DESCRIPTION
Verapamil hydrochloride extended-release tablets, USP is a calcium ion influx inhibitor (slow-channel blocker or calcium ion antagonist). Verapamil hydrochloride extended-release tablets, USP is available for oral administration as light blue, oval shaped, scored, film coated tablets containing 240 mg of verapamil hydrochloride USP; as light blue, capsule shaped, bevelled edged, scored, film coated tablets containing 180 mg of verapamil hydrochloride USP. The tablets are designed for sustained release of the drug in the gastrointestinal tract; sustained-release characteristics are not altered when the tablet is divided in half.
The structural formula of verapamil HCl USP is:
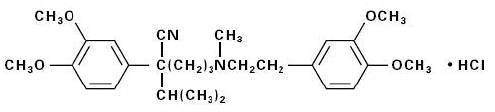 |
|||
| C27H38N2O4 ∙ HCl | M.W.=491.08 | ||
| Benzeneacetonitrile, α-[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino] propyl]-3,4-dimethoxy-α-(1-methylethyl) hydrochloride | |||
Verapamil HCl, USP is an almost white, crystalline powder, practically free of odor, with a bitter taste. It is soluble in water, chloroform, and methanol. Verapamil HCl, USP is not chemically related to other cardioactive drugs.
Inactive ingredients include colloidal silicon dioxide, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinylpyrrolidone, sodium alginate, and film coating contains FD&C Blue No. 1 Brilliant Blue FCF aluminum lake, hypromellose, iron oxide yellow, titanium dioxide and triacetin.
Verapamil hydrochloride extended-release tablets USP, 180 mg and 240 mg meet USP Dissolution Test 3.
CLINICAL PHARMACOLOGY
Verapamil HCl is a calcium ion influx inhibitor (slow-channel blocker or calcium ion antagonist) that exerts its pharmacologic effects by modulating the influx of ionic calcium across the cell membrane of the arterial smooth muscle as well as in conductile and contractile myocardial cells.
Mechanism of action
Essential hypertension
Verapamil exerts antihypertensive effects by decreasing systemic vascular resistance, usually without orthostatic decreases in blood pressure or reflex tachycardia; bradycardia (rate less than 50 beats/min) is uncommon (1.4%). During isometric or dynamic exercise, verapamil hydrochloride does not alter systolic cardiac function in patients with normal ventricular function.
Verapamil hydrochloride does not alter total serum calcium levels. However, one report suggested that calcium levels above the normal range may alter the therapeutic effect of verapamil hydrochloride.
Other pharmacologic actions of verapamil hydrochloride include the following
Verapamil hydrochloride dilates the main coronary arteries and coronary arterioles, both in normal and ischemic regions, and is a potent inhibitor of coronary artery spasm, whether spontaneous or ergonovine-induced. This property increases myocardial oxygen delivery in patients with coronary artery spasm and is responsible for the effectiveness of verapamil hydrochloride in vasospastic (Prinzmetal's or variant) as well as unstable angina at rest. Whether this effect plays any role in classical effort angina is not clear, but studies of exercise tolerance have not shown an increase in the maximum exercise rate–pressure product, a widely accepted measure of oxygen utilization. This suggests that, in general, relief of spasm or dilation of coronary arteries is not an important factor in classical angina.
Verapamil hydrochloride regularly reduces the total systemic resistance (afterload) against which the heart works both at rest and at a given level of exercise by dilating peripheral arterioles.
Electrical activity through the AV node depends, to a significant degree, upon calcium influx through the slow channel. By decreasing the influx of calcium, verapamil hydrochloride prolongs the effective refractory period within the AV node and slows AV conduction in a rate-related manner.
Normal sinus rhythm is usually not affected, but in patients with sick sinus syndrome, verapamil hydrochloride may interfere with sinus-node impulse generation and may induce sinus arrest or sinoatrial block. Atrioventricular block can occur in patients without preexisting conduction defects (see WARNINGS).
Verapamil hydrochloride does not alter the normal atrial action potential or intraventricular conduction time, but depresses amplitude, velocity of depolarization, and conduction in depressed atrial fibers. Verapamil hydrochloride may shorten the antegrade effective refractory period of the accessory bypass tract. Acceleration of ventricular rate and/or ventricular fibrillation has been reported in patients with atrial flutter or atrial fibrillation and a coexisting accessory AV pathway following administration of verapamil (see WARNINGS).
Verapamil hydrochloride has a local anesthetic action that is 1.6 times that of procaine on an equimolar basis. It is not known whether this action is important at the doses used in man.
