FULL PRESCRIBING INFORMATION
WARNING: DISTANT SPREAD OF TOXIN EFFECT
Postmarketing reports indicate that the effects of BOTOX and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These may include asthenia, generalized muscle weakness, diplopia, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, particularly in those patients who have an underlying condition that would predispose them to these symptoms. In unapproved uses and in approved indications, cases of spread of effect have been reported at doses comparable to those used to treat cervical dystonia and spasticity and at lower doses [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
1.1 Adult Bladder Dysfunction
Overactive Bladder
BOTOX for injection is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and frequency, in adults who have an inadequate response to or are intolerant of an anticholinergic medication.
Detrusor Overactivity associated with a Neurologic Condition
BOTOX is indicated for the treatment of urinary incontinence due to detrusor overactivity associated with a neurologic condition (e.g., SCI, MS) in adults who have an inadequate response to or are intolerant of an anticholinergic medication.
1.2 Pediatric Detrusor Overactivity Associated with a Neurologic Condition
BOTOX is indicated for the treatment of neurogenic detrusor overactivity (NDO) in pediatric patients 5 years of age and older who have an inadequate response to or are intolerant of anticholinergic medication.
1.3 Chronic Migraine
BOTOX is indicated for the prophylaxis of headaches in adult patients with chronic migraine (≥15 days per month with headache lasting 4 hours a day or longer).
Limitations of Use
Safety and effectiveness have not been established for the prophylaxis of episodic migraine (14 headache days or fewer per month) in seven placebo-controlled studies.
1.4 Spasticity
BOTOX is indicated for the treatment of spasticity in patients 2 years of age and older.
Limitations of Use
BOTOX has not been shown to improve upper extremity functional abilities, or range of motion at a joint affected by a fixed contracture.
1.5 Cervical Dystonia
BOTOX is indicated for the treatment of adults with cervical dystonia, to reduce the severity of abnormal head position and neck pain associated with cervical dystonia.
1.6 Primary Axillary Hyperhidrosis
BOTOX is indicated for the treatment of severe primary axillary hyperhidrosis that is inadequately managed with topical agents.
Limitations of Use
The safety and effectiveness of BOTOX for hyperhidrosis in other body areas have not been established. Weakness of hand muscles and blepharoptosis may occur in patients who receive BOTOX for palmar hyperhidrosis and facial hyperhidrosis, respectively. Patients should be evaluated for potential causes of secondary hyperhidrosis (e.g., hyperthyroidism) to avoid symptomatic treatment of hyperhidrosis without the diagnosis and/or treatment of the underlying disease.
Safety and effectiveness of BOTOX have not been established for the treatment of axillary hyperhidrosis in pediatric patients under age 18.
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Safe Use
The potency Units of BOTOX (onabotulinumtoxinA) for injection are specific to the preparation and assay method utilized. They are not interchangeable with other preparations of botulinum toxin products and, therefore, units of biological activity of BOTOX cannot be compared to nor converted into units of any other botulinum toxin products assessed with any other specific assay method [see Warnings and Precautions (5.2) and Description (11)].
Indication specific dosage and administration recommendations should be followed. When initiating treatment, the lowest recommended dose should be used. In treating adult patients for one or more indications, the maximum cumulative dose should not exceed 400 Units, in a 3-month interval. In pediatric patients, the total dose should not exceed the lower of 10 Units/kg body weight or 340 Units, in a 3-month interval [see Dosage and Administration (2.7)].
The safe and effective use of BOTOX depends upon proper storage of the product, selection of the correct dose, and proper reconstitution and administration techniques. An understanding of standard electromyographic techniques is also required for treatment of strabismus, upper or lower limb spasticity, and may be useful for the treatment of cervical dystonia. Physicians administering BOTOX must understand the relevant neuromuscular and structural anatomy of the area involved and any alterations to the anatomy due to prior surgical procedures and disease, especially when injecting near the lungs.
Do not use BOTOX and contact AbbVie (1-800-678-1605) if:
- the tamper evident features on the carton appear to be broken or compromised, or
- the U.S. License number 1889 is not present on the vial label and carton labeling [see How Supplied/Storage and Handling (16)].
2.2 Preparation and Dilution Technique
Prior to injection, reconstitute each vacuum-dried vial of BOTOX with only sterile, preservative-free 0.9% Sodium Chloride Injection, USP. Draw up the proper amount of diluent in the appropriate size syringe (see Table 1, or for specific instructions for detrusor overactivity associated with a neurologic condition, see Section 2.3), and slowly inject the diluent into the vial. Discard the vial if a vacuum does not pull the diluent into the vial. Gently mix BOTOX with the diluent by rotating the vial. Record the date and time of reconstitution on the space on the label. BOTOX should be administered within 24 hours after reconstitution. During this time period, unused reconstituted BOTOX should be stored in a refrigerator (2° to 8°C) for up to 24 hours until time of use. BOTOX vials are for single-dose only. Discard any unused portion.
| Diluent* Added to 100 Unit Vial | Resulting Dose Units per 0.1 mL | Diluent* Added to 200 Unit Vial | Resulting Dose Units per 0.1 mL |
| 1 mL 2 mL 4 mL 8 mL 10 mL | 10 Units 5 Units 2.5 Units 1.25 Units 1 Unit | 1 mL 2 mL 4 mL 8 mL 10 mL | 20 Units 10 Units 5 Units 2.5 Units 2 Units |
*Preservative-free 0.9% Sodium Chloride Injection, USP Only
**For Detrusor Overactivity associated with a Neurologic Condition Dilution, see Section 2.3
Note: These dilutions are calculated for an injection volume of 0.1 mL. A decrease or increase in the BOTOX dose is also possible by administering a smaller or larger injection volume - from 0.05 mL (50% decrease in dose) to 0.15 mL (50% increase in dose).
An injection of BOTOX is prepared by drawing into an appropriately sized sterile syringe an amount of the properly reconstituted toxin slightly greater than the intended dose. Air bubbles in the syringe barrel are expelled and the syringe is attached to an appropriate injection needle. Patency of the needle should be confirmed. A new, sterile needle and syringe should be used to enter the vial on each occasion for removal of BOTOX.
Reconstituted BOTOX should be clear, colorless, and free of particulate matter. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration and whenever the solution and the container permit.
2.3 Adult Bladder Dysfunction
General
Patients must not have a urinary tract infection (UTI) at the time of treatment. Prophylactic antibiotics, except aminoglycosides, [see Drug Interactions (7.1)] should be administered 1-3 days pre-treatment, on the treatment day, and 1-3 days post-treatment to reduce the likelihood of procedure-related UTI.
Patients should discontinue anti-platelet therapy at least 3 days before the injection procedure. Patients on anti-coagulant therapy need to be managed appropriately to decrease the risk of bleeding.
Appropriate caution should be exercised when performing a cystoscopy.
Overactive Bladder
An intravesical instillation of diluted local anesthetic with or without sedation may be used prior to injection, per local site practice. If a local anesthetic instillation is performed, the bladder should be drained and irrigated with sterile saline before injection.
The recommended dose is 100 Units of BOTOX, and is the maximum recommended dose. The recommended dilution is 100 Units/10 mL with preservative-free 0.9% Sodium Chloride Injection, USP (see Table 1). Dispose of any unused saline.
Reconstituted BOTOX (100 Units/10 mL) is injected into the detrusor muscle via a flexible or rigid cystoscope, avoiding the trigone. The bladder should be instilled with enough saline to achieve adequate visualization for the injections, but over-distension should be avoided.
The injection needle should be filled (primed) with approximately 1 mL of reconstituted BOTOX prior to the start of injections (depending on the needle length) to remove any air.
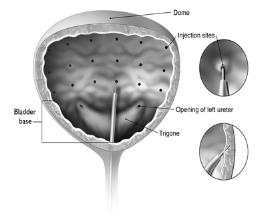
The needle should be inserted approximately 2 mm into the detrusor, and 20 injections of 0.5 mL each (total volume of 10 mL) should be spaced approximately 1 cm apart (see Figure 1). For the final injection, approximately 1 mL of sterile normal saline should be injected so that the remaining BOTOX in the needle is delivered to the bladder. After the injections are given, patients should demonstrate their ability to void prior to leaving the clinic. The patient should be observed for at least 30 minutes post-injection and until a spontaneous void has occurred.
Patients should be considered for reinjection when the clinical effect of the previous injection has diminished (median time until patients qualified for the second treatment of BOTOX in double-blind, placebo-controlled clinical studies was 169 days [~24 weeks]), but no sooner than 12 weeks from the prior bladder injection.
Figure 1: Injection Pattern for Intradetrusor Injections for Treatment of Overactive Bladder and Detrusor Overactivity Associated with a Neurologic Condition
Detrusor Overactivity associated with a Neurologic Condition
An intravesical instillation of diluted local anesthetic with or without sedation, or general anesthesia may be used prior to injection, per local site practice. If a local anesthetic instillation is performed, the bladder should be drained and irrigated with sterile saline before injection.
The recommended dose is 200 Units of BOTOX per treatment, and should not be exceeded.
200 Unit Vial of BOTOX
- Reconstitute a 200 Unit vial of BOTOX with 6 mL of preservative-free 0.9% Sodium Chloride Injection, USP and mix the vial gently.
- Draw 2 mL from the vial into each of three 10 mL syringes.
- Complete the reconstitution by adding 8 mL of preservative-free 0.9% Sodium Chloride Injection, USP into each of the 10 mL syringes, and mix gently. This will result in three 10 mL syringes each containing 10 mL (~67 Units in each), for a total of 200 Units of reconstituted BOTOX.
- Use immediately after reconstitution in the syringe. Dispose of any unused saline.
100 Unit Vial of BOTOX
- Reconstitute two 100 Unit vials of BOTOX, each with 6 mL of preservative-free 0.9% Sodium Chloride Injection, USP and mix the vials gently.
- Draw 4 mL from each vial into each of two 10 mL syringes. Draw the remaining 2 mL from each vial into a third 10 mL syringe for a total of 4 mL in each syringe.
- Complete the reconstitution by adding 6 mL of preservative-free 0.9% Sodium Chloride Injection, USP into each of the 10 mL syringes, and mix gently. This will result in three 10 mL syringes each containing 10 mL (~67 Units in each), for a total of 200 Units of reconstituted BOTOX.
- Use immediately after reconstitution in the syringe. Dispose of any unused saline.
Reconstituted BOTOX (200 Units/30 mL) is injected into the detrusor muscle via a flexible or rigid cystoscope, avoiding the trigone. The bladder should be instilled with enough saline to achieve adequate visualization for the injections, but over-distension should be avoided.
The injection needle should be filled (primed) with approximately 1 mL of reconstituted BOTOX prior to the start of injections (depending on the needle length) to remove any air.
The needle should be inserted approximately 2 mm into the detrusor, and 30 injections of 1 mL (~6.7 Units) each (total volume of 30 mL) should be spaced approximately 1 cm apart (see Figure 1). For the final injection, approximately 1 mL of sterile normal saline should be injected so that the remaining BOTOX in the needle is delivered to the bladder. After the injections are given, the saline used for bladder wall visualization should be drained. The patient should be observed for at least 30 minutes post-injection.
Patients should be considered for re-injection when the clinical effect of the previous injection diminishes (median time to qualification for re-treatment in the double-blind, placebo-controlled clinical studies was 295-337 days [42-48 weeks] for BOTOX 200 Units), but no sooner than 12 weeks from the prior bladder injection.
2.4 Pediatric Detrusor Overactivity Associated with a Neurologic Condition
Patients must not have a urinary tract infection (UTI) at the time of treatment. Oral prophylactic antibiotics, except aminoglycosides, [see Drug Interactions (7.1)] should be administered 1-3 days pre-treatment, on the treatment day, and 1-3 days post-treatment to reduce the likelihood of procedure-related UTI. Alternatively, for patients receiving general anesthesia (or conscious sedation) for the treatment of detrusor overactivity associated with a neurologic condition, one dose of IV prophylactic antibiotics, except aminoglycosides, [see Drug Interactions (7.1)] may be administered prior to treatment administration on the day of treatment.
Patients should discontinue anti-platelet therapy at least 3 days before the injection procedure. Patients on anti-coagulant therapy need to be managed appropriately to decrease the risk of bleeding.
Appropriate caution should be exercised when performing a cystoscopy.
- In patients 5 years to less than 12 years of age: Consider general anesthesia (or conscious sedation) prior to injection, per local site practice.
- In patients 12 years of age or older: Consider an intravesical instillation of diluted local anesthetic with or without sedation, or general anesthesia prior to injection, per local site practice.
At a minimum, consider a diluted instillation of local anesthetic for all age groups. If a local anesthetic instillation is performed, drain and irrigate the bladder with sterile saline before injection.
If patient’s body weight is greater than or equal to 34 kg, the recommended dosage is 200 Units of BOTOX per treatment administered as an intradetrusor injection after dilution:
- Reconstitute BOTOX to result in 20 Units BOTOX/mL in the vial(s):
◦ BOTOX 200 Unit vial: add 10 mL of preservative-free 0.9% Sodium Chloride Injection, USP and mix the vial gently.
◦ BOTOX 100 Unit vials: add 5 mL of preservative-free 0.9% Sodium Chloride Injection, USP to each of two 100 Unit vials of BOTOX and mix the vials gently.
- Draw 10 mL from the vial(s) into one 10 mL dosing syringe.
- Use immediately after reconstitution in the syringe. Dispose of any unused saline.
If patient’s body weight is less than 34 kg, the recommended dosage is 6 Units/kg body weight administered as a bladder injection after dilution (refer to Table 2):
- Reconstitute BOTOX to result in 20 Units BOTOX/mL in the vial(s):
◦ BOTOX 200 Unit vial: add 10 mL of preservative-free 0.9% Sodium Chloride Injection, USP and mix the vial gently.
◦ BOTOX 100 Unit vial(s): add 5 mL of preservative-free 0.9% Sodium Chloride Injection, USP to one 100 Unit vial of BOTOX (if final dose is less than or equal to 100 U) or to each of two 100 Unit vials of BOTOX (if final dose is greater than 100 U) and mix the vial(s) gently.
- Refer to Table 2 for dilution instructions (i.e., the amount of reconstituted BOTOX and additional diluent to draw into one 10 mL dosing syringe).
- Use BOTOX immediately after reconstitution in the syringe. Dispose of any unused preservative-free 0.9% Sodium Chloride Injection, USP.
| Body Weight
(kg) | Volume of reconstituted BOTOX and Diluent* (mL) to draw into dosing syringe to achieve a final volume of 10 mL | Final dose of BOTOX in dosing syringe | |
| BOTOX
(mL) | Diluent*
(mL) | ||
| 12 to less than 14 | 3.6 | 6.4 | 72 Units |
| 14 to less than 16 | 4.2 | 5.8 | 84 Units |
| 16 to less than 18 | 4.8 | 5.2 | 96 Units |
| 18 to less than 20 | 5.4 | 4.6 | 108 Units |
| 20 to less than 22 | 6 | 4 | 120 Units |
| 22 to less than 24 | 6.6 | 3.4 | 132 Units |
| 24 to less than 26 | 7.2 | 2.8 | 144 Units |
| 26 to less than 28 | 7.8 | 2.2 | 156 Units |
| 28 to less than 30 | 8.4 | 1.6 | 168 Units |
| 30 to less than 32 | 9 | 1 | 180 Units |
| 32 to less than 34 | 9.6 | 0.4 | 192 Units |
*Preservative-free 0.9% Sodium Chloride Injection, USP Only
Reconstituted BOTOX is injected into the detrusor muscle via a flexible or rigid cystoscope, avoiding the trigone. The bladder should be instilled with enough saline to achieve adequate visualization for the injections, but over-distension should be avoided.
The injection needle should be filled (primed) with approximately 1 mL of reconstituted BOTOX prior to the start of injections (depending on the needle length) to remove any air.
The needle should be inserted approximately 2 mm into the detrusor, and 20 injections of 0.5 mL each (total volume of 10 mL) should be spaced approximately 1 cm apart (see Figure 1). For the final injection, approximately 1 mL of sterile normal saline should be injected so that the remaining BOTOX in the needle is delivered to the bladder. After the injections are given, the saline used for bladder wall visualization should be drained. The patient should be observed for at least 30 minutes post-injection.
Patients should be considered for re-injection when the clinical effect of the previous injection diminishes (median time to qualification for re-treatment in the double-blind, parallel group clinical study was 207 days [30 weeks] for BOTOX 200 Units), but no sooner than 12 weeks from the prior bladder injection.
2.5 Chronic Migraine
The recommended dilution is 200 Units/4 mL or 100 Units/2 mL, with a final concentration of 5 Units per 0.1 mL (see Table 1). The recommended dose for treating chronic migraine is 155 Units administered intramuscularly using a sterile 30-gauge, 0.5 inch needle as 0.1 mL (5 Units) injections per each site. Injections should be divided across 7 specific head/neck muscle areas as specified in the diagrams and Table 3 below. A one inch needle may be needed in the neck region for patients with thick neck muscles. With the exception of the procerus muscle, which should be injected at one site (midline), all muscles should be injected bilaterally with half the number of injection sites administered to the left, and half to the right side of the head and neck. The recommended re-treatment schedule is every 12 weeks.
