|
|
|
| Notify patient's physician managing intrathecal baclofen injection therapy. | ||
|
| ||
| Continue to monitor closely for symptom recurrence. | ||
|
| ||
| Report incident to Medtronic, Inc. | ||

Lioresal Intrathecal (baclofen injection) underdose/withdrawal
Consult the patient's medical record or with the patient's physician to confirm the drug or drug concentration within the pump reservoir.
Symptoms of underdose
Pruritus without rash, hypotension, paresthesia, fever, and altered mental state. Priapism may develop or recur if treatment with intrathecal baclofen is interrupted.
Symptoms of withdrawal
Exaggerated rebound spasticity and muscle rigidity, rhabdomyolysis, and multiple organ failure. The condition may resemble autonomic dysreflexia, infection (sepsis), malignant hyperthermia, and neuroleptic-malignant syndrome.
Actions:
See Table 2.
|
| Initiate life-sustaining measures if indicated. |
|
|
If a patient receiving intrathecal baclofen injection presents with the signs and symptoms suggestive of withdrawal the following is consistent with that suggested by a panel of therapy-experienced clinicians convened to explore this issue.*†
|
|
|
| Report incident to Medtronic, Inc. |
Emergency procedure to empty pump reservoir
Equipment
- 22-gauge noncoring needle
- 20-mL syringe
- 3-way stopcock or extension set with clamp
- Antiseptic agent
- Assemble the needle, syringe, and stopcock or extension set.
- Locate the pump by palpation. The reservoir fill port is located in the CENTER of the pump. If you have difficulty identifying the pump features, you may seek assistance from another clinician. If deemed necessary by the clinician, x-ray and fluoroscopy can be used to assist in locating or determining the orientation of the pump.
- Prepare the injection site by cleansing the area using an antiseptic agent.
- Gently insert the 22-gauge noncoring needle into the center of the reservoir fill port until the needle touches the bottom of the reservoir fill port (Figure 6).
During proper needle insertion, you will feel the needle:
- pass through the patient's skin and subcutaneous tissue,
- hit the silicone septum, (Scar tissue, if present, can feel similar to the septum.)
- pass through the septum, and
- hit the metal bottom of the reservoir fill port.
(The top of the pump is metal and hitting the top of the pump can feel similar to hitting the bottom of the reservoir fill port.)
If excessive resistance is encountered during needle insertion, reassess placement. Do not force the needle. The feel of abnormal resistance during the procedure may be an indication that the needle is not in the center of the reservoir fill port.
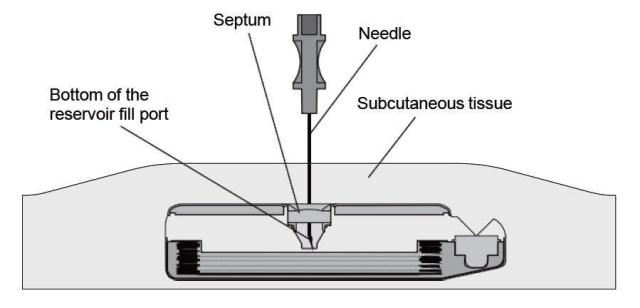
Figure 6. View inside of a SynchroMed programmable pump while the needle is fully and properly inserted.
- Open the clamp or stopcock and slowly withdraw the fluid from the reservoir into the empty syringe.
- Depending on pump reservoir volume, more than one syringe may be needed to empty the pump. Close the clamp or stopcock when changing syringes.
- Completely empty the pump. When the pump is empty, the bubbles will stop forming, and negative pressure in the syringe can be felt.
- Remove the needle from the reservoir fill port.
- Record in patient chart the amount of fluid emptied from the pump reservoir.
Special notice
The Medtronic 856X Refill Kit is designed to be used for refilling Medtronic implantable programmable infusion pumps with the exception of Medtronic MiniMed infusion pumps. Medtronic cannot warrant or guarantee the refill kit because, despite the exercise of all due care in design, component selection, manufacture, and testing prior to sale, the components of the refill kit may be easily damaged before or during use by improper handling or other intervening acts.
Medtronic® Neuromodulation MODEL 856X REFILL KIT LIMITED WARRANTY1
- This Limited Warranty provides the following assurance to the purchaser of the Medtronic Model 856X packaged herein, hereafter referred to as the “Product”:
- Should the Product fail to function within normal tolerances due to a defect in materials or workmanship prior to its "Use By" date, Medtronic will at its option: (a) issue a credit to the purchaser equal to the Purchase Price, as defined in Subsection A(2), against the purchase of the replacement Product or provide a functionally comparable replacement Product at no charge.
- As used herein, Purchase Price shall mean the lesser of the net invoiced price of the original, or current functionally comparable, or replacement Product.
- To qualify for the Limited Warranty set forth in Section A(1), the following conditions must be met:
- The Product must be used prior to its "Use By" date.
- The unused portion of the Product must be returned to Medtronic within thirty (30) days after discovery of the defect and shall be the Property of Medtronic.
- The Product must not have been altered or subjected to misuse, abuse or accident.
- The Product must be used in accordance with the labeling and instructions for use provided with the Product.
- This Limited Warranty is limited to its express terms. In particular:
- Except as expressly provided by this Limited Warranty,
MEDTRONIC IS NOT RESPONSIBLE FOR ANY DIRECT, INCIDENTAL OR CONSEQUENTIAL DAMAGES BASED ON ANY DEFECT, FAILURE OR MALFUNCTION OF THE PRODUCT, WHETHER THE CLAIM IS BASED ON WARRANTY, CONTRACT, TORT OR OTHERWISE. - This Limited Warranty is made only to the purchaser who uses the Product. AS TO ALL OTHERS, MEDTRONIC MAKES NO WARRANTY, EXPRESS OR IMPLIED, INCLUDING, BUT NOT LIMITED TO, ANY IMPLIED WARRANTY OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE WHETHER ARISING FROM STATUTE, COMMON LAW, CUSTOM OR OTHERWISE. NO EXPRESS OR IMPLIED WARRANTY TO THE PATIENT SHALL EXTEND BEYOND THE PERIOD SPECIFIED IN A(1) ABOVE. THIS LIMITED WARRANTY SHALL BE THE EXCLUSIVE REMEDY AVAILABLE TO ANY PERSON.
- The exclusions and limitations set out above are not intended to, and should not be construed so as to contravene mandatory provisions of applicable law. If any part or term of this Limited Warranty is held to be illegal, unenforceable or in conflict with applicable law by a court of competent jurisdiction, the validity of the remaining portions of the Limited Warranty shall not be affected, and all rights and obligations shall be construed and enforced as if this Limited Warranty did not contain the particular part or term held to be invalid. This Limited Warranty gives the patient specific legal rights. The patient may also have other rights which vary from state to state.
- No person has any authority to bind Medtronic to any representation, condition or warranty except this Limited Warranty.
- Except as expressly provided by this Limited Warranty,
1This Limited Warranty is provided by Medtronic, Inc., 710 Medtronic Parkway, Minneapolis, MN 55432-5604. It applies only in the United States. Areas outside the United States should contact their local Medtronic representative for exact terms of the Limited Warranty
Manufacturer
Medtronic, Inc.
710 Medtronic Parkway
Minneapolis, MN 55432-5604
USA
www.medtronic.com
Tel. 1-763-505-5000
Toll-free 1-800-328-0810
Fax 1-763-505-1000
© Medtronic, Inc. 2016
All Rights Reserved
M956884A003