Pharmacokinetics and metabolism
With the immediate-release formulation, more than 90% of the orally administered dose of verapamil hydrochloride is absorbed. Because of rapid biotransformation of verapamil during its first pass through the portal circulation, bioavailability ranges from 20% to 35%. Peak plasma concentrations are reached between 1 and 2 hours after oral administration. Chronic oral administration of 120 mg of verapamil HCl every 6 hours resulted in plasma levels of verapamil ranging from 125 to 400 ng/mL, with higher values reported occasionally. A nonlinear correlation between the verapamil dose administered and verapamil plasma level does exist. In early dose titration with verapamil, a relationship exists between verapamil plasma concentration and prolongation of the PR interval. However, during chronic administration this relationship may disappear. The mean elimination half-life in single-dose studies ranged from 2.8 to 7.4 hours. In these same studies, after repetitive dosing, the half-life increased to a range from 4.5 to 12.0 hours (after less than 10 consecutive doses given 6 hours apart). Half-life of verapamil may increase during titration. No relationship has been established between the plasma concentration of verapamil and a reduction in blood pressure.
Aging may affect the pharmacokinetics of verapamil. Elimination half-life may be prolonged in the elderly. In multiple-dose studies under fasting conditions, the bioavailability, measured by AUC, of verapamil hydrochloride extended-release tablets was similar to verapamil hydrochloride tablets (immediate release); rates of absorption were of course different.
In a randomized, single-dose, crossover study using healthy volunteers, administration of 240 mg verapamil hydrochloride extended-release tablets with food produced peak plasma verapamil concentrations of 79 ng/mL; time to peak plasma verapamil concentration of 7.71 hours; and AUC (0–24 hr) of 841 ng∙hr/mL. When verapamil hydrochloride extended-release tablets was administered to fasting subjects, peak plasma verapamil concentration was 164 ng/mL; time to peak plasma verapamil concentration was 5.21 hours; and AUC (0–24 hr) was 1,478 ng∙hr/mL. Similar results were demonstrated for plasma norverapamil. Food thus produces decreased bioavailability (AUC) but a narrower peak-to-trough ratio. Good correlation of dose and response is not available, but controlled studies of verapamil hydrochloride extended-release tablets have shown effectiveness of doses similar to the effective doses of verapamil hydrochloride tablets (immediate release).
In healthy men, orally administered verapamil hydrochloride undergoes extensive metabolism in the liver. Twelve metabolites have been identified in plasma; all except norverapamil are present in trace amounts only. Norverapamil can reach steady-state plasma concentrations approximately equal to those of verapamil itself. The cardiovascular activity of norverapamil appears to be approximately 20% that of verapamil. Approximately 70% of an administered dose is excreted as metabolites in the urine and 16% or more in the feces within 5 days. About 3% to 4% is excreted in the urine as unchanged drug. Approximately 90% is bound to plasma proteins. In patients with hepatic insufficiency, metabolism of immediate-release verapamil is delayed and elimination half-life prolonged up to 14 to 16 hours (see PRECAUTIONS); the volume of distribution is increased and plasma clearance reduced to about 30% of normal. Verapamil clearance values suggest that patients with liver dysfunction may attain therapeutic verapamil plasma concentrations with one third of the oral daily dose required for patients with normal liver function.
After four weeks of oral dosing (120 mg q.i.d.), verapamil and norverapamil levels were noted in the cerebrospinal fluid with estimated partition coefficient of 0.06 for verapamil and 0.04 for norverapamil.
In ten healthy males, administration of oral verapamil (80 mg every 8 hours for 6 days) and a single oral dose of ethanol (0.8 g/kg) resulted in a 17% increase in mean peak ethanol concentrations (106.45 ± 21.40 to 124.23 ± 24.74 mg∙hr/dL) compared to placebo. The area under the blood ethanol concentration versus time curve (AUC over 12 hours) increased by 30% (365.67 ± 93.52 to 475.07 ± 97.24 mg∙hr/dL). Verapamil AUCs were positively correlated (r=0.71) to increased ethanol blood AUC values (see PRECAUTIONS, Drug interactions).
Hemodynamics and myocardial metabolism
Verapamil hydrochloride reduces afterload and myocardial contractility. Improved left ventricular diastolic function in patients with Idiopathic Hypertrophic Subaortic Stenosis (IHSS) and those with coronary heart disease has also been observed with verapamil hydrochloride. In most patients, including those with organic cardiac disease, the negative inotropic action of verapamil hydrochloride is countered by reduction of afterload, and cardiac index is usually not reduced. However, in patients with severe left ventricular dysfunction (eg, pulmonary wedge pressure above 20 mm Hg or ejection fraction less than 30%), or in patients taking beta-adrenergic blocking agents or other cardiodepressant drugs, deterioration of ventricular function may occur (see PRECAUTIONS, Drug interactions).