Diagrams 1-4: Recommended Injection Sites (A through G) for Chronic Migraine
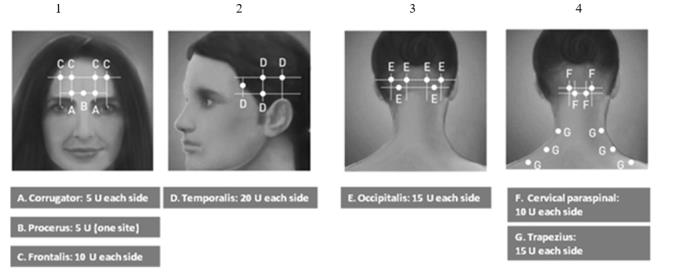
| Head/Neck Area | Recommended Dose (Number of Sitesa) |
| Frontalisb | 20 Units divided in 4 sites |
| Corrugatorb | 10 Units divided in 2 sites |
| Procerus | 5 Units in 1 site |
| Occipitalisb | 30 Units divided in 6 sites |
| Temporalisb | 40 Units divided in 8 sites |
| Trapeziusb | 30 Units divided in 6 sites |
| Cervical Paraspinal Muscle Groupb | 20 Units divided in 4 sites |
| Total Dose: | 155 Units divided in 31 sites |
a Each IM injection site = 0.1 mL = 5 Units BOTOX
b Dose distributed bilaterally
2.6 Adult Spasticity
General
Dosing in initial and sequential treatment sessions should be tailored to the individual based on the size, number and location of muscles involved, severity of spasticity, the presence of local muscle weakness, the patient’s response to previous treatment, or adverse event history with BOTOX.
The recommended dilution is 200 Units/4 mL or 100 Units/2 mL with preservative-free 0.9% Sodium Chloride Injection, USP (see Table 1). The lowest recommended starting dose should be used, and no more than 50 Units per site should generally be administered. An appropriately sized needle (e.g., 25-30 gauge) may be used for superficial muscles, and a longer 22 gauge needle may be used for deeper musculature. Localization of the involved muscles with techniques such as needle electromyographic guidance, nerve stimulation, or ultrasound is recommended.
Repeat BOTOX treatment may be administered when the effect of a previous injection has diminished, but generally no sooner than 12 weeks after the previous injection. The degree and pattern of muscle spasticity at the time of re-injection may necessitate alterations in the dose of BOTOX and muscles to be injected.
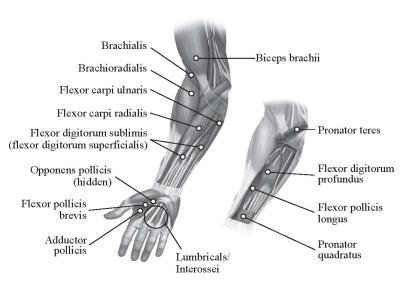
Adult Upper Limb Spasticity
In clinical trials, doses ranging from 75 Units to 400 Units were divided among selected muscles (see Table 4 and Figure 2) at a given treatment session.
| Muscle | Recommended Dose
Total Dosage (Number of Sites) |
| Biceps Brachii | 60 Units to 200 Units divided in 2 to 4 sites |
| Brachioradialis | 45 Units to 75 Units divided in 1 to 2 sites |
| Brachialis | 30 Units to 50 Units divided in 1 to 2 sites |
| Pronator Teres | 15 Units to 25 Units in 1 site |
| Pronator Quadratus | 10 Units to 50 Units in 1 site |
| Flexor Carpi Radialis | 12.5 Units to 50 Units in 1 site |
| Flexor Carpi Ulnaris | 12.5 Units to 50 Units in 1 site |
| Flexor Digitorum Profundus | 30 Units to 50 Units in 1 site |
| Flexor Digitorum Sublimis | 30 Units to 50 Units in 1 site |
| Lumbricals/Interossei | 5 Units to 10 Units in 1 site |
| Adductor Pollicis | 20 Units in 1 site |
| Flexor Pollicis Longus | 20 Units in 1 site |
| Flexor pollicis brevis/ Opponens pollicis | 5 Units to 25 Units in 1 site |
Figure 2: Injection Sites for Adult Upper Limb Spasticity
Adult Lower Limb Spasticity
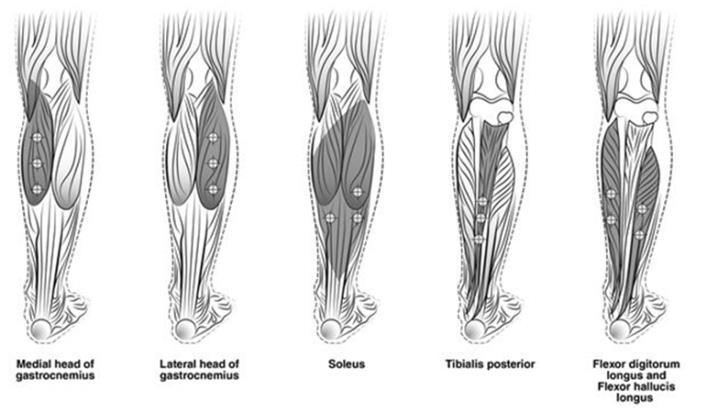
The recommended dose for treating adult lower limb spasticity is 300 Units to 400 Units divided among 5 muscles (gastrocnemius, soleus, tibialis posterior, flexor hallucis longus and flexor digitorum longus) (see Table 5 and Figure 3).
| Muscle | Recommended Dose
Total Dosage (Number of Sites) |
| Gastrocnemius medial head | 75 Units divided in 3 sites |
| Gastrocnemius lateral head | 75 Units divided in 3 sites |
| Soleus | 75 Units divided in 3 sites |
| Tibialis Posterior | 75 Units divided in 3 sites |
| Flexor hallucis longus | 50 Units divided in 2 sites |
| Flexor digitorum longus | 50 Units divided in 2 sites |
Figure 3: Injection Sites for Adult Lower Limb Spasticity
2.7 Pediatric Spasticity
General
Localization of the involved muscles with techniques such as needle electromyographic guidance, nerve stimulation, or ultrasound is recommended. When treating both lower limbs or the upper and lower limbs in combination, the total dose should not exceed the lower of 10 Units/kg body weight or 340 Units, in a 3-month interval [see Boxed Warning and Warnings and Precautions (5.1, 5.6)]. Additional general adult spasticity dosing information is also applicable to pediatric spasticity patients [see Dosage and Administration (2.6)].
Pediatric Upper Limb Spasticity
The recommended dose for treating pediatric upper limb spasticity is 3 Units/kg to 6 Units/kg divided among the affected muscles (see Table 6 and Figure 4). The total dose of BOTOX administered per treatment session in the upper limb should not exceed 6 Units/kg or 200 Units, whichever is lower.
| Muscle | Recommended Dose and
Number of Sites |
| Biceps Brachii | 1.5 Units/kg to 3 Units/kg divided in 4 sites |
| Brachialis | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Brachioradialis | 0.5 Units/kg to 1 Unit/kg divided in 2 sites |
| Flexor Carpi Radialis | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Flexor Carpi Ulnaris | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Flexor Digitorum Profundus | 0.5 Units/kg to 1 Unit/kg divided in 2 sites |
| Flexor Digitorum Sublimis | 0.5 Units/kg to 1 Unit/kg divided in 2 sites |
Figure 4: Injection Sites for Pediatric Upper Limb Spasticity
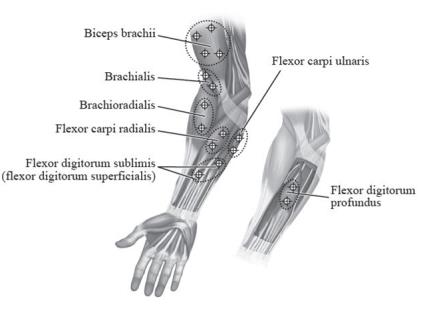
Pediatric Lower Limb Spasticity
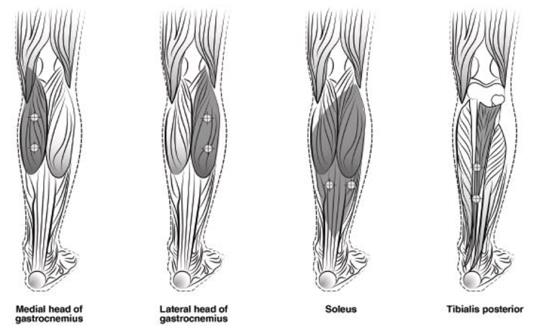
The recommended dose for treating pediatric lower limb spasticity is 4 Units/kg to 8 Units/kg divided among the affected muscles (see Table 7 and Figure 5). The total dose of BOTOX administered per treatment session in the lower limb should not exceed 8 Units/kg or 300 Units, whichever is lower.
| Muscle | Recommended Dose
Total Dosage (Number of Sites) |
| Gastrocnemius medial head | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Gastrocnemius lateral head | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Soleus | 1 Unit/kg to 2 Units/kg divided in 2 sites |
| Tibialis Posterior | 1 Unit/kg to 2 Units/kg divided in 2 sites |
Figure 5: Injection Sites for Pediatric Lower Limb Spasticity
2.8 Cervical Dystonia
A double-blind, placebo-controlled study enrolled patients who had extended histories of receiving and tolerating BOTOX injections, with prior individualized adjustment of dose. The mean BOTOX dose administered to patients in this study was 236 Units (25th to 75th percentile range of 198 Units to 300 Units). The BOTOX dose was divided among the affected muscles [see Clinical Studies (14.7)].
Dosing in initial and sequential treatment sessions should be tailored to the individual patient based on the patient’s head and neck position, localization of pain, muscle hypertrophy, patient response, and adverse event history. The initial dose for a patient without prior use of BOTOX should be at a lower dose, with subsequent dosing adjusted based on individual response. Limiting the total dose injected into the sternocleidomastoid muscle to 100 Units or less may decrease the occurrence of dysphagia [see Warnings and Precautions (5.1, 5.5, 5.6)].
The recommended dilution is 200 Units/2 mL, 200 Units/4 mL, 100 Units/1 mL, or 100 Units/2 mL with preservative-free 0.9% Sodium Chloride Injection, USP, depending on volume and number of injection sites desired to achieve treatment objectives (see Table 1). In general, no more than 50 Units per site should be administered using a sterile needle (e.g., 25-30 gauge) of an appropriate length. Localization of the involved muscles with electromyographic guidance may be useful.
Clinical improvement generally begins within the first two weeks after injection with maximum clinical benefit at approximately six weeks post-injection. In the double-blind, placebo-controlled study most subjects were observed to have returned to pre-treatment status by 3 months post-treatment.
2.9 Primary Axillary Hyperhidrosis
The recommended dose is 50 Units per axilla. The hyperhidrotic area to be injected should be defined using standard staining techniques, e.g., Minor’s Iodine-Starch Test. The recommended dilution is 100 Units/4 mL with preservative-free 0.9% Sodium Chloride Injection, USP (see Table 1). Using a sterile 30 gauge needle, 50 Units of BOTOX (2 mL) is injected intradermally in 0.1 to 0.2 mL aliquots to each axilla evenly distributed in multiple sites (10-15) approximately 1-2 cm apart.
Repeat injections for hyperhidrosis should be administered when the clinical effect of a previous injection diminishes.
Instructions for the Minor’s Iodine-Starch Test Procedure:
Patients should shave underarms and abstain from use of over-the-counter deodorants or antiperspirants for 24 hours prior to the test. Patient should be resting comfortably without exercise or hot drinks for approximately 30 minutes prior to the test. Dry the underarm area and then immediately paint it with iodine solution. Allow the area to dry, then lightly sprinkle the area with starch powder. Gently blow off any excess starch powder. The hyperhidrotic area will develop a deep blue-black color over approximately 10 minutes.
Each injection site has a ring of effect of up to approximately 2 cm in diameter. To minimize the area of no effect, the injection sites should be evenly spaced as shown in Figure 6.
Figure 6: Injection Pattern for Primary Axillary Hyperhidrosis
Each dose is injected to a depth of approximately 2 mm and at a 45° angle to the skin surface, with the bevel side up to minimize leakage and to ensure the injections remain intradermal. If injection sites are marked in ink, do not inject BOTOX directly through the ink mark to avoid a permanent tattoo effect.
2.10 Blepharospasm
For blepharospasm, reconstituted BOTOX is injected using a sterile, 27-30 gauge needle without electromyographic guidance. The initial recommended dose is 1.25 Units-2.5 Units (0.05 mL to 0.1 mL volume at each site) injected into the medial and lateral pre-tarsal orbicularis oculi of the upper lid and into the lateral pre-tarsal orbicularis oculi of the lower lid. Avoiding injection near the levator palpebrae superioris may reduce the complication of ptosis. Avoiding medial lower lid injections, and thereby reducing diffusion into the inferior oblique, may reduce the complication of diplopia. Ecchymosis occurs easily in the soft eyelid tissues. This can be prevented by applying pressure at the injection site immediately after the injection.
The recommended dilution to achieve 1.25 Units is 100 Units/8 mL; for 2.5 Units it is 100 Units/4 mL (see Table 1).
In general, the initial effect of the injections is seen within three days and reaches a peak at one to two weeks post-treatment. Each treatment lasts approximately three months, following which the procedure can be repeated. At repeat treatment sessions, the dose may be increased up to two-fold if the response from the initial treatment is considered insufficient, usually defined as an effect that does not last longer than two months. However, there appears to be little benefit obtainable from injecting more than 5 Units per site. Some tolerance may be found when BOTOX is used in treating blepharospasm if treatments are given any more frequently than every three months, and is rare to have the effect be permanent.
The cumulative dose of BOTOX treatment for blepharospasm in a 30-day period should not exceed 200 Units.
2.11 Strabismus
BOTOX is intended for injection into extraocular muscles utilizing the electrical activity recorded from the tip of the injection needle as a guide to placement within the target muscle. Injection without surgical exposure or electromyographic guidance should not be attempted. Physicians should be familiar with electromyographic technique.
To prepare the eye for BOTOX injection, it is recommended that several drops of a local anesthetic and an ocular decongestant be given several minutes prior to injection.
The volume of BOTOX injected for treatment of strabismus should be between 0.05-0.15 mL per muscle.
The initial listed doses of the reconstituted BOTOX [see Dosage and Administration (2.2)] typically create paralysis of the injected muscles beginning one to two days after injection and increasing in intensity during the first week. The paralysis lasts for 2-6 weeks and gradually resolves over a similar time period. Overcorrections lasting over six months have been rare. About one half of patients will require subsequent doses because of inadequate paralytic response of the muscle to the initial dose, or because of mechanical factors such as large deviations or restrictions, or because of the lack of binocular motor fusion to stabilize the alignment.
Initial Doses in Units
Use the lower listed doses for treatment of small deviations. Use the larger doses only for large deviations.
- For vertical muscles, and for horizontal strabismus of less than 20 prism diopters: 1.25 Units-2.5 Units in any one muscle.
- For horizontal strabismus of 20 prism diopters to 50 prism diopters: 2.5 Units-5 Units in any one muscle.
- For persistent VI nerve palsy of one month or longer duration: 1.25 Units-2.5 Units in the medial rectus muscle.
Subsequent Doses for Residual or Recurrent Strabismus
- It is recommended that patients be re-examined 7-14 days after each injection to assess the effect of that dose.
- Patients experiencing adequate paralysis of the target muscle that require subsequent injections should receive a dose comparable to the initial dose.
- Subsequent doses for patients experiencing incomplete paralysis of the target muscle may be increased up to two-fold compared to the previously administered dose.
- Subsequent injections should not be administered until the effects of the previous dose have dissipated as evidenced by substantial function in the injected and adjacent muscles.
- The maximum recommended dose as a single injection for any one muscle is 25 Units.
The recommended dilution to achieve 1.25 Units is 100 Units/8 mL; for 2.5 Units it is 100 Units/4 mL (see Table 1).
3 DOSAGE FORMS AND STRENGTHS
For Injection: sterile 100 Units or 200 Units vacuum-dried powder in single-dose vials for reconstitution only with sterile, preservative-free 0.9% Sodium Chloride Injection, USP prior to injection.
4 CONTRAINDICATIONS
BOTOX is contraindicated:
- In patients who are hypersensitive to any botulinum toxin product or to any of the components in the formulation [see Warnings and Precautions (5.4)].
- In the presence of infection at the proposed injection site(s).
- For intradetrusor injection in patients with a urinary tract infection; or in patients with urinary retention or post-void residual (PVR) urine volume >200 mL who are not routinely performing clean intermittent self-catheterization (CIC) [see Warnings and Precautions (5.12, 5.13)].
5 WARNINGS AND PRECAUTIONS
5.1 Spread of Toxin Effect
Postmarketing safety data from BOTOX and other approved botulinum toxins suggest that botulinum toxin effects may, in some cases, be observed beyond the site of local injection. The symptoms are consistent with the mechanism of action of botulinum toxin and may include asthenia, generalized muscle weakness, diplopia, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence, and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death related to spread of toxin effects. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, and particularly in those patients who have an underlying condition that would predispose them to these symptoms. In unapproved uses and in approved indications, symptoms consistent with spread of toxin effect have been reported at doses comparable to or lower than doses used to treat cervical dystonia and spasticity. Patients or caregivers should be advised to seek immediate medical care if swallowing, speech or respiratory disorders occur.
No definitive serious adverse event reports of distant spread of toxin effect associated with BOTOX for blepharospasm at the recommended dose (30 Units and below), severe primary axillary hyperhidrosis at the recommended dose (100 Units), strabismus, or for chronic migraine at the labeled doses have been reported.
5.2 Lack of Interchangeability between Botulinum Toxin Products
The potency Units of BOTOX are specific to the preparation and assay method utilized. They are not interchangeable with other preparations of botulinum toxin products and, therefore, units of biological activity of BOTOX cannot be compared to nor converted into units of any other botulinum toxin products assessed with any other specific assay method [see Description (11)].
5.3 Serious Adverse Reactions with Unapproved Use
Serious adverse reactions, including excessive weakness, dysphagia, and aspiration pneumonia, with some adverse reactions associated with fatal outcomes, have been reported in patients who received BOTOX injections for unapproved uses. In these cases, the adverse reactions were not necessarily related to distant spread of toxin, but may have resulted from the administration of BOTOX to the site of injection and/or adjacent structures. In several of the cases, patients had pre-existing dysphagia or other significant disabilities. There is insufficient information to identify factors associated with an increased risk for adverse reactions associated with the unapproved uses of BOTOX. The safety and effectiveness of BOTOX for unapproved uses have not been established.
5.4 Hypersensitivity Reactions
Serious and/or immediate hypersensitivity reactions have been reported. These reactions include anaphylaxis, serum sickness, urticaria, soft tissue edema, and dyspnea. If such a reaction occurs, further injection of BOTOX should be discontinued and appropriate medical therapy immediately instituted. One fatal case of anaphylaxis has been reported in which lidocaine was used as the diluent, and consequently the causal agent cannot be reliably determined.
5.5 Increased Risk of Clinically Significant Effects with Pre-Existing Neuromuscular Disorders
Individuals with peripheral motor neuropathic diseases, amyotrophic lateral sclerosis or neuromuscular junction disorders (e.g., myasthenia gravis or Lambert-Eaton syndrome) should be monitored when given botulinum toxin. Patients with known or unrecognized neuromuscular disorders or neuromuscular junction disorders may be at increased risk of clinically significant effects including generalized muscle weakness, diplopia, ptosis, dysphonia, dysarthria, severe dysphagia and respiratory compromise from therapeutic doses of BOTOX [see Warnings and Precautions (5.1, 5.6)].
5.6 Dysphagia and Breathing Difficulties
Treatment with BOTOX and other botulinum toxin products can result in swallowing or breathing difficulties. Patients with pre-existing swallowing or breathing difficulties may be more susceptible to these complications. In most cases, this is a consequence of weakening of muscles in the area of injection that are involved in breathing or oropharyngeal muscles that control swallowing or breathing [see Warnings and Precautions (5.1)].
Deaths as a complication of severe dysphagia have been reported after treatment with botulinum toxin. Dysphagia may persist for several months, and require use of a feeding tube to maintain adequate nutrition and hydration. Aspiration may result from severe dysphagia and is a particular risk when treating patients in whom swallowing or respiratory function is already compromised.
Treatment with botulinum toxins may weaken neck muscles that serve as accessory muscles of ventilation. This may result in a critical loss of breathing capacity in patients with respiratory disorders who may have become dependent upon these accessory muscles. There have been postmarketing reports of serious breathing difficulties, including respiratory failure.
Patients with smaller neck muscle mass and patients who require bilateral injections into the sternocleidomastoid muscle for the treatment of cervical dystonia have been reported to be at greater risk for dysphagia. Limiting the dose injected into the sternocleidomastoid muscle may reduce the occurrence of dysphagia. Injections into the levator scapulae may be associated with an increased risk of upper respiratory infection and dysphagia.
Patients treated with botulinum toxin may require immediate medical attention should they develop problems with swallowing, speech or respiratory disorders. These reactions can occur within hours to weeks after injection with botulinum toxin [see Warnings and Precautions (5.1)].
5.7 Pulmonary Effects of BOTOX in Patients with Compromised Respiratory Status Treated for Spasticity or for Detrusor Overactivity Associated with a Neurologic Condition
Patients with compromised respiratory status treated with BOTOX for spasticity should be monitored closely. In a double-blind, placebo-controlled, parallel group study in adult patients treated for upper limb spasticity with stable reduced pulmonary function (defined as FEV1 40-80% of predicted value and FEV1/FVC ≤ 0.75), the event rate in change of Forced Vital Capacity (FVC) ≥15% or ≥20% was generally greater in patients treated with BOTOX than in patients treated with placebo (see Table 8).
Table 8: Event Rate Per Patient Treatment Cycle Among Adult Upper Limb Spasticity Patients with Reduced Lung Function Who Experienced at Least a 15% or 20% Decrease in FVC From Baseline at Week 1, 6, 12 Post-injection with Up to Two Treatment Cycles with BOTOX or Placebo
|
| BOTOX
360 Units | BOTOX
240 Units | Placebo | |||
| ≥15% | ≥20% | ≥15% | ≥20% | ≥15% | ≥20% | |
| Week 1 | 4% | 0% | 3% | 0% | 7% | 3% |
| Week 6 | 7% | 4% | 4% | 2% | 2% | 2% |
| Week 12 | 10% | 5% | 2% | 1% | 4% | 1% |
Differences from placebo were not statistically significant
In adult spasticity patients with reduced lung function, upper respiratory tract infections were also reported more frequently as adverse reactions in patients treated with BOTOX than in patients treated with placebo [see Warnings and Precautions (5.10)].
In a double-blind, placebo-controlled, parallel group study in adult patients with detrusor overactivity associated with a neurologic condition and restrictive lung disease of neuromuscular etiology [defined as FVC 50-80% of predicted value in patients with spinal cord injury between C5 and C8, or MS] the event rate in change of Forced Vital Capacity ≥15% or ≥20% was generally greater in patients treated with BOTOX than in patients treated with placebo (see Table 9).
Table 9: Number and Percent of Patients Experiencing at Least a 15% or 20% Decrease in FVC From Baseline at Week 2, 6, 12 Post-Injection with BOTOX or Placebo
| BOTOX
200 Units | Placebo | |||
| ≥15% | ≥20% | ≥15% | ≥20% | |
| Week 2 | 0/15 (0%) | 0/15 (0%) | 1/11 (9%) | 0/11 (0%) |
| Week 6 | 2/13 (15%) | 1/13 (8%) | 0/12 (0%) | 0/12 (0%) |
| Week 12 | 0/12(0%) | 0/12 (0%) | 0/7 (0%) | 0/7 (0%) |
5.8 Corneal Exposure and Ulceration in Patients Treated with BOTOX for Blepharospasm
Reduced blinking from BOTOX injection of the orbicularis muscle can lead to corneal exposure, persistent epithelial defect, and corneal ulceration, especially in patients with VII nerve disorders. Vigorous treatment of any epithelial defect should be employed. This may require protective drops, ointment, therapeutic soft contact lenses, or closure of the eye by patching or other means.
5.9 Retrobulbar Hemorrhages in Patients Treated with BOTOX for Strabismus
During the administration of BOTOX for the treatment of strabismus, retrobulbar hemorrhages sufficient to compromise retinal circulation have occurred. It is recommended that appropriate instruments to decompress the orbit be accessible.
5.10 Bronchitis and Upper Respiratory Tract Infections in Patients Treated for Spasticity
Bronchitis was reported more frequently as an adverse reaction in adult patients treated for upper limb spasticity with BOTOX (3% at 251 Units-360 Units total dose), compared to placebo (1%). In adult patients with reduced lung function treated for upper limb spasticity, upper respiratory tract infections were also reported more frequently as adverse reactions in patients treated with BOTOX (11% at 360 Units total dose; 8% at 240 Units total dose) compared to placebo (6%). In adult patients treated for lower limb spasticity, upper respiratory tract infections were reported more frequently as an adverse reaction in patients treated with BOTOX (2% at 300 Units to 400 Units total dose) compared to placebo (1%). In pediatric patients treated for upper limb spasticity, upper respiratory tract infections were reported more frequently as an adverse reaction in patients treated with BOTOX (17% at 6 Units/kg and 10% at 3 Units/kg) compared to placebo (9%). In pediatric patients treated for lower limb spasticity, upper respiratory tract infection was not reported with an incidence greater than placebo.
5.11 Autonomic Dysreflexia in Patients Treated for Detrusor Overactivity Associated with a Neurologic Condition
Autonomic dysreflexia associated with intradetrusor injections of BOTOX could occur in patients treated for detrusor overactivity associated with a neurologic condition and may require prompt medical therapy. In clinical trials, the incidence of autonomic dysreflexia was greater in adult patients treated with BOTOX 200 Units compared with placebo (1.5% versus 0.4%, respectively).
5.12 Urinary Tract Infections in Patients with Overactive Bladder
BOTOX increases the incidence of urinary tract infection [see Adverse Reactions (6.1)]. Clinical trials for overactive bladder excluded patients with more than 2 UTIs in the past 6 months and those taking antibiotics chronically due to recurrent UTIs. Use of BOTOX for the treatment of overactive bladder in such patients and in patients with multiple recurrent UTIs during treatment should only be considered when the benefit is likely to outweigh the potential risk.
5.13 Urinary Retention in Adults Treated for Bladder Dysfunction
Due to the risk of urinary retention, treat only patients who are willing and able to initiate catheterization post-treatment, if required, for urinary retention.
In patients who are not catheterizing, post-void residual (PVR) urine volume should be assessed within 2 weeks post-treatment and periodically as medically appropriate up to 12 weeks, particularly in patients with multiple sclerosis or diabetes mellitus. Depending on patient symptoms, institute catheterization if PVR urine volume exceeds 200 mL and continue until PVR falls below 200 mL. Instruct patients to contact their physician if they experience difficulty in voiding as catheterization may be required.
The incidence and duration of urinary retention is described below for adult patients with overactive bladder and detrusor overactivity associated with a neurologic condition who received BOTOX or placebo injections.
Overactive Bladder
In double-blind, placebo-controlled trials in patients with OAB, the proportion of subjects who initiated clean intermittent catheterization (CIC) for urinary retention following treatment with BOTOX or placebo is shown in Table 10. The duration of post-injection catheterization for those who developed urinary retention is also shown.
| Timepoint | BOTOX 100 Units
(N=552) | Placebo
(N=542) |
| Proportion of Patients Catheterizing for Urinary Retention | ||
| At any time during complete treatment cycle | 6.5% (n=36) | 0.4% (n=2) |
| Duration of Catheterization for Urinary Retention (Days) | ||
| Median | 63 | 11 |
| Min, Max | 1, 214 | 3, 18 |
Patients with diabetes mellitus treated with BOTOX were more likely to develop urinary retention than those without diabetes, as shown in Table 11.
| Patients with Diabetes | Patients without Diabetes | |||
| BOTOX 100 Units
(N=81) | Placebo
(N=69) | BOTOX 100 Units (N=526) | Placebo
(N=516) |
|
| Urinary retention | 12.3% (n=10) | 0 | 6.3% (n=33) | 0.6% (n=3) |
Adult Detrusor Overactivity associated with a Neurologic Condition
In two double-blind, placebo-controlled trials in adult patients with detrusor overactivity associated with a neurologic condition (NDO-1 and NDO-2), the proportion of subjects who were not using clean intermittent catheterization (CIC) prior to injection and who subsequently required catheterization for urinary retention following treatment with BOTOX 200 Units or placebo is shown in Table 12. The duration of post-injection catheterization for those who developed urinary retention is also shown.
| Timepoint | BOTOX 200 Units
(N=108) | Placebo
(N=104) |
| Proportion of Patients Catheterizing for Urinary Retention | ||
| At any time during complete treatment cycle | 30.6% (n=33) | 6.7% (n=7) |
| Duration of Catheterization for Urinary Retention (Days) | ||
| Median | 289 | 358 |
| Min, Max | 1, 530 | 2, 379 |
Among adult patients not using CIC at baseline, those with Multiple Sclerosis (MS) were more likely to require CIC post-injection than those with Spinal Cord Injury (SCI) (see Table 13).
| Timepoint | MS | SCI | ||
| BOTOX 200 Units
(N=86) | Placebo
(N=88) | BOTOX 200 Units
(N=22) | Placebo
(N=16) |
|
| At any time during complete treatment cycle | 31% (n=27) | 5% (n=4) | 27% (n=6) | 19% (n=3) |
A placebo-controlled, double-blind post-approval 52 week study with BOTOX 100 Units (Study NDO-3) was conducted in non-catheterizing adult MS patients with urinary incontinence due to detrusor overactivity associated with a neurologic condition. Catheterization for urinary retention was initiated in 15.2% (10/66) of patients following treatment with BOTOX 100 Units versus 2.6% (2/78) on placebo at any time during the complete treatment cycle. The median duration of post-injection catheterization for those who developed urinary retention was 64 days for BOTOX 100 Units and 2 days for placebo.
5.14 Human Albumin and Transmission of Viral Diseases
This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases and variant Creutzfeldt-Jakob disease (vCJD). There is a theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD), but if that risk actually exists, the risk of transmission would also be considered extremely remote. No cases of transmission of viral diseases, CJD or vCJD have ever been identified for licensed albumin or albumin contained in other licensed products.
6 ADVERSE REACTIONS
The following adverse reactions to BOTOX (onabotulinumtoxinA) for injection are discussed in greater detail in other sections of the labeling:
- Spread of Toxin Effects [see Warnings and Precautions (5.1)]
- Serious Adverse Reactions with Unapproved Use [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Contraindications (4) and Warnings and Precautions (5.4)]
- Increased Risk of Clinically Significant Effects with Pre-Existing Neuromuscular Disorders [see Warnings and Precautions (5.5)]
- Dysphagia and Breathing Difficulties [see Warnings and Precautions (5.6)]
- Pulmonary Effects of BOTOX in Patients with Compromised Respiratory Status Treated for Spasticity or for Detrusor Overactivity Associated with a Neurologic Condition [see Warnings and Precautions (5.7)]
- Corneal Exposure and Ulceration in Patients Treated with BOTOX for Blepharospasm [see Warnings and Precautions (5.8)]
- Retrobulbar Hemorrhages in Patients Treated with BOTOX for Strabismus [see Warnings and Precautions (5.9)]
- Bronchitis and Upper Respiratory Tract Infections in Patients Treated for Spasticity [see Warnings and Precautions (5.10)]
- Autonomic Dysreflexia in Patients Treated for Detrusor Overactivity Associated with a Neurologic Condition [see Warnings and Precautions (5.11)]
- Urinary Tract Infections in Patients with Overactive Bladder [see Warnings and Precautions (5.12)]
- Urinary Retention in Patients Treated for Bladder Dysfunction [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
BOTOX and BOTOX Cosmetic contain the same active ingredient in the same formulation, but with different labeled Indications and Usage. Therefore, adverse reactions observed with the use of BOTOX Cosmetic also have the potential to be observed with the use of BOTOX.
In general, adverse reactions occur within the first week following injection of BOTOX and, while generally transient, may have a duration of several months or longer. Localized pain, infection, inflammation, tenderness, swelling, erythema, and/or bleeding/bruising may be associated with the injection. Symptoms associated with flu-like symptoms (e.g., nausea, fever, myalgia) have been reported after treatment. Needle-related pain and/or anxiety may result in vasovagal responses (including syncope, hypotension), which may require appropriate medical therapy.
Local weakness of the injected muscle(s) represents the expected pharmacological action of botulinum toxin. However, weakness of nearby muscles may also occur due to spread of toxin [see Warnings and Precautions (5.1)].
Overactive Bladder
Table 14 presents the most frequently reported adverse reactions in double-blind, placebo-controlled clinical trials for overactive bladder occurring within 12 weeks of the first BOTOX treatment.
| Adverse Reactions | BOTOX
100 Units (N=552) % | Placebo
(N=542) % |
| Urinary tract infection Dysuria Urinary retention Bacteriuria Residual urine volume* | 18 9 6 4 3 | 6 7 0 2 0 |
*Elevated PVR not requiring catheterization. Catheterization was required for PVR ≥350 mL regardless of symptoms, and for PVR ≥200 mL to <350 mL with symptoms (e.g., voiding difficulty).
A higher incidence of urinary tract infection was observed in patients with diabetes mellitus treated with BOTOX 100 Units and placebo than in patients without diabetes, as shown in Table 15.
| Patients with Diabetes | Patients without Diabetes | |||
| BOTOX 100 Units
(N=81) % | Placebo
(N=69) % | BOTOX 100 Units (N=526)
% | Placebo
(N=516) % |
|
| Urinary tract infection (UTI) | 31 | 12 | 26 | 10 |
The incidence of UTI increased in patients who experienced a maximum post-void residual (PVR) urine volume ≥200 mL following BOTOX injection compared to those with a maximum PVR <200 mL following BOTOX injection, 44% versus 23%, respectively.
No change was observed in the overall safety profile with repeat dosing during an open-label, uncontrolled extension trial.