INDICATIONS AND USAGE
Verapamil hydrochloride extended-release tablets is indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including this drug.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
CONTRAINDICATIONS
Verapamil HCl tablets are contraindicated in:
- Severe left ventricular dysfunction (see WARNINGS)
- Hypotension (systolic pressure less than 90 mm Hg) or cardiogenic shock
- Sick sinus syndrome (except in patients with a functioning artificial ventricular pacemaker)
- Second- or third-degree AV block (except in patients with a functioning artificial ventricular pacemaker)
- Patients with atrial flutter or atrial fibrillation and an accessory bypass tract (eg, Wolff-Parkinson-White, Lown-Ganong-Levine syndromes) (see WARNINGS)
- Patients with known hypersensitivity to verapamil hydrochloride
WARNINGS
Heart failure
Verapamil has a negative inotropic effect, which in most patients is compensated by its afterload reduction (decreased systemic vascular resistance) properties without a net impairment of ventricular performance. In clinical experience with 4,954 patients, 87 (1.8%) developed congestive heart failure or pulmonary edema. Verapamil should be avoided in patients with severe left ventricular dysfunction (eg, ejection fraction less than 30%) or moderate to severe symptoms of cardiac failure and in patients with any degree of ventricular dysfunction if they are receiving a beta-adrenergic blocker (see PRECAUTIONS, Drug interactions). Patients with milder ventricular dysfunction should, if possible, be controlled with optimum doses of digitalis and/or diuretics before verapamil treatment. (Note interactions with digoxin under PRECAUTIONS)
Hypotension
Occasionally, the pharmacologic action of verapamil may produce a decrease in blood pressure below normal levels, which may result in dizziness or symptomatic hypotension. The incidence of hypotension observed in 4,954 patients enrolled in clinical trials was 2.5%. In hypertensive patients, decreases in blood pressure below normal are unusual. Tilt-table testing (60 degrees) was not able to induce orthostatic hypotension.
Elevated liver enzymes
Elevations of transaminases with and without concomitant elevations in alkaline phosphatase and bilirubin have been reported. Such elevations have sometimes been transient and may disappear even in the face of continued verapamil treatment. Several cases of hepatocellular injury related to verapamil have been proven by rechallenge; half of these had clinical symptoms (malaise, fever, and/or right upper quadrant pain) in addition to elevation of SGOT, SGPT, and alkaline phosphatase. Periodic monitoring of liver function in patients receiving verapamil is therefore prudent.
Accessory bypass tract (Wolff-Parkinson-White or Lown-Ganong-Levine)
Some patients with paroxysmal and/or chronic atrial fibrillation or atrial flutter and a coexisting accessory AV pathway have developed increased antegrade conduction across the accessory pathway bypassing the AV node, producing a very rapid ventricular response or ventricular fibrillation after receiving intravenous verapamil (or digitalis). Although a risk of this occurring with oral verapamil has not been established, such patients receiving oral verapamil may be at risk and its use in these patients is contraindicated (see CONTRAINDICATIONS). Treatment is usually DC-cardioversion. Cardioversion has been used safely and effectively after oral verapamil hydrochloride tablets.
Atrioventricular block
The effect of verapamil on AV conduction and the SA node may cause asymptomatic first-degree AV block and transient bradycardia, sometimes accompanied by nodal escape rhythms. PR-interval prolongation is correlated with verapamil plasma concentrations, especially during the early titration phase of therapy. Higher degrees of AV block, however, were infrequently (0.8%) observed. Marked first-degree block or progressive development to second- or third-degree AV block, requires a reduction in dosage or, in rare instances, discontinuation of verapamil HCl and institution of appropriate therapy, depending upon the clinical situation.
Patients with hypertrophic cardiomyopathy (IHSS)
In 120 patients with hypertrophic cardiomyopathy (most of them refractory or intolerant to propranolol) who received therapy with verapamil at doses up to 720 mg/day, a variety of serious adverse effects were seen. Three patients died in pulmonary edema; all had severe left ventricular outflow obstruction and a past history of left ventricular dysfunction. Eight other patients had pulmonary edema and/or severe hypotension; abnormally high (greater than 20 mm Hg) pulmonary wedge pressure and a marked left ventricular outflow obstruction were present in most of these patients. Concomitant administration of quinidine (see PRECAUTIONS, Drug interactions) preceded the severe hypotension in 3 of the 8 patients (2 of whom developed pulmonary edema). Sinus bradycardia occurred in 11% of the patients, second-degree AV block in 4%, and sinus arrest in 2%. It must be appreciated that this group of patients had a serious disease with a high mortality rate. Most adverse effects responded well to dose reduction, and only rarely did verapamil use have to be discontinued.