Adult Detrusor Overactivity associated with a Neurologic Condition
Table 16 presents the most frequently reported adverse reactions in the double-blind, placebo-controlled studies within 12 weeks of injection for patients with detrusor overactivity associated with a neurologic condition treated with BOTOX 200 Units.
| Adverse Reactions | BOTOX 200 Units
(N=262) % | Placebo
(N=272) % |
| Urinary tract infection Urinary retention Hematuria | 24 17 4 | 17 3 3 |
The following adverse reactions with BOTOX 200 Units were reported at any time following initial injection and prior to re-injection or study exit (median duration of exposure was 44 weeks): urinary tract infections (49%), urinary retention (17%), constipation (4%), muscular weakness (4%), dysuria (4%), fall (3%), gait disturbance (3%), and muscle spasm (2%).
In the Multiple Sclerosis (MS) patients enrolled in the double-blind, placebo-controlled trials, the MS exacerbation annualized rate (i.e., number of MS exacerbation events per patient-year) was 0.23 for BOTOX and 0.20 for placebo.
No change was observed in the overall safety profile with repeat dosing.
Table 17 presents the most frequently reported adverse reactions in a placebo-controlled, double-blind post-approval 52 week study with BOTOX 100 Units (Study NDO-3) conducted in MS patients with urinary incontinence due to detrusor overactivity associated with a neurologic condition. These patients were not adequately managed with at least one anticholinergic agent and not catheterized at baseline. The table below presents the most frequently reported adverse reactions within 12 weeks of injection.
| Adverse Reactions | BOTOX
100 Units (N=66) % | Placebo
(N=78) % |
| Urinary tract infection Bacteriuria Urinary retention Dysuria Residual urine volume* | 26 9 15 5 17 | 6 5 1 1 1 |
* Elevated PVR not requiring catheterization. Catheterization was required for PVR ≥350 mL regardless of symptoms, and for PVR ≥200 mL to <350 mL with symptoms (e.g., voiding difficulty).
The following adverse events with BOTOX 100 Units were reported at any time following initial injection and prior to re-injection or study exit (median duration of exposure was 51 weeks): urinary tract infections (39%), bacteriuria (18%), urinary retention (17%), residual urine volume* (17%), dysuria (9%), and hematuria (5%).
No difference in the MS exacerbation annualized rate (i.e., number of MS exacerbating events per patient-year) was observed (BOTOX =0, placebo =0.07).
Pediatric Detrusor Overactivity associated with a Neurologic Condition
Table 18 presents the most frequently reported adverse reactions in Study 191622-120, a double-blind, parallel-group study conducted in pediatric patients with detrusor overactivity associated with a neurologic condition. These patients were not adequately managed with at least one anticholinergic agent and were using clean intermittent catheterization at baseline [see Clinical Studies (14.3)]. The table below presents the most frequently reported adverse reactions during the 12 weeks following intradetrusor administration of BOTOX 200 Units.
| Adverse Reactions | BOTOX 200 Unit
(N=30) |
| Urinary tract infection | 2 (7%) |
| Bacteriuria | 6 (20%) |
| Leukocyturia | 2 (7%) |
| Hematuria | 1 (3%) |
No change was observed in the overall safety profile with repeat dosing.
The most common adverse reactions in patients who received BOTOX 6 U/kg and less than a total dose of 200 U in Study 191622-120 were urinary tract infection (UTI), bacteriuria and hematuria.
Chronic Migraine
In double-blind, placebo-controlled chronic migraine efficacy trials (Study 1 and Study 2), the discontinuation rate was 12% in the BOTOX treated group and 10% in the placebo-treated group. Discontinuations due to an adverse event were 4% in the BOTOX group and 1% in the placebo group. The most frequent adverse events leading to discontinuation in the BOTOX group were neck pain, headache, worsening migraine, muscular weakness and eyelid ptosis.
The most frequently reported adverse reactions following injection of BOTOX for chronic migraine appear in Table 19.
| Adverse Reactions | BOTOX
155 Units-195 Units (N=687) % | Placebo
(N=692) % |
|
| Nervous system disorders Headache Migraine Facial paresis |
5 4 2 |
3 3 0 |
|
| Eye disorders Eyelid ptosis |
4 |
<1 |
|
| Infections and Infestations Bronchitis |
3 |
2 |
|
| Musculoskeletal and connective tissue disorders Neck pain Musculoskeletal stiffness Muscular weakness Myalgia Musculoskeletal pain Muscle spasms |
9 4 4 3 3 2 |
3 1 <1 1 1 1 |
|
| General disorders and administration site conditions Injection site pain |
3 |
2 |
|
| Vascular Disorders Hypertension |
2 |
1 |
Other adverse reactions that occurred more frequently in the BOTOX group compared to the placebo group at a frequency less than 1% and potentially BOTOX related include: vertigo, dry eye, eyelid edema, dysphagia, eye infection, and jaw pain. Severe worsening of migraine requiring hospitalization occurred in approximately 1% of BOTOX treated patients in Study 1 and Study 2, usually within the first week after treatment, compared to 0.3% of placebo-treated patients.
Adult Upper Limb Spasticity
The most frequently reported adverse reactions following injection of BOTOX for adult upper limb spasticity appear in Table 20.
| Adverse Reactions | BOTOX
251 Units - 360 Units (N=115) % | BOTOX
150 Units - 250 Units (N=188) % | BOTOX
<150 Units (N=54) % | Placebo
(N=182) % |
| Gastrointestinal disorder Nausea | 3 | 2 | 2 | 1 |
| General disorders and administration site conditions Fatigue | 3 | 2 | 2 | 0 |
| Infections and infestations Bronchitis | 3 | 2 | 0 | 1 |
| Musculoskeletal and connective tissue disorders Pain in extremity Muscular weakness | 6 0 | 5 4 | 9 2 | 4 1 |
Twenty-two adult patients, enrolled in double-blind placebo controlled studies, received 400 Units or higher of BOTOX for treatment of upper limb spasticity. In addition, 44 adults received 400 Units of BOTOX or higher for four consecutive treatments over approximately one year for treatment of upper limb spasticity. The type and frequency of adverse reactions observed in patients treated with 400 Units of BOTOX were similar to those reported in patients treated for upper limb spasticity with 360 Units of BOTOX.
Adult Lower Limb Spasticity
The most frequently reported adverse reactions following injection of BOTOX for adult lower limb spasticity appear in Table 21. Two hundred thirty-one patients enrolled in a double-blind placebo controlled study (Study 7) received 300 Units to 400 Units of BOTOX, and were compared with 233 patients who received placebo. Patients were followed for an average of 91 days after injection.
| Adverse Reactions | BOTOX
(N=231) % | Placebo
(N=233) % |
| Musculoskeletal and connective tissue disorders Arthralgia Back pain Myalgia | 3 3 2 | 1 2 1 |
| Infections and infestations Upper respiratory tract infection | 2 | 1 |
| General disorders and administration site conditions Injection site pain | 2 | 1 |
Pediatric Upper Limb Spasticity
The most frequently reported adverse reactions following injection of BOTOX in pediatric patients 2 to 17 years of age with upper limb spasticity appear in Table 22. In a double-blind, placebo-controlled trial (Study 1), 78 patients were treated with 3 Units/kg of BOTOX, and 77 patients received 6 Units/kg to a maximum dose of 200 Units of BOTOX, and were compared to 79 patients who received placebo [see Clinical Studies (14.6)]. Patients were followed for an average of 91 days after injection.
| Adverse Reactions | BOTOX 6 Units/kg
(N=77) % | BOTOX 3 Units/kg
(N=78) % | Placebo
(N=79) % |
| Infections and infestations Upper respiratory tract infection* | 17 | 10 | 9 |
| General disorders and administration site conditions Injection site pain | 4 | 3 | 1 |
| Gastrointestinal disorders Nausea Constipation | 4 3 | 0 0 | 0 1 |
| Respiratory, thoracic and mediastinal disorders Rhinorrhea Nasal congestion | 4 3 | 0 0 | 1 1 |
| Nervous system disorders Seizure** | 5 | 1 | 0 |
*Includes upper respiratory tract infection and viral upper respiratory tract infection
**Includes seizure and partial seizure
Pediatric Lower Limb Spasticity
The most frequently reported adverse reactions following injection of BOTOX in pediatric patients 2 to 17 years of age with lower limb spasticity appear in Table 23. In a double-blind, placebo-controlled trial (Study 2), 126 patients were treated with 4 Units/kg of BOTOX, and 128 patients received 8 Units/kg to a maximum dose of 300 Units of BOTOX, and were compared to 128 patients who received placebo [see Clinical Studies (14.6)]. Patients were followed for an average of 89 days after injection.
| Adverse Reactions | BOTOX
8 Units/kg (N=128) % | BOTOX
4 Units/kg (N=126) % | Placebo
(N=128) % |
| General disorders and administration site conditions Injection site erythema Injection site pain | 2 2 | 0 2 | 0 0 |
| Respiratory, thoracic and mediastinal disorders Oropharyngeal pain | 2 | 0 | 1 |
| Injury, poisoning and procedural complications Ligament sprain Skin abrasion | 2 2 | 1 0 | 0 0 |
| Metabolism and nutrition disorders Decreased appetite | 2 | 0 | 0 |
Cervical Dystonia
In cervical dystonia patients evaluated for safety in double-blind and open-label studies following injection of BOTOX, the most frequently reported adverse reactions were dysphagia (19%), upper respiratory infection (12%), neck pain (11%), and headache (11%).
Other events reported in 2-10% of patients in any one study in decreasing order of incidence include: increased cough, flu syndrome, back pain, rhinitis, dizziness, hypertonia, soreness at injection site, asthenia, oral dryness, speech disorder, fever, nausea, and drowsiness. Stiffness, numbness, diplopia, ptosis, and dyspnea have been reported.
Dysphagia and symptomatic general weakness may be attributable to an extension of the pharmacology of BOTOX resulting from the spread of the toxin outside the injected muscles [see Warnings and Precautions (5.1, 5.6)].
The most common severe adverse reaction associated with the use of BOTOX injection in patients with cervical dystonia is dysphagia with about 20% of these cases also reporting dyspnea [see Warnings and Precautions (5.1, 5.6)]. Most dysphagia is reported as mild or moderate in severity. However, it may be associated with more severe signs and symptoms [see Warnings and Precautions (5.6)].
Additionally, reports in the literature include a case of a female patient who developed brachial plexopathy two days after injection of 120 Units of BOTOX for the treatment of cervical dystonia, and reports of dysphonia in patients who have been treated for cervical dystonia.
Primary Axillary Hyperhidrosis
The most frequently reported adverse reactions (3-10% of adult patients) following injection of BOTOX in double-blind studies included injection site pain and hemorrhage, non-axillary sweating, infection, pharyngitis, flu syndrome, headache, fever, neck or back pain, pruritus, and anxiety.
The data reflect 346 patients exposed to BOTOX 50 Units and 110 patients exposed to BOTOX 75 Units in each axilla.
Blepharospasm
In a study of blepharospasm patients who received an average dose per eye of 33 Units (injected at 3 to 5 sites) of the currently manufactured BOTOX, the most frequently reported adverse reactions were ptosis (21%), superficial punctate keratitis (6%), and eye dryness (6%).
Other events reported in prior clinical studies in decreasing order of incidence include: irritation, tearing, lagophthalmos, photophobia, ectropion, keratitis, diplopia, entropion, diffuse skin rash, and local swelling of the eyelid skin lasting for several days following eyelid injection.
In two cases of VII nerve disorder, reduced blinking from BOTOX injection of the orbicularis muscle led to serious corneal exposure, persistent epithelial defect, corneal ulceration and a case of corneal perforation. Focal facial paralysis, syncope, and exacerbation of myasthenia gravis have also been reported after treatment of blepharospasm.
Strabismus
Extraocular muscles adjacent to the injection site can be affected, causing vertical deviation, especially with higher doses of BOTOX. The incidence rates of these adverse effects in 2058 adults who received a total of 3650 injections for horizontal strabismus was 17%.
The incidence of ptosis has been reported to be dependent on the location of the injected muscles, 1% after inferior rectus injections, 16% after horizontal rectus injections and 38% after superior rectus injections.
In a series of 5587 injections, retrobulbar hemorrhage occurred in 0.3% of cases.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to onabotulinumtoxinA in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In a long term, open-label study evaluating 326 cervical dystonia patients treated for an average of 9 treatment sessions with the current formulation of BOTOX, 4 (1.2%) patients had positive antibody tests. All 4 of these patients responded to BOTOX therapy at the time of the positive antibody test. However, 3 of these patients developed clinical resistance after subsequent treatment, while the fourth patient continued to respond to BOTOX therapy for the remainder of the study.
One patient among the 445 hyperhidrosis patients (0.2%), two patients among the 380 adult upper limb spasticity patients (0.5%), and no patients among 406 migraine patients with analyzed specimens developed the presence of neutralizing antibodies.
In one Phase 3 study and the open-label extension study in patients with pediatric lower limb spasticity, neutralizing antibodies developed in 2 of 264 patients (0.8%) treated with BOTOX for up to 5 treatment cycles. Both patients continued to experience clinical benefit following subsequent BOTOX treatments.
In overactive bladder patients with analyzed specimens from the two phase 3 studies and the open-label extension study, neutralizing antibodies developed in 0 of 954 patients (0.0%) while receiving BOTOX 100 Unit doses and 3 of 260 patients (1.2%) after subsequently receiving at least one 150 Unit dose. Response to subsequent BOTOX treatment was not different following seroconversion in these three patients.
In detrusor overactivity associated with neurologic condition patients with analyzable specimens in the adult drug development program (including the open-label extension study), neutralizing antibodies developed in 3 of 300 patients (1.0%) after receiving only BOTOX 200 Unit doses and 5 of 258 patients (1.9%) after receiving at least one 300 Unit dose. Following development of neutralizing antibodies in these 8 patients, 4 continued to experience clinical benefit, 2 did not experience clinical benefit, and the effect on the response to BOTOX in the remaining 2 patients is not known. In 99 pediatric patients who had a negative baseline result for binding antibodies or neutralizing antibodies and had at least one evaluable post-baseline value from one randomized double-blind study and one double-blind extension study, no patients developed neutralizing antibodies after receiving 50 Units to 200 Units of BOTOX.
The data reflect the patients whose test results were considered positive for neutralizing activity to BOTOX in a mouse protection assay or negative based on a screening ELISA assay or mouse protection assay.
Formation of neutralizing antibodies to botulinum toxin type A may reduce the effectiveness of BOTOX treatment by inactivating the biological activity of the toxin. The critical factors for neutralizing antibody formation have not been well characterized. The results from some studies suggest that BOTOX injections at more frequent intervals or at higher doses may lead to greater incidence of antibody formation. The potential for antibody formation may be minimized by injecting with the lowest effective dose given at the longest feasible intervals between injections.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of BOTOX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions include: abdominal pain; alopecia, including madarosis; anorexia; brachial plexopathy; denervation/muscle atrophy; diarrhea; dry eye; eyelid edema (following periocular injection); hyperhidrosis; hypoacusis; hypoaesthesia; localized muscle twitching; malaise; Mephisto sign; paresthesia; peripheral neuropathy; radiculopathy; erythema multiforme, dermatitis psoriasiform, and psoriasiform eruption; strabismus; tinnitus; and visual disturbances.
There have been spontaneous reports of death, sometimes associated with dysphagia, pneumonia, and/or other significant debility or anaphylaxis, after treatment with botulinum toxin [see Warnings and Precautions (5.4, 5.6)].
There have also been reports of adverse events involving the cardiovascular system, including arrhythmia and myocardial infarction, some with fatal outcomes. Some of these patients had risk factors including cardiovascular disease. The exact relationship of these events to the botulinum toxin injection has not been established.
New onset or recurrent seizures have also been reported, typically in patients who are predisposed to experiencing these events. The exact relationship of these events to the botulinum toxin injection has not been established.
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Other Agents Interfering with Neuromuscular Transmission
Co-administration of BOTOX and aminoglycosides or other agents interfering with neuromuscular transmission (e.g., curare-like compounds) should only be performed with caution as the effect of the toxin may be potentiated.
7.2 Anticholinergic Drugs
Use of anticholinergic drugs after administration of BOTOX may potentiate systemic anticholinergic effects.
7.3 Other Botulinum Neurotoxin Products
The effect of administering different botulinum neurotoxin products at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no studies or adequate data from postmarketing surveillance on the developmental risk associated with use of BOTOX in pregnant women. In animal studies, administration of BOTOX during pregnancy resulted in adverse effects on fetal growth (decreased fetal weight and skeletal ossification) at clinically relevant doses, which were associated with maternal toxicity [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated populations is unknown.
Data
Animal Data
When BOTOX (4, 8, or 16 Units/kg) was administered intramuscularly to pregnant mice or rats two times during the period of organogenesis (on gestation days 5 and 13), reductions in fetal body weight and decreased fetal skeletal ossification were observed at the two highest doses. The no-effect dose for developmental toxicity in these studies (4 Units/kg) is approximately equal to the human dose of 400 Units, on a body weight basis (Units/kg).
When BOTOX was administered intramuscularly to pregnant rats (0.125, 0.25, 0.5, 1, 4, or 8 Units/kg) or rabbits (0.063, 0.125, 0.25, or 0.5 Units/kg) daily during the period of organogenesis (total of 12 doses in rats, 13 doses in rabbits), reduced fetal body weights and decreased fetal skeletal ossification were observed at the two highest doses in rats and at the highest dose in rabbits. These doses were also associated with significant maternal toxicity, including abortions, early deliveries, and maternal death. The developmental no-effect doses in these studies of 1 Unit/kg in rats and 0.25 Units/kg in rabbits are less than the human dose of 400 Units, based on Units/kg.
When pregnant rats received single intramuscular injections (1, 4, or 16 Units/kg) at three different periods of development (prior to implantation, implantation, or organogenesis), no adverse effects on fetal development were observed. The developmental no-effect level for a single maternal dose in rats (16 Units/kg) is approximately 2 times the human dose of 400 Units, based on Units/kg.
8.2 Lactation
Risk Summary
There are no data on the presence of BOTOX in human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for BOTOX and any potential adverse effects on the breastfed infant from BOTOX or from the underlying maternal conditions.
8.4 Pediatric Use
Detrusor Overactivity associated with a Neurologic Condition
The safety and effectiveness of BOTOX for detrusor overactivity associated with a neurologic condition have been established in pediatric patients 5 years of age and older who have an inadequate response to or are intolerant of anticholinergic medication. Use of BOTOX in this patient population is based on the results of a randomized, double-blind, parallel group trial in 113 pediatric patients 5 to 17 years of age (inclusive) with detrusor overactivity associated with a neurologic condition (Study 191622-120) and a long-term, multicenter, double-blind, long-term extension trial (Study 191622-121) [see Clinical Studies (14.3)]. The most common adverse reactions in this population were urinary tract infection, bacteriuria, hematuria, and leukocyturia [see Adverse Reactions (6.1)].
The safety and effectiveness of BOTOX have not been established in patients with NDO younger than 5 years of age.
Overactive Bladder
The safety and effectiveness of BOTOX for the treatment of overactive bladder have not been established in pediatric patients.
Efficacy was not demonstrated in a multicenter, randomized, double-blind, parallel-group, multiple-dose clinical study which was conducted to evaluate the efficacy and safety of BOTOX in pediatric patients aged 12 to 17 years with overactive bladder. Fifty-five patients who had an inadequate response to or were intolerant of at least one anticholinergic medication were treated with BOTOX. There was not a statistically significant difference in the mean change from baseline in the daily average frequency of daytime urinary incontinence episodes (primary efficacy endpoint) at week 12 post-treatment when a medium and high dose were each compared to a low dose of BOTOX. The adverse reactions in pediatric patients treated with BOTOX were comparable with the known safety profile in adults with overactive bladder.
Prophylaxis of Headaches in Chronic Migraine
Safety and effectiveness in patients below the age of 18 years have not been established.
In a 12-week, multicenter, double-blind, placebo-controlled clinical trial, 123 adolescent patients (ages 12 to below 18 years) with chronic migraine were randomized to receive BOTOX 74 Units, BOTOX 155 Units, or placebo, for one injection cycle. This trial did not establish the efficacy of BOTOX, compared with placebo, for the prophylaxis of headaches in adolescents with chronic migraine.
Spasticity
Safety and effectiveness have been established in pediatric patients 2 to 17 years of age [see Warnings and Precautions (5.1), Adverse Reactions (6.1), and Clinical Studies (14.6)]. The safety and effectiveness of BOTOX have been established by evidence from adequate and well-controlled studies of BOTOX in patients 2 to 17 years of age with upper and lower limb spasticity.
Safety and effectiveness in pediatric patients below the age of 2 years have not been established [see Boxed Warning and Warnings and Precautions (5.1)].
Axillary Hyperhidrosis
Safety and effectiveness in patients below the age of 18 years have not been established.
Cervical Dystonia
Safety and effectiveness in pediatric patients below the age of 16 years have not been established.
Blepharospasm and Strabismus
Safety and effectiveness in pediatric patients below the age of 12 years have not been established.
Juvenile Animal Data
In a study in which juvenile rats received intramuscular injection of BOTOX (0, 8, 16, or 24 Units/kg) every other week from postnatal day 21 for 12 weeks, changes in bone size/geometry associated with decreased bone density and bone mass were observed at all doses, in association with limb disuse, decreased muscle contraction, and decreased body weight gain. Impairment of fertility and male reproductive organ histopathology (degeneration of seminiferous tubules of the testis) were observed at the mid and high doses. Bone and male reproductive organ effects showed evidence of reversibility after dosing cessation. The no-effect dose for adverse developmental effects in juvenile animals (8 Units/kg) is similar to the human dose (400 Units) on a body weight (kg) basis.
8.5 Geriatric Use
Of the 2145 adult patients in placebo-controlled clinical studies of BOTOX for the treatment of spasticity, 33.5% were 65 or older, and 7.7% were 75 years of age or older. No overall differences in safety were observed between elderly patients and adult patients younger than 65 years of age.
In clinical studies of BOTOX across other indications, no overall differences in safety were observed between elderly patients and younger adult patients, with the exception of Overactive Bladder (see below). Other reported clinical experience has not identified differences in responses between the elderly and younger adult patients, but greater sensitivity of some older individuals cannot be ruled out.
Overactive Bladder
Of 1242 overactive bladder patients in placebo-controlled clinical studies of BOTOX, 41.4% were 65 years of age or older, and 14.7% were 75 years of age or older. Adverse reactions of UTI and urinary retention were more common in patients 65 years of age or older in both placebo and BOTOX groups compared to younger patients (see Table 24). Otherwise, there were no overall differences in the safety profile following BOTOX treatment between patients aged 65 years and older compared to adult patients younger than 65 years of age in these studies.
| <65 Years | 65 to 74 Years | ≥75 Years | ||||
| Adverse Reactions | BOTOX
100 Units (N=344) % |
Placebo (N=348) % | BOTOX
100 Units (N=169) % |
Placebo (N=151) % | BOTOX
100 Units (N=94) % |
Placebo (N=86) % |
| Urinary tract infection | 21 | 7 | 30 | 13 | 38 | 19 |
| Urinary retention | 6 | 0.6 | 8 | 0 | 9 | 1 |
Observed effectiveness was comparable between these age groups in placebo-controlled clinical studies.
10 OVERDOSAGE
Excessive doses of BOTOX (onabotulinumtoxinA) for injection may be expected to produce neuromuscular weakness with a variety of symptoms.
Symptoms of overdose are likely not to be present immediately following injection. Should accidental injection or oral ingestion occur or overdose be suspected, the person should be medically supervised for several weeks for signs and symptoms of systemic muscular weakness which could be local, or distant from the site of injection [see Boxed Warning and Warnings and Precautions (5.1, 5.6)]. These patients should be considered for further medical evaluation and appropriate medical therapy immediately instituted, which may include hospitalization.
If the musculature of the oropharynx and esophagus are affected, aspiration may occur which may lead to development of aspiration pneumonia. If the respiratory muscles become paralyzed or sufficiently weakened, intubation and assisted respiration may be necessary until recovery takes place. Supportive care could involve the need for a tracheostomy and/or prolonged mechanical ventilation, in addition to other general supportive care.
In the event of overdose, antitoxin raised against botulinum toxin is available from the Centers for Disease Control and Prevention (CDC) in Atlanta, GA. However, the antitoxin will not reverse any botulinum toxin-induced effects already apparent by the time of antitoxin administration. In the event of suspected or actual cases of botulinum toxin poisoning, please contact your local or state Health Department to process a request for antitoxin through the CDC. If you do not receive a response within 30 minutes, please contact the CDC directly at 1-770-488-7100. More information can be obtained at http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5232a8.htm.
11 DESCRIPTION
OnabotulinumtoxinA is a sterile, vacuum-dried purified botulinum toxin type A, produced from fermentation of Hall strain Clostridium botulinum type A, and intended for intramuscular, intradetrusor and intradermal use. It is purified from the culture solution by dialysis and a series of acid precipitations to a complex consisting of the neurotoxin, and several accessory proteins. The complex is dissolved in sterile sodium chloride solution containing Albumin Human and is sterile filtered (0.2 microns) prior to filling and vacuum-drying.
The primary release procedure for BOTOX uses a cell-based potency assay to determine the potency relative to a reference standard. The assay is specific to AbbVie’s products BOTOX and BOTOX Cosmetic. One Unit of BOTOX corresponds to the calculated median intraperitoneal lethal dose (LD50) in mice. Due to specific details of this assay such as the vehicle, dilution scheme, and laboratory protocols, Units of biological activity of BOTOX cannot be compared to nor converted into Units of any other botulinum toxin or any toxin assessed with any other specific assay method. The specific activity of BOTOX is approximately 20 Units/nanogram of neurotoxin protein complex.
Each vial of BOTOX (onabotulinumtoxinA) for injection contains either 100 Units of Clostridium botulinum type A neurotoxin complex, 0.5 mg of Albumin Human, and 0.9 mg of sodium chloride; or 200 Units of Clostridium botulinum type A neurotoxin complex, 1 mg of Albumin Human, and 1.8 mg of sodium chloride in a sterile, vacuum-dried form without a preservative.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
BOTOX blocks neuromuscular transmission by binding to acceptor sites on motor or autonomic nerve terminals, entering the nerve terminals, and inhibiting the release of acetylcholine. This inhibition occurs as the neurotoxin cleaves SNAP-25, a protein integral to the successful docking and release of acetylcholine from vesicles situated within nerve endings. When injected intramuscularly at therapeutic doses, BOTOX produces partial chemical denervation of the muscle resulting in a localized reduction in muscle activity. In addition, the muscle may atrophy, axonal sprouting may occur, and extrajunctional acetylcholine receptors may develop. There is evidence that reinnervation of the muscle may occur, thus slowly reversing muscle denervation produced by BOTOX.
When injected intradermally, BOTOX produces temporary chemical denervation of the sweat gland resulting in local reduction in sweating.
Following intradetrusor injection, BOTOX affects the efferent pathways of detrusor activity via inhibition of acetylcholine release.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long term studies in animals have not been performed to evaluate the carcinogenic potential of BOTOX.
Mutagenesis
BOTOX was negative in a battery of in vitro (microbial reverse mutation assay, mammalian cell mutation assay, and chromosomal aberration assay) and in vivo (micronucleus assay) genetic toxicology assays.
Impairment of Fertility
In fertility studies of BOTOX (4, 8, or 16 Units/kg) in which either male or female rats were injected intramuscularly prior to mating and on the day of mating (3 doses, 2 weeks apart for males: 2 doses, 2 weeks apart for females) to untreated animals, reduced fertility was observed in males at the intermediate and high doses and in females at the high dose. The no-effect doses for reproductive toxicity (4 Units/kg in males, 8 Units/kg in females) are approximately equal to the human dose of 400 Units, on a body weight basis (Units/kg).
13.2 Animal Toxicology and/or Pharmacology
In a study to evaluate inadvertent peribladder administration, bladder stones were observed in 1 of 4 male monkeys that were injected with a total of 6.8 Units/kg divided into the prostatic urethra and proximal rectum (single administration). No bladder stones were observed in male or female monkeys following injection of up to 36 Units/kg (~12X the highest human bladder dose) directly to the bladder as either single or 4 repeat dose injections or in female rats for single injections up to 100 Units/kg (~33X the highest human bladder dose [200 Units], based on Units/kg).
14 CLINICAL STUDIES
14.1 Overactive Bladder (OAB)
Two double-blind, placebo-controlled, randomized, multi-center, 24-week clinical studies were conducted in patients with OAB with symptoms of urge urinary incontinence, urgency, and frequency (Studies OAB-1 and OAB-2). Patients needed to have at least 3 urinary urgency incontinence episodes and at least 24 micturitions in 3 days to enter the studies. A total of 1105 patients, whose symptoms had not been adequately managed with anticholinergic therapy (inadequate response or intolerable side effects), were randomized to receive either 100 Units of BOTOX (n=557), or placebo (n=548). Patients received 20 injections of study drug (5 Units of BOTOX or placebo) spaced approximately 1 cm apart into the detrusor muscle.
In both studies, significant improvements compared to placebo in the primary efficacy variable of change from baseline in daily frequency of urinary incontinence episodes were observed for BOTOX 100 Units at the primary time point of week 12. Significant improvements compared to placebo were also observed for the secondary efficacy variables of daily frequency of micturition episodes and volume voided per micturition. These primary and secondary variables are shown in Table 25 and Table 26, and Figure 7 and Figure 8.
| BOTOX
100 Units (N=278) |
Placebo (N=272) |
Treatment Difference |
p-value |
|
| Daily Frequency of Urinary Incontinence Episodesa | ||||
| Mean Baseline | 5.5 | 5.1 | ||
| Mean Change* at Week 2 | -2.6 | -1.0 | -1.6 | |
| Mean Change* at Week 6 | -2.8 | -1.0 | -1.8 | |
| Mean Change* at Week 12** | -2.5 | -0.9 | -1.6 (-2.1, -1.2) | <0.001 |
| Daily Frequency of Micturition Episodesb | ||||
| Mean Baseline | 12.0 | 11.2 | ||
| Mean Change† at Week 12** | -1.9 | -0.9 | -1.0 (-1.5, -0.6) | <0.001 |
| Volume Voided per Micturitionb (mL) | ||||
| Mean Baseline | 156 | 161 | ||
| Mean Change† at Week 12** | 38 | 8 | 30 (17, 43) | <0.001 |
* Least squares (LS) mean change, treatment difference and p-value are based on an ANCOVA model with baseline value as covariate and treatment group and investigator as factors. Last observation carried forward (LOCF) values were used to analyze the primary efficacy variable.
† LS mean change, treatment difference and p-value are based on an ANCOVA model with baseline value as covariate and stratification factor, treatment group and investigator as factors.
** Primary timepoint
a Primary variable
b Secondary variable
|
| BOTOX
100 Units (N=275) |
Placebo (N=269) |
Treatment Difference |
p-value |
| Daily Frequency of Urinary Incontinence Episodesa | ||||
| Mean Baseline | 5.5 | 5.7 | ||
| Mean Change* at Week 2 | -2.7 | -1.1 | -1.6 | |
| Mean Change* at Week 6 | -3.1 | -1.3 | -1.8 | |
| Mean Change* at Week 12** | -3.0 | -1.1 | -1.9 (-2.5, -1.4) | <0.001 |
| Daily Frequency of Micturition Episodesb | ||||
| Mean Baseline | 12.0 | 11.8 | ||
| Mean Change† at Week 12** | -2.3 | -0.6 | -1.7 (-2.2, -1.3) | <0.001 |
| Volume Voided per Micturitionb (mL) | ||||
| Mean Baseline | 144 | 153 | ||
| Mean Change† at Week 12** | 40 | 10 | 31 (20, 41) | <0.001 |
* LS mean change, treatment difference and p-value are based on an ANCOVA model with baseline value as covariate and treatment group and investigator as factors. LOCF values were used to analyze the primary efficacy variable.
† LS mean change, treatment difference and p-value are based on an ANCOVA model with baseline value as covariate and stratification factor, treatment group and investigator as factors.
** Primary timepoint
a Primary variable
b Secondary variable
Figure 7: Mean Change from Baseline in Daily Frequency of Urinary Incontinence Episodes Following Intradetrusor Injection in Study OAB-1
Figure 8: Mean Change from Baseline in Daily Frequency of Urinary Incontinence Episodes Following Intradetrusor Injection in Study OAB-2
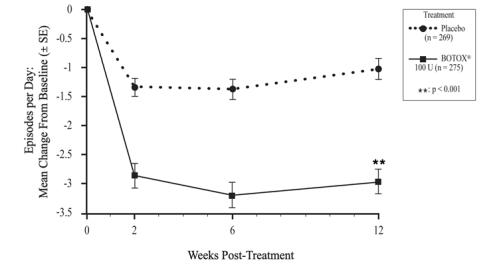
The median duration of response in Study OAB-1 and OAB-2, based on patient qualification for re-treatment, was 19-24 weeks for the BOTOX 100 Unit dose group compared to 13 weeks for placebo. To qualify for re-treatment, at least 12 weeks must have passed since the prior treatment, post-void residual urine volume must have been less than 200 mL and patients must have reported at least 2 urinary incontinence episodes over 3 days.
14.2 Adult Detrusor Overactivity Associated with a Neurologic Condition
Two double-blind, placebo-controlled, randomized, multi-center clinical studies were conducted in patients with urinary incontinence due to detrusor overactivity associated with a neurologic condition who were either spontaneously voiding or using catheterization (Studies NDO-1 and NDO-2). A total of 691 spinal cord injury (T1 or below) or multiple sclerosis patients, who had an inadequate response to or were intolerant of at least one anticholinergic medication, were enrolled. These patients were randomized to receive either 200 Units of BOTOX (n=227), 300 Units of BOTOX (n=223), or placebo (n=241).
In both studies, significant improvements compared to placebo in the primary efficacy variable of change from baseline in weekly frequency of incontinence episodes were observed for BOTOX (200 Units) at the primary efficacy time point at week 6. Increases in maximum cystometric capacity and reductions in maximum detrusor pressure during the first involuntary detrusor contraction were also observed. These primary and secondary endpoints are shown in Table 27 and Table 28, and Figure 9 and Figure 10.
No additional benefit of BOTOX 300 Units over 200 Units was demonstrated.
| BOTOX 200 Units | Placebo | Treatment
Difference* | p-value* | |
| Weekly Frequency of Urinary Incontinence
Episodesa | ||||
| N | 134 | 146 | ||
| Mean Baseline | 32.3 | 28.3 | ||
| Mean Change* at Week 2 | -15.3 | -10.0 | -5.3 | ─ |
| Mean Change* at Week 6** | -19.9 | -10.6 | -9.2 | p<0.001 |
| (-13.1, -5.3) | ||||
| Mean Change* at Week 12 | -19.8 | -8.8 | -11.0 | ─ |
| Maximum Cystometric Capacityb (mL) | ||||
| N | 123 | 129 | ||
| Mean Baseline | 253.8 | 259.1 | ||
| Mean Change* at Week 6** | 135.9 | 12.1 | 123.9 | p<0.001 |
| (89.1, 158.7) | ||||
| Maximum Detrusor Pressure during First
Involuntary Detrusor Contractionb (cmH2O) | ||||
| N | 41 | 103 | ||
| Mean Baseline | 63.1 | 57.4 | ||
| Mean Change* at Week 6** | -28.1 | -3.7 | -24.4 | ─ |
* LS mean change, treatment difference and p-value are based on an analysis using an ANCOVA model with baseline weekly endpoint as covariate and treatment group, etiology at study entry (spinal cord injury or multiple sclerosis), concurrent anticholinergic therapy at screening, and investigator as factors. LOCF values were used to analyze the primary efficacy variable.
** Primary timepoint
a Primary endpoint
b Secondary endpoint
| BOTOX 200 Units | Placebo
| Treatment
Difference* | p-value* | |
| Weekly Frequency of Urinary Incontinence Episodesa
| ||||
| N | 91 | 91 | ||
| Mean Baseline | 32.7 | 36.8 | ||
| Mean Change* at Week 2 | -18.0 | -7.9 | -10.1 | ─ |
| Mean Change* at Week 6** | -19.6 | -10.8 | -8.8 | p=0.003 |
| (-14.5, -3.0) | ||||
| Mean Change* at Week 12 | -19.6 | -10.7 | -8.9 | ─ |
| Maximum Cystometric Capacityb (mL) | ||||
| N | 88 | 85 | ||
| Mean Baseline | 239.6 | 253.8 | ||
| Mean Change* at Week 6** | 150.8 | 2.8 | 148.0 | p<0.001 |
| (101.8, 194.2) | ||||
| Maximum Detrusor Pressure during First Involuntary Detrusor Contractionb (cmH2O) | ||||
| N | 29 | 68 | ||
| Mean Baseline | 65.6 | 43.7 | ||
| Mean Change* at Week 6** | -28.7 | 2.1 | -30.7 | ─ |
* LS mean change, treatment difference and p-value are based on an analysis using an ANCOVA model with baseline weekly endpoint as covariate and treatment group, etiology at study entry (spinal cord injury or multiple sclerosis), concurrent anticholinergic therapy at screening, and investigator as factors. LOCF values were used to analyze the primary efficacy variable.
** Primary timepoint
a Primary endpoint
b Secondary endpoint
Figure 9: Mean Change from Baseline in Weekly Frequency of Urinary Incontinence Episodes During Treatment Cycle 1 in Study NDO-1
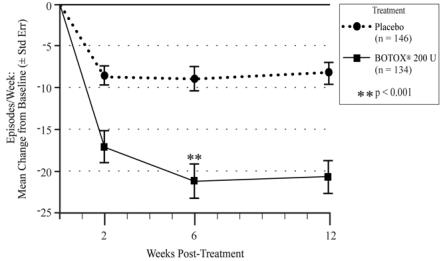
Figure 10: Mean Change from Baseline in Weekly Frequency of Urinary Incontinence Episodes During Treatment Cycle 1 in Study NDO-2
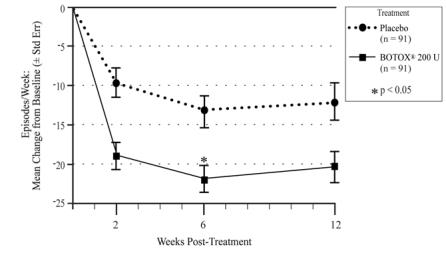
The median duration of response in study NDO-1 and NDO-2, based on patient qualification for re-treatment was 295-337 days (42-48 weeks) for the 200 Units dose group compared to 96-127 days (13-18 weeks) for placebo. Re-treatment was based on loss of effect on incontinence episode frequency (50% of effect in Study NDO-1; 70% of effect in Study NDO-2).
A placebo-controlled, double-blind randomized post-approval 52 week study (Study NDO-3) was conducted in MS patients with urinary incontinence due to neurogenic detrusor overactivity who were not adequately managed with at least one anticholinergic agent and not catheterizing at baseline. These patients were randomized to receive either 100 Units of BOTOX (n=66) or placebo (n=78).
Significant improvements compared to placebo in the primary efficacy variable of change from baseline in daily frequency of incontinence episodes were observed for BOTOX (100 Units) at the primary efficacy time point at week 6. Increases in maximum cystometric capacity and reductions in maximum detrusor pressure during the first involuntary detrusor contraction were also observed. These primary and secondary endpoints are shown in Table 29.
| BOTOX 100 Units | Placebo
| Treatment
Difference* | p-value* | |
| Daily Frequency of Urinary Incontinence Episodesa | ||||
| N | 66 | 78 | ||
| Mean Baseline | 4.2 | 4.3 | ||
| Mean Change* at Week 2 | -2.9 | -1.2 | -1.7 | ─ |
| Mean Change* at Week 6** | -3.4 | -1.1 | -2.3 | p<0.001 |
| (-3.0, -1.7) | ||||
| Mean Change* at Week 12 | -2.7 | -1.0 | -1.8 | ─ |
| Maximum Cystometric Capacityb (mL) | ||||
| N | 62 | 72 | ||
| Mean Baseline | 248.9 | 245.5 | ||
| Mean Change* at Week 6** | 134.4 | 3.5 | 130.9 | p<0.001 |
| (94.8, 167.0) | ||||
| Maximum Detrusor Pressure during First Involuntary Detrusor Contractionb (cmH2O) | ||||
| N | 25 | 51 | ||
| Mean Baseline | 42.4 | 39.0 | ||
| Mean Change* at Week 6** | -19.2 | 2.7 | -21.9 | |
| (-37.5, -6.3) |
* LS mean change, treatment difference and p-value are based on an analysis using an ANCOVA model with baseline daily endpoint as covariate and treatment group and propensity score stratification as factors. LOCF values were used to analyze the primary efficacy variable.
** Primary timepoint
a Primary endpoint
b Secondary endpoint
The median duration of response in study NDO-3, based on patient qualification for re-treatment was 362 days (52 weeks) for the BOTOX 100 Units dose group compared to 88 days (13 weeks) for placebo. To qualify for re-treatment, at least 12 weeks must have passed since the prior treatment, post-void residual urine volume must have been less than 200 mL and patients must have reported at least 2 urinary incontinence episodes over 3 days with no more than 1 incontinence-free day.
14.3 Pediatric Detrusor Overactivity Associated with a Neurologic Condition
Study 191622-120 (NCT01852045) was a multicenter, randomized, double-blind, parallel-group clinical study conducted in patients 5 to 17 years of age with urinary incontinence due to detrusor overactivity associated with a neurologic condition and using clean intermittent catheterization. A total of 113 patients (including 99 with spinal dysraphism such as spina bifida, 13 with spinal cord injury and 1 with transverse myelitis) who had an inadequate response to or were intolerant of at least one anticholinergic medication were enrolled. The median age was 11 years (range: 5 to 17 years), 49% were female; 75% were White, 10% were Black. These patients were randomized to 50 Units, 100 Units or 200 Units, not to exceed 6 Units/kg body weight. Patients receiving less than the randomized dose due to the 6 Units/kg maximum, were assigned to the nearest dose group for analysis. The sample size for BOTOX 50 Units, 100 Units, and 200 Units were 38, 45 and 30, respectively. Prior to treatment administration, patients received anesthesia based on age and local site practice. One hundred and nine patients (97.3%) received general anesthesia or conscious sedation and 3 patients (2.7%) received local anesthesia.
The study results demonstrated within group improvements in the primary efficacy variable of change from baseline in daytime urinary incontinence episodes (normalized to 12 hours) at the primary efficacy time point (Week 6) for all 3 BOTOX treatment groups. Additional benefits were seen with BOTOX 200 Units for measures related to reducing maximum bladder pressure when compared to 50 Units. The decrease in maximum detrusor pressure (MDP) during the storage phase (MDP defined as the highest value in the Pdet channel during the storage phase [e.g., the greater of the following: the maximum Pdet during the highest amplitude IDC, the maximum Pdet during a terminal detrusor contraction, the Pdet at the end of filling, or the highest Pdet at any other time during the storage phase]) for BOTOX 200 Units at Week 6 was greater than the decrease observed for 50 Units. Within group improvements for the primary and secondary endpoints for the 200 Units dose group are shown in Table 30.
The efficacy of BOTOX 6 U/kg for pediatric patients with NDO weighing less than 34 kg was comparable to that of BOTOX 200 U.
| BOTOX 200 U
N=30 |
|
| Daily average frequency of daytime urinary incontinence episodesa | |
| Mean Baseline | 3.7 |
| Mean Change* at Week 2 (95% CI) | -1.1 (-1.7, -0.6) |
| Mean Change* at Week 6** (95% CI) | -1.3 (-1.8, -0.9) |
| Mean Change* at Week 12 (95% CI) | -0.9 (-1.5, -0.4) |
| Urine Volume at First Morning Catheterization (mL)ᵇ | |
| Mean Baseline | 187.7 |
| Mean Change* at Week 2 (95% CI) | 63.2 (27.9, 98.6) |
| Mean Change* at Week 6** (95% CI) | 87.5 (52.1, 122.8) |
| Mean Change* at Week 12 (95% CI) | 45.2 (10.0, 80.5) |
| Maximum Detrusor Pressure (PdetMax) During the Storage Phase (cm H2O)ᵇ | |
| Mean Baseline | 56.7 |
| Mean Change* at Week 6** (95% CI) | -27.3 (-36.4, -18.2) |
| Maximum Cystometric Capacity (mL) (MCC)ᵇ | |
| Mean Baseline | 202.3 |
| Mean Change* at Week 6** (95% CI) | 63.6 (29.0, 98.1) |
CI = Confidence Interval
* LSmean change and 95% CI are based on an ANCOVA model with baseline value as covariate and treatment group, age (< 12 years or >= 12 years), baseline daytime urinary incontinence episodes (<= 6 or > 6) and anticholinergic therapy (yes/no) at baseline as factors.
** Primary timepoint
a Primary endpoint
b Secondary endpoint
The median duration of response in this study, based on patient qualification for re-treatment was 207 days (30 weeks) for BOTOX 200 Units dose group. To qualify for re-treatment, patients must have reported at least 2 urinary incontinence episodes over 2 days and at least 12 weeks have passed from the prior bladder injection.
14.4 Chronic Migraine
BOTOX was evaluated in two randomized, multi-center, 24-week, 2 injection cycle, placebo-controlled double-blind studies. Study 1 and Study 2 included chronic migraine adults who were not using any concurrent headache prophylaxis, and during a 28-day baseline period had >15 headache days lasting 4 hours or more, with >50% being migraine/probable migraine. In both studies, patients were randomized to receive placebo or 155 Units to 195 Units BOTOX injections every 12 weeks for the 2-cycle, double-blind phase. Patients were allowed to use acute headache treatments during the study. BOTOX treatment demonstrated statistically significant and clinically meaningful improvements from baseline compared to placebo for key efficacy variables (see Table 31).
|
Efficacy per 28 days | Study 1 | Study 2 | ||
| BOTOX
(N=341) | Placebo
(N=338) | BOTOX
(N=347) | Placebo
(N=358) |
|
| Change from baseline in frequency of headache days | -7.8* | -6.4 | -9.2* | -6.9 |
| Change from baseline in total cumulative hours of headache on headache days | -107* | -70 | -134* | -95 |
* Significantly different from placebo (p<0.05)
Patients treated with BOTOX had a significantly greater mean decrease from baseline in the frequency of headache days at most timepoints from Week 4 to Week 24 in Study 1 (Figure 11), and all timepoints from Week 4 to Week 24 in Study 2 (Figure 12), compared to placebo-treated patients.
Figure 11: Mean Change from Baseline in Number of Headache Days for Study 1
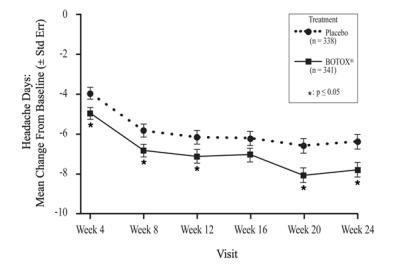
Figure 12: Mean Change from Baseline in Number of Headache Days for Study 2
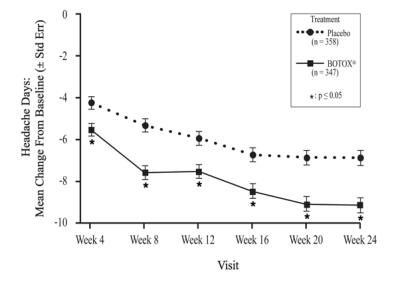
14.5 Adult Spasticity
Adult Upper Limb Spasticity
The efficacy of BOTOX for the treatment of adult upper limb spasticity was evaluated in several randomized, multi-center, double-blind, placebo-controlled studies (Studies 1 through 6).
Study 1 included 126 adult patients (64 BOTOX and 62 placebo) with upper limb spasticity (Ashworth score of at least 3 for wrist flexor tone and at least 2 for finger flexor tone) who were at least 6 months post-stroke. BOTOX (a total dose of 200 Units to 240 Units) and placebo were injected intramuscularly (IM) into the flexor digitorum profundus, flexor digitorum sublimis, flexor carpi radialis, flexor carpi ulnaris, and if necessary into the adductor pollicis and flexor pollicis longus (see Table 32). Use of an EMG/nerve stimulator was recommended to assist in proper muscle localization for injection. Patients were followed for 12 weeks.
|
Muscles Injected | Volume
(mL) | BOTOX
(Units) | Number of Injection Sites |
| Wrist
Flexor Carpi Radialis | 1 | 50 | 1 |
| Flexor Carpi Ulnaris | 1 | 50 | 1 |
| Finger
Flexor Digitorum Profundus | 1 | 50 | 1 |
| Flexor Digitorum Sublimis | 1 | 50 | 1 |
| Thumb
Adductor Pollicisa | 0.4 | 20 | 1 |
| Flexor Pollicis Longusa | 0.4 | 20 | 1 |
a Injected only if spasticity is present in this muscle
The primary efficacy variable was wrist flexors muscle tone at week 6, as measured by the Ashworth score. The Ashworth Scale is a 5-point scale with grades of 0 [no increase in muscle tone] to 4 [limb rigid in flexion or extension]. It is a clinical measure of the force required to move an extremity around a joint, with a reduction in score clinically representing a reduction in the force needed to move a joint (i.e., improvement in spasticity).
Key secondary endpoints included Physician Global Assessment, finger flexors muscle tone, and thumb flexors tone at Week 6. The Physician Global Assessment evaluated the response to treatment in terms of how the patient was doing in his/her life using a scale from -4 = very marked worsening to +4 = very marked improvement. Study 1 results on the primary endpoint and the key secondary endpoints are shown in Table 33.
| BOTOX (N=64) | Placebo (N=62) | |
| Median Change from Baseline in Wrist Flexor Muscle Tone on the Ashworth Scale†a | -2.0* | 0.0 |
| Median Change from Baseline in Finger Flexor Muscle Tone on the Ashworth Scale††b | -1.0* | 0.0 |
| Median Change from Baseline in Thumb Flexor Muscle Tone on the Ashworth Scale††c | -1.0 | -1.0 |
| Median Physician Global Assessment of Response to Treatment†† | 2.0* | 0.0 |
† Primary endpoint at Week 6
†† Secondary endpoints at Week 6
* Significantly different from placebo (p<0.05)
a BOTOX injected into both the flexor carpi radialis and ulnaris muscles
b BOTOX injected into the flexor digitorum profundus and flexor digitorum sublimis muscles
c BOTOX injected into the adductor pollicis and flexor pollicis longus muscles
Study 2 compared 3 doses of BOTOX with placebo and included 91 adult patients [BOTOX 360 Units (N=21), BOTOX 180 Units (N=23), BOTOX 90 Units (N=21), and placebo (N=26)] with upper limb spasticity (expanded Ashworth score of at least 2 for elbow flexor tone and at least 3 for wrist flexor tone) who were at least 6 weeks post-stroke. BOTOX and placebo were injected with EMG guidance into the flexor digitorum profundus, flexor digitorum sublimis, flexor carpi radialis, flexor carpi ulnaris, and biceps brachii (see Table 34).
| Total Dose | |||||
| Muscles Injected | BOTOX low dose
(90 Units) | BOTOX mid dose
(180 Units) | BOTOX high dose
(360 Units) | Volume (mL)
per site | Injection Sites
(n) |
| Wrist
Flexor Carpi Ulnaris | 10 Units | 20 Units | 40 Units | 0.4 | 1 |
| Flexor Carpi Radialis | 15 Units | 30 Units | 60 Units | 0.6 | 1 |
| Finger
Flexor Digitorum Profundus | 7.5 Units | 15 Units | 30 Units | 0.3 | 1 |
| Flexor Digitorum Sublimis | 7.5 Units | 15 Units | 30 Units | 0.3 | 1 |
| Elbow
Biceps Brachii | 50 Units | 100 Units | 200 Units | 0.5 | 4 |
The primary efficacy variable in Study 2 was the wrist flexor tone at Week 6 as measured by the expanded Ashworth Scale. The expanded Ashworth Scale uses the same scoring system as the Ashworth Scale, but allows for half-point increments.
Key secondary endpoints in Study 2 included Physician Global Assessment, finger flexors muscle tone, and elbow flexors muscle tone at Week 6. Study 2 results on the primary endpoint and the key secondary endpoints at Week 6 are shown in Table 35.
| BOTOX
low dose (90 Units) (N=21) | BOTOX
mid dose (180 Units) (N=23) | BOTOX
high dose (360 Units) (N=21) | Placebo
(N=26) |
|
| Median Change from Baseline in Wrist Flexor Muscle Tone on the Ashworth Scale†b | -1.5* | -1.0* | -1.5* | -1.0 |
| Median Change from Baseline in Finger Flexor Muscle Tone on the Ashworth Scale††c | -0.5 | -0.5 | -1.0 | -0.5 |
| Median Change from Baseline in Elbow Flexor Muscle Tone on the Ashworth Scale††d | -0.5 | -1.0* | -0.5a | -0.5 |
| Median Physician Global Assessment of Response to Treatment | 1.0* | 1.0* | 1.0* | 0.0 |
† Primary endpoint at Week 6
†† Secondary endpoints at Week 6
* Significantly different from placebo (p<0.05)
a p=0.053
b Total dose of BOTOX injected into both the flexor carpi radialis and ulnaris muscles
c Total dose of BOTOX injected into the flexor digitorum profundus and flexor digitorum sublimis muscles
d Dose of BOTOX injected into biceps brachii muscle
Study 3 compared 3 doses of BOTOX with placebo and enrolled 88 adult patients [BOTOX 360 Units (N=23), BOTOX 180 Units (N=23), BOTOX 90 Units (N=23), and placebo (N=19)] with upper limb spasticity (expanded Ashworth score of at least 2 for elbow flexor tone and at least 3 for wrist flexor tone and/or finger flexor tone) who were at least 6 weeks post-stroke. BOTOX and placebo were injected with EMG guidance into the flexor digitorum profundus, flexor digitorum sublimis, flexor carpi radialis, flexor carpi ulnaris, and biceps brachii (see Table 34).
The primary efficacy variable in Study 3 was wrist and elbow flexor tone as measured by the expanded Ashworth score. A key secondary endpoint was assessment of finger flexors muscle tone. Study 3 results on the primary endpoint at Week 4 are shown in Table 36.
| BOTOX low dose (90 Units)
(N=23) | BOTOX mid dose
(180 Units) (N=21) | BOTOX high dose (360 Units)
(N=22) | Placebo
(N=19) |
|
| Median Change from Baseline in Wrist Flexor Muscle Tone on the Ashworth Scale†b | -1.0 | -1.0 | -1.5* | -0.5 |
| Median Change from Baseline in Finger Flexor Muscle Tone on the Ashworth Scale††c | -1.0 | -1.0 | -1.0* | -0.5 |
| Median Change from Baseline in Elbow Flexor Muscle Tone on the Ashworth Scale†d | -0.5 | -0.5 | -1.0* | -0.5 |
† Primary endpoint at Week 4
†† Secondary endpoints at Week 4
* Significantly different from placebo (p<0.05)
b Total dose of BOTOX injected into both the flexor carpi radialis and ulnaris muscles
c Total dose of BOTOX injected into the flexor digitorum profundus and flexor digitorum sublimis muscles
d Dose of BOTOX injected into biceps brachii muscle
Study 4 (NCT01153815) included 170 adult patients (87 BOTOX and 83 placebo) with upper limb spasticity who were at least 6 months post-stroke. In Study 4, patients received 20 Units of BOTOX into the adductor pollicis and flexor pollicis longus (total BOTOX dose = 40 Units in thumb muscles) or placebo (see Table 37). Study 5 (NCT00460564) included 109 patients with upper limb spasticity who were at least 6 months post-stroke. In Study 5, adult patients received 15 Units (low dose) or 20 Units (high dose) of BOTOX into the adductor pollicis and flexor pollicis longus under EMG guidance (total BOTOX low dose = 30 Units, total BOTOX high dose = 40 Units), or placebo (see Table 37). The duration of follow-up in Study 4 and Study 5 was 12 weeks.
| Study 4 | Study 5 | ||||||
| Muscles
Injected | BOTOX
(Units) | Volume
(mL) | BOTOX
low dose (Units) | BOTOX
high dose (Units) | Volume
low dose (mL) | Volume
high dose (mL) | Number of Injection Sites for Studies 4 and 5 |
| Thumb
Adductor Pollicis | 20 | 0.4 | 15 | 20 | 0.3 | 0.4 | 1 |
| Flexor Pollicis Longus | 20 | 0.4 | 15 | 20 | 0.3 | 0.4 | 1 |
The results of Study 4 for the change from Baseline to Week 6 in thumb flexor tone measured by modified Ashworth Scale (MAS) and overall treatment response by Physician Global Assessment at week 6 are presented in Table 38. The MAS uses a similar scoring system as the Ashworth Scale.
| BOTOX
(N=66) | Placebo
(N=57) |
|
| Median Change from Baseline in Thumb Flexor Muscle Tone on the modified Ashworth Scale††a | -1.0* | 0.0 |
| Median Physician Global Assessment of Response to Treatment†† | 2.0* | 0.0 |
†† Secondary endpoints at Week 6
* Significantly different from placebo (p<0.001)
a BOTOX injected into the adductor pollicis and flexor pollicis longus muscles
In Study 5, the results of the change from Baseline to Week 6 in thumb flexor tone measured by modified Ashworth Scale and Clinical Global Impression (CGI) of functional assessment scale assessed by the physician using an 11-point Numeric Rating Scale [-5 worst possible function to +5 best possible function] are presented in Table 39.
| BOTOX
low dose (30 Units) (N=14) | Placebo
low dose (N=9) | BOTOX
high dose (40 Units) (N=43) | Placebo
high dose (N=23) |
|
| Median Change from Baseline in Thumb Flexor Muscle Tone on the modified Ashworth Scale†††a | -1.0 | -1.0 | -0.5* | 0.0 |
| Median Change from Baseline in Clinical Global Impression Score by Physician †† | 1.0 | 0.0 | 2.0* | 0.0 |
†† Secondary endpoint at Week 6
††† Other endpoint at Week 6
* Significantly different from placebo (p<0.010)
a BOTOX injected into the adductor pollicis and flexor pollicis longus muscles
Study 6 (NCT03261167) enrolled 124 post-stroke adult patients with upper limb spasticity. In Study 6, 61 patients received 160 Units BOTOX divided among 3 elbow flexors (biceps brachii, brachioradialis, and brachialis) and 63 patients received placebo (see Table 40). EMG, nerve stimulation, or ultrasound techniques were recommended to assist in proper muscle localization for injections. The duration of follow-up was 12 weeks.
| Muscles Injected | BOTOX
160 U (Units) | Volume
(mL) | Number of Injection Sites |
| Elbow
Biceps Brachii | 70 | 1.4 | 2 |
| Brachioradialis | 45 | 0.9 | 1 |
| Brachialis | 45 | 0.9 | 1 |
The change from baseline in elbow flexor tone measured by modified Ashworth Scale at Week 6 is presented in Table 41.
| BOTOX
160 U (N=61) | Placebo
(N=63) |
|
| Mean Change from Baseline in Elbow Flexor Muscle Tone on the modified Ashworth Scale at Week 6 | -1.09* | -0.71 |
*nominal p value <0.05
Adult Lower Limb Spasticity
The efficacy and safety of BOTOX for the treatment of adult lower limb spasticity was evaluated in Study 7, a randomized, multi-center, double-blind, placebo-controlled study. Study 7 included 468 post-stroke adult patients (233 BOTOX and 235 placebo) with ankle spasticity (modified Ashworth Scale ankle score of at least 3) who were at least 3 months post-stroke. A total dose of 300 Units of BOTOX or placebo were injected intramuscularly and divided between the gastrocnemius, soleus, and tibialis posterior, with optional injection into the flexor hallucis longus, flexor digitorum longus, flexor digitorum brevis, extensor hallucis, and rectus femoris (see Table 42) with up to an additional 100 Units (400 Units total dose). The use of electromyographic guidance or nerve stimulation was required to assist in proper muscle localization for injections. Patients were followed for 12 weeks.
|
Muscles Injected | BOTOX (Units) | Number of
Injection Sites |
| Mandatory Ankle Muscles
Gastrocnemius (medial head) | 75 | 3 |
| Gastrocnemius (lateral head) | 75 | 3 |
| Soleus | 75 | 3 |
| Tibialis Posterior | 75 | 3 |
| Optional Muscles
Flexor Hallucis Longus | 50 | 2 |
| Flexor Digitorum Longus | 50 | 2 |
| Flexor Digitorum Brevis | 25 | 1 |
| Extensor Hallucis | 25 | 1 |
| Rectus Femoris | 100 | 4 |
The co-primary endpoints were the average of the change from baseline in modified Ashworth Scale (MAS) ankle score at Week 4 and Week 6, and the average of the Physician Global Assessment of Response (CGI) at Week 4 and Week 6. The CGI evaluated the response to treatment in terms of how the patient was doing in his/her life using a 9-point scale from -4=very marked worsening to +4=very marked improvement.
Statistically significant between-group differences for BOTOX over placebo were demonstrated for the co-primary efficacy measures of MAS and CGI (see Table 43).
| BOTOX
300 to 400 Units (N=233) | Placebo
(N=235) |
|
| Mean Change from Baseline in Ankle Plantar Flexors on the modified Ashworth Scale | ||
| Week 4 and 6 Average | -0.8* | -0.6 |
| Mean Clinical Global Impression Score by Investigator | ||
| Week 4 and 6 Average | 0.9* | 0.7 |
* Significantly different from placebo (p<0.05)
Compared to placebo, significant improvements in MAS change from baseline for ankle plantar flexors (see Figure 13) and CGI (see Figure 14) were observed at Week 2, Week 4, and Week 6 for patients treated with BOTOX.
Figure 13: Modified Ashworth Scale Ankle Score for Study 7 – Mean Change from Baseline by Visit
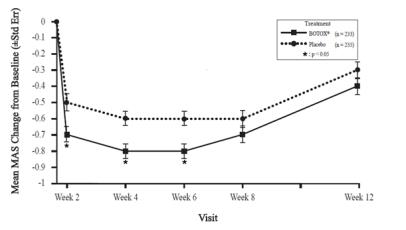
Figure 14: Clinical Global Impression by Physician for Study 7 – Mean Scores by Visit
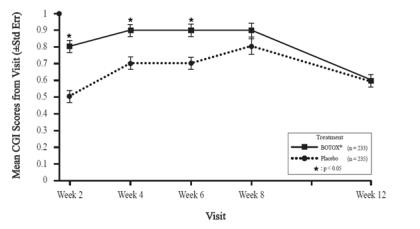
14.6 Pediatric Spasticity
Pediatric Upper Limb Spasticity
The efficacy and safety of BOTOX for the treatment of upper limb spasticity in pediatric patients 2 to 17 years of age was evaluated in Study 1 (NCT01603602), a randomized, multi-center, double-blind, placebo-controlled study. Study 1 included 234 pediatric patients (78 BOTOX 3 Units/kg, 77 BOTOX 6 Units/kg, and 79 placebo) with upper limb spasticity (modified Ashworth Scale elbow or wrist score of at least 2) because of cerebral palsy or stroke. A total dose of 3 Units/kg BOTOX (maximum 100 Units), 6 Units/kg BOTOX (maximum 200 Units), or placebo was injected intramuscularly and divided between the elbow or wrist and finger muscles (see Table 44). Electromyographic guidance, nerve stimulation, or ultrasound techniques were used to assist in muscle localization for injections. Patients were followed for 12 weeks after injection.
| Muscles Injected | BOTOX 3 Units/kg* (maximum Units per muscle) | BOTOX 6 Units/kg** (maximum Units per muscle) | Number of Injection Sites |
| Elbow Flexor Muscles | |||
| Biceps | 1.5 Units/kg (50 Units) | 3 Units/kg (100 Units) | 4 |
| Brachialis | 1 Unit/kg (30 Units) | 2 Units/kg (60 Units) | 2 |
| Brachioradialis | 0.5 Units/kg (20 Units) | 1 Unit/kg (40 Units) | 2 |
| Wrist and Finger Muscles | |||
| Flexor carpi radialis | 1 Unit/kg (25 Units) | 2 Units/kg (50 Units) | 2 |
| Flexor carpi ulnaris | 1 Unit/kg (25 Units) | 2 Units/kg (50 Units) | 2 |
| Flexor digitorum profundus | 0.5 Units/kg (25 Units) | 1 Unit/kg (50 Units) | 2 |
| Flexor digitorum sublimis | 0.5 Units/kg (25 Units) | 1 Unit/kg (50 Units) | 2 |
* Did not exceed a total dose of 100 Units
** Did not exceed a total dose of 200 Units
The co-primary endpoints were the average of the change from baseline in modified Ashworth Scale (MAS) principal muscle group score (elbow or wrist) at Week 4 and Week 6, and the average of the Clinical Global Impression of Overall Change by Physician (CGI) at Week 4 and Week 6. The CGI evaluated the response to treatment in terms of how the patient was doing in his/her life using a 9-point scale (-4=very marked worsening to +4=very marked improvement).
Compared to placebo, significant improvements in MAS change from baseline were observed at all timepoints for BOTOX-treated patients (see Table 45, Figure 15 and Figure 16). Although CGI scores numerically favored BOTOX over placebo, the difference was not statistically significant.
| BOTOX 3 Units/kg
(N=78) | BOTOX 6 Units/kg
(N=77) | Placebo
(N=79) |
|
| Mean Change from Baseline in Principal Muscle Group (Elbow or Wrist) on the modified Ashworth Scale | |||
| Week 4 and 6 Average | -1.92* | -1.87* | -1.21 |
| Mean Clinical Global Impression Score | |||
| Week 4 and 6 Average | 1.88 | 1.87 | 1.66 |
*Nominal p value <0.05
Figure 15: Modified Ashworth Scale Score for Study 1 (Pediatric Upper Limb Spasticity, Modified Intent-To-Treat Population) – Mean Change from Baseline by Visit
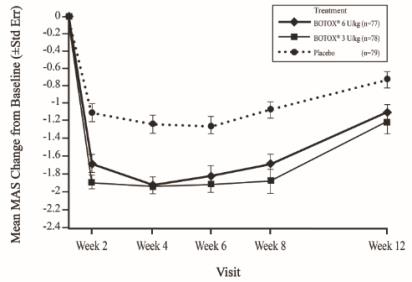
Figure 16: Clinical Global Impression of Overall Change for Study 1 (Pediatric Upper Limb Spasticity, Modified Intent-To-Treat Population) – Mean Scores by Visit
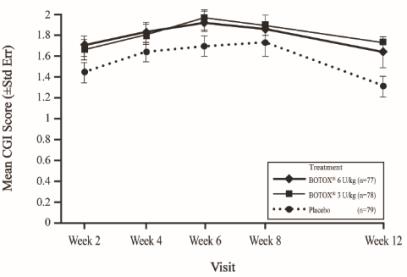
Pediatric Lower Limb Spasticity
The efficacy and safety of BOTOX for the treatment of lower limb spasticity in pediatric patients 2 to 17 years of age was evaluated in Study 2 (NCT01603628), a randomized, multi-center, double-blind, placebo-controlled study. Study 2 included 381 pediatric patients (125 BOTOX 4 Units/kg, 127 BOTOX 8 Units/kg, and 129 placebo) with lower limb spasticity (modified Ashworth Scale ankle score of at least 2) because of cerebral palsy. A total dose of 4 Units/kg BOTOX (maximum 150 Units), 8 Units/kg BOTOX (maximum 300 Units), or placebo was injected intramuscularly and divided between the gastrocnemius, soleus, and tibialis posterior (see Table 46). Electromyographic guidance, nerve stimulation, or ultrasound techniques were used to assist in muscle localization for injections. Patients were followed for 12 weeks after injection.
| Muscles Injected | BOTOX 4 Units/kg* (maximum Units per muscle) | BOTOX 8 Units/kg** (maximum Units per muscle) | Number of
Injection Sites |
| Mandatory Ankle Muscles
Gastrocnemius medial head | 1 Unit/kg (37.5 Units) | 2 Units/kg (75 Units) | 2 |
| Gastrocnemius lateral head | 1 Unit/kg (37.5 Units) | 2 Units/kg (75 Units) | 2 |
| Soleus | 1 Unit/kg (37.5 Units) | 2 Units/kg (75 Units) | 2 |
| Tibialis Posterior | 1 Unit/kg (37.5 Units) | 2 Units/kg (75 Units) | 2 |
* did not exceed a total dose of 150 Units
** did not exceed a total dose of 300 Units
The co-primary endpoints were the average of the change from baseline in modified Ashworth Scale (MAS) ankle score at Week 4 and Week 6, and the average of the Clinical Global Impression of Overall Change by Physician (CGI) at Week 4 and Week 6. The CGI evaluated the response to treatment in terms of how the patient was doing in his/her life using a 9-point scale (-4=very marked worsening to +4=very marked improvement).
Statistically significant differences between BOTOX and placebo were demonstrated for the MAS and CGI for the 8 Units/kg dose only (see Table 47).
| BOTOX 4 Units/kg
(N = 125) | BOTOX 8 Units/kg
(N=127) | Placebo
(N=129) |
|
| Mean Change from Baseline in Plantar Flexors on the modified Ashworth Scale | |||
| Week 4 and 6 Average | -1.01** | -1.06* | -0.80 |
| Mean Clinical Global Impression Score | |||
| Week 4 and 6 Average | 1.49 | 1.65* | 1.36 |
* Significantly different from placebo (p<0.05)
** Nominal p value <0.05
Compared to placebo, improvements in mean change from baseline for the MAS, and mean CGI score for lower limb spasticity were observed at timepoints up to Week 12 for BOTOX-treated patients (see Figure 17 and Figure 18).
Figure 17: Modified Ashworth Scale Ankle Score for Study 2 (Pediatric Lower Limb Spasticity, Modified Intent-To-Treat Population) – Mean Change from Baseline by Visit
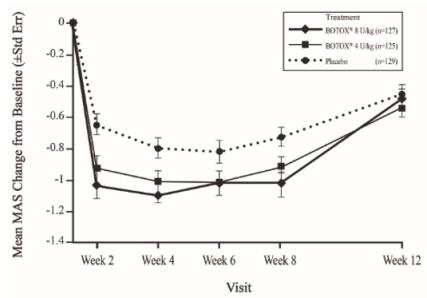
Figure 18: Clinical Global Impression of Overall Change for Study 2 (Pediatric Lower Limb Spasticity, Modified Intent-To-Treat Population) – Mean Scores by Visit
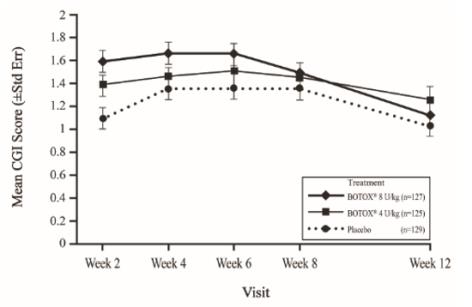
14.7 Cervical Dystonia
A randomized, multi-center, double-blind, placebo-controlled study of the treatment of cervical dystonia was conducted. This study enrolled adult patients with cervical dystonia and a history of having received BOTOX in an open label manner with perceived good response and tolerable side effects. Patients were excluded if they had previously received surgical or other denervation treatment for their symptoms or had a known history of neuromuscular disorder. Subjects participated in an open label enrichment period where they received their previously employed dose of BOTOX. Only patients who were again perceived as showing a response were advanced to the randomized evaluation period. The muscles in which the blinded study agent injections were to be administered were determined on an individual patient basis.
There were 214 subjects evaluated for the open label period, of which 170 progressed into the randomized, blinded treatment period (88 in the BOTOX group, 82 in the placebo group). Patient evaluations continued for at least 10 weeks post-injection. The primary outcome for the study was a dual endpoint, requiring evidence of both a change in the Cervical Dystonia Severity Scale (CDSS) and an increase in the percentage of patients showing any improvement on the Physician Global Assessment Scale at 6 weeks after the injection session. The CDSS quantifies the severity of abnormal head positioning and was newly devised for this study. CDSS allots 1 point for each 5 degrees (or part thereof) of head deviation in each of the three planes of head movement (range of scores up to theoretical maximum of 54). The Physician Global Assessment Scale is a 9 category scale scoring the physician’s evaluation of the patients’ status compared to baseline, ranging from –4 to +4 (very marked worsening to complete improvement), with 0 indicating no change from baseline and +1 slight improvement. Pain is also an important symptom of cervical dystonia and was evaluated by separate assessments of pain frequency and severity on scales of 0 (no pain) to 4 (constant in frequency or extremely severe in intensity). Study results on the primary endpoints and the pain-related secondary endpoints are shown in Table 48.
| Placebo (N=82) | BOTOX (N=88) | 95% CI on Difference | |
| Baseline CDSS | 9.3 | 9.2 | |
| Change in CDSS
at Week 6 | -0.3 | -1.3 | (-2.3, 0.3)[a,b] |
| % Patients with Any Improvement on Physician Global Assessment | 31% | 51% | (5%, 34%)[a] |
| Pain Intensity Baseline | 1.8 | 1.8 | |
| Change in Pain Intensity at Week 6 | -0.1 | -0.4 | (-0.7, -0.2)[c] |
| Pain Frequency Baseline | 1.9 | 1.8 | |
| Change in Pain Frequency at Week 6 | -0.0 | -0.3 | (-0.5, -0.0)[c] |
[a] Confidence intervals are constructed from the analysis of covariance table with treatment and investigational site as main effects, and baseline CDSS as a covariate.
[b] These values represent the prospectively planned method for missing data imputation and statistical test. Sensitivity analyses indicated that the 95% confidence interval excluded the value of no difference between groups and the p-value was less than 0.05. These analyses included several alternative missing data imputation methods and non-parametric statistical tests.
[c] Confidence intervals are based on the t-distribution.
Exploratory analyses of this study suggested that the majority of patients who had shown a beneficial response by week 6 had returned to their baseline status by 3 months after treatment. Exploratory analyses of subsets by patient sex and age suggest that both sexes receive benefit, although female patients may receive somewhat greater amounts than male patients. There is a consistent treatment-associated effect between subsets greater than and less than age 65. There were too few non-Caucasian patients enrolled to draw any conclusions regarding relative efficacy in racial subsets.
In this study the median total BOTOX dose in patients randomized to receive BOTOX (N=88) was 236 Units, with 25th to 75th percentile ranges of 198 Units to 300 Units. Of these 88 patients, most received injections to 3 or 4 muscles; 38 received injections to 3 muscles, 28 to 4 muscles, 5 to 5 muscles, and 5 to 2 muscles. The dose was divided amongst the affected muscles in quantities shown in Table 49. The total dose and muscles selected were tailored to meet individual patient needs.
| Muscle | Number of Patients Treated in this Muscle (N=88) | Mean % Dose per Muscle | Mid-Range of % Dose per Muscle* |
| Splenius capitis/cervicis | 83 | 38 | 25-50 |
| Sternocleidomastoid | 77 | 25 | 17-31 |
| Levator scapulae | 52 | 20 | 16-25 |
| Trapezius | 49 | 29 | 18-33 |
| Semispinalis | 16 | 21 | 13-25 |
| Scalene | 15 | 15 | 6-21 |
| Longissimus | 8 | 29 | 17-41 |
* The mid-range of dose is calculated as the 25th to 75th percentiles.
There were several randomized studies conducted prior to the double-blind, placebo-controlled study, which were supportive but not adequately designed to assess or quantitatively estimate the efficacy of BOTOX.
14.8 Primary Axillary Hyperhidrosis
The efficacy and safety of BOTOX for the treatment of primary axillary hyperhidrosis were evaluated in two randomized, multi-center, double-blind, placebo-controlled studies. Study 1 included adult patients with persistent primary axillary hyperhidrosis who scored 3 or 4 on a Hyperhidrosis Disease Severity Scale (HDSS) and who produced at least 50 mg of sweat in each axilla at rest over 5 minutes. HDSS is a 4-point scale with 1 = “underarm sweating is never noticeable and never interferes with my daily activities”; to 4 = “underarm sweating is intolerable and always interferes with my daily activities”. A total of 322 patients were randomized in a 1:1:1 ratio to treatment in both axillae with either 50 Units of BOTOX, 75 Units of BOTOX, or placebo. Patients were evaluated at 4-week intervals. Patients who responded to the first injection were re-injected when they reported a re-increase in HDSS score to 3 or 4 and produced at least 50 mg sweat in each axilla by gravimetric measurement, but no sooner than 8 weeks after the initial injection.
Study responders were defined as patients who showed at least a 2-grade improvement from baseline value on the HDSS 4 weeks after both of the first two treatment sessions or had a sustained response after their first treatment session and did not receive re-treatment during the study. Spontaneous resting axillary sweat production was assessed by weighing a filter paper held in the axilla over a period of 5 minutes (gravimetric measurement). Sweat production responders were those patients who demonstrated a reduction in axillary sweating from baseline of at least 50% at week 4.
In the three study groups the percentage of patients with baseline HDSS score of 3 ranged from 50% to 54% and from 46% to 50% for a score of 4. The median amount of sweat production (averaged for each axilla) was 102 mg, 123 mg, and 114 mg for the placebo, 50 Units and 75 Units groups respectively.
The percentage of responders based on at least a 2-grade decrease from baseline in HDSS or based on a >50% decrease from baseline in axillary sweat production was greater in both BOTOX groups than in the placebo group (p<0.001), but was not significantly different between the two BOTOX doses (see Table 50).
Duration of response was calculated as the number of days between injection and the date of the first visit at which patients returned to 3 or 4 on the HDSS scale. The median duration of response following the first treatment in BOTOX treated patients with either dose was 201 days. Among those who received a second BOTOX injection, the median duration of response was similar to that observed after the first treatment.
In study 2, 320 adults with bilateral axillary primary hyperhidrosis were randomized to receive either 50 Units of BOTOX (n=242) or placebo (n=78). Treatment responders were defined as subjects showing at least a 50% reduction from baseline in axillary sweating measured by gravimetric measurement at 4 weeks. At week 4 post-injection, the percentages of responders were 91% (219/242) in the BOTOX group and 36% (28/78) in the placebo group, p<0.001. The difference in percentage of responders between BOTOX and placebo was 55% (95% CI=43.3, 65.9).
|
Treatment Response | BOTOX
50 Units (N=104) | BOTOX
75 Units (N=110) | Placebo
(N=108) | BOTOX
50-placebo (95% CI) | BOTOX
75-placebo (95% CI) |
| HDSS Score change ≥2 (n)a | 55% (57) | 49% (54) | 6% (6) | 49.3% (38.8, 59.7) | 43% (33.2, 53.8) |
| >50% decrease in axillary sweat production % (n) | 81% (84) | 86% (94) | 41% (44) | 40% (28.1, 52.0) | 45% (33.3, 56.1) |
a Patients who showed at least a 2-grade improvement from baseline value on the HDSS 4 weeks after both of the first two treatment sessions or had a sustained response after their first treatment session and did not receive re-treatment during the study.
14.9 Blepharospasm
Botulinum toxin has been investigated for use in patients with blepharospasm in several studies. In an open label, historically controlled study, 27 patients with essential blepharospasm were injected with 2 Units of BOTOX at each of six sites on each side. Twenty-five of the 27 patients treated with botulinum toxin reported improvement within 48 hours. One patient was controlled with a higher dosage at 13 weeks post initial injection and one patient reported mild improvement but remained functionally impaired.
In another study, 12 patients with blepharospasm were evaluated in a double-blind, placebo-controlled study. Patients receiving botulinum toxin (n=8) improved compared with the placebo group (n=4). The effects of the treatment lasted a mean of 12 weeks.
One thousand six hundred eighty-four patients with blepharospasm who were evaluated in an open label trial showed clinical improvement as evaluated by measured eyelid force and clinically observed intensity of lid spasm, lasting an average of 12 weeks prior to the need for re-treatment.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
BOTOX (onabotulinumtoxinA) for injection is a sterile, vacuum-dried powder supplied in a single-dose vial in the following sizes:
100 Units NDC 0023-1145-01
200 Units NDC 0023-3921-02
BOTOX cartons have features to alert users if contents may have been compromised. Each BOTOX vial label and carton also contains the U.S. License number: 1889 [see Dosage and Administration (2.1)].
Do not use the product and contact AbbVie for additional information at 1-800-678-1605 if the labeling is not described as above.
16.2 Storage and Handling
Unopened vials of BOTOX should be stored in a refrigerator between 2° to 8°C (36º to 46ºF) for up to 36 months. Do not use after the expiration date on the vial. Reconstituted BOTOX may be stored in a refrigerator (2° to 8°C) for up to 24 hours until time of use [see Dosage and Administration (2.2)].
17 PATIENT COUNSELING INFORMATION
Advise the patient or caretaker to read the FDA-approved patient labeling (Medication Guide).
Swallowing, Speaking or Breathing Difficulties, or Other Unusual Symptoms
Advise patients or their caretaker(s) to inform their doctor or pharmacist if they develop any unusual symptoms (including difficulty with swallowing, speaking, or breathing), or if any existing symptom worsens [see Boxed Warning and Warnings and Precautions (5.1, 5.6)].
Ability to Operate Machinery or Vehicles
Advise patients or their caretaker(s) that if loss of strength, muscle weakness, blurred vision, dizziness, or drooping eyelids occur, they should avoid driving a car or engaging in other potentially hazardous activities.
Voiding Symptoms after Bladder Injections
After bladder injections for urinary incontinence, advise patients to contact their physician if they experience difficulties in voiding or burning sensation upon voiding.
Manufactured by: AbbVie Inc.
1 N Waukegan Rd. North Chicago, IL. 60064
U.S. License Number 1889
© 2023 AbbVie.
All rights reserved.
BOTOX and its design are trademarks of Allergan Inc., an AbbVie company
Patented. See: https://www.abbvie.com/patents.html
v11.0USPI1145
| MEDICATION GUIDE
BOTOX® BOTOX® Cosmetic (Boe-tox) (onabotulinumtoxinA) for injection, for intramuscular, intradetrusor, or intradermal use |
| What is the most important information I should know about BOTOX and BOTOX Cosmetic?
BOTOX and BOTOX Cosmetic may cause serious side effects that can be life threatening, including:
These problems could make it unsafe for you to drive a car or do other dangerous activities. See "What should I avoid while receiving BOTOX or BOTOX Cosmetic?" There has not been a confirmed serious case of spread of toxin effect away from the injection site when BOTOX has been used at the recommended dose to treat chronic migraine, severe underarm sweating, blepharospasm, or strabismus, or when BOTOX Cosmetic has been used at the recommended dose to treat frown lines, crow’s feet lines, and/or forehead lines. |
| What are BOTOX and BOTOX Cosmetic?
BOTOX is a prescription medicine that is injected into muscles and used:
|
BOTOX Cosmetic is a prescription medicine for adults that is injected into muscles and used for a short period of time (temporary) to improve the look of:
|
It is not known whether BOTOX is safe and effective in people younger than:
It is not known whether BOTOX and BOTOX Cosmetic are safe and effective to prevent headaches in people with migraine who have 14 or fewer headache days each month (episodic migraine). It is not known whether BOTOX and BOTOX Cosmetic are safe and effective for severe sweating anywhere other than your armpits. It is not known if BOTOX Cosmetic is safe and effective for use more than 1 time every 3 months. |
| Who should not receive BOTOX or BOTOX Cosmetic?
Do not receive BOTOX or BOTOX Cosmetic if you:
|
| What should I tell my doctor before receiving BOTOX or BOTOX Cosmetic?
Tell your doctor about all your medical conditions, including if you:
Especially tell your doctor if you:
Know the medicines you take. Keep a list of your medicines with you to show your doctor and pharmacist each time you get a new medicine. |
How will I receive BOTOX or BOTOX Cosmetic?
|
| What should I avoid while receiving BOTOX or BOTOX Cosmetic?
BOTOX and BOTOX Cosmetic may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours to weeks of taking BOTOX or BOTOX Cosmetic. If this happens, do not drive a car, operate machinery, or do other dangerous activities. See "What is the most important information I should know about BOTOX and BOTOX Cosmetic?" |
| What are the possible side effects of BOTOX and BOTOX Cosmetic?
BOTOX and BOTOX Cosmetic can cause serious side effects. See "What is the most important information I should know about BOTOX and BOTOX Cosmetic?" Other side effects of BOTOX and BOTOX Cosmetic include:
These are not all the possible side effects of BOTOX and BOTOX Cosmetic. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
| General information about the safe and effective use of BOTOX and BOTOX Cosmetic:
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. This Medication Guide summarizes the most important information about BOTOX and BOTOX Cosmetic. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about BOTOX and BOTOX Cosmetic that is written for health professionals. |
| What are the ingredients in BOTOX and BOTOX Cosmetic?
Active ingredient: onabotulinumtoxinA Inactive ingredients: human albumin and sodium chloride |
| Manufactured by: AbbVie Inc. 1 N Waukegan Rd. North Chicago, IL. 60064 U.S. License Number 1889 © 2023 AbbVie. All rights reserved. BOTOX and its design are trademarks of Allergan Inc., an AbbVie company. Patented. See: https://www.abbvie.com/patents.html |
 |
| v7.0MG1145 |
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 11/2023