PRECAUTIONS
General
Use in patients with impaired hepatic function
Since verapamil is highly metabolized by the liver, it should be administered cautiously to patients with impaired hepatic function. Severe liver dysfunction prolongs the elimination half-life of immediate-release verapamil to about 14 to 16 hours; hence, approximately 30% of the dose given to patients with normal liver function should be administered to these patients. Careful monitoring for abnormal prolongation of the PR interval or other signs of excessive pharmacologic effects (see OVERDOSAGE) should be carried out.
Use in patients with attenuated (decreased) neuromuscular transmission
It has been reported that verapamil decreases neuromuscular transmission in patients with Duchenne's muscular dystrophy, and that verapamil prolongs recovery from the neuromuscular blocking agent vecuronium. It may be necessary to decrease the dosage of verapamil when it is administered to patients with attenuated neuromuscular transmission.
Use in patients with impaired renal function
About 70% of an administered dose of verapamil is excreted as metabolites in the urine. Verapamil is not removed by hemodialysis. Until further data are available, verapamil should be administered cautiously to patients with impaired renal function. These patients should be carefully monitored for abnormal prolongation of the PR interval or other signs of overdosage (see OVERDOSAGE).
Drug interactions
Cytochrome inducers/inhibitors
In vitro metabolic studies indicate that verapamil is metabolized by cytochrome P450 CYP3A4, CYP1A2, CYP2C8, CYP2C9, and CYP2C18. Clinically significant interactions have been reported with inhibitors of CYP3A4 (e.g., erythromycin, ritonavir) causing elevation of plasma levels of verapamil while inducers of CYP3A4 (e.g., rifampin) have caused a lowering of plasma levels of verapamil.
HMG-CoA reductase inhibitors
The use of HMG-CoA reductase inhibitors that are CYP3A4 substrates in combination with verapamil has been associated with reports of myopathy/rhabdomyolysis.
Co-administration of multiple doses of 10 mg of verapamil with 80 mg simvastatin resulted in exposure to simvastatin 2.5-fold that following simvastatin alone. Limit the dose of simvastatin in patients on verapamil to 10 mg daily. Limit the daily dose of lovastatin to 40 mg. Lower starting and maintenance doses of other CYP3A4 substrates (e.g., atorvastatin) may be required as verapamil may increase the plasma concentration of these drugs.
Ivabradine
Concurrent use of verapamil increases exposure to ivabradine and may exacerbate bradycardia and conduction disturbances. Avoid co-administration of verapamil and ivabradine.
Beta-blockers
Concomitant therapy with beta-adrenergic blockers and verapamil may result in additive negative effects on heart rate, atrioventricular conduction and/or cardiac contractility. The combination of sustained-release verapamil and beta-adrenergic blocking agents has not been studied. However, there have been reports of excessive bradycardia and AV block, including complete heart block, when the combination has been used for the treatment of hypertension. For hypertensive patients, the risks of combined therapy may outweigh the potential benefits. The combination should be used only with caution and close monitoring.
Asymptomatic bradycardia (36 beats/min) with a wandering atrial pacemaker has been observed in a patient receiving concomitant timolol (a beta-adrenergic blocker) eyedrops and oral verapamil.
A decrease in metoprolol and propranolol clearance has been observed when either drug is administered concomitantly with verapamil. A variable effect has been seen when verapamil and atenolol were given together.
Digitalis
Clinical use of verapamil in digitalized patients has shown the combination to be well tolerated if digoxin doses are properly adjusted. However, chronic verapamil treatment can increase serum digoxin levels by 50% to 75% during the first week of therapy, and this can result in digitalis toxicity. In patients with hepatic cirrhosis the influence of verapamil on digoxin kinetics is magnified. Verapamil may reduce total body clearance and extrarenal clearance of digitoxin by 27% and 29%, respectively. Maintenance and digitalization doses should be reduced when verapamil is administered, and the patient should be carefully monitored to avoid over- or under- digitalization. Whenever over-digitalization is suspected, the daily dose of digitalis should be reduced or temporarily discontinued. On discontinuation of verapamil hydrochloride tablets use, the patient should be reassessed to avoid under-digitalization.
Antihypertensive agents
Verapamil administered concomitantly with oral antihypertensive agents (eg, vasodilators, angiotensin-converting enzyme inhibitors, diuretics, beta-blockers) will usually have an additive effect on lowering blood pressure. Patients receiving these combinations should be appropriately monitored. Concomitant use of agents that attenuate alpha-adrenergic function with verapamil may result in a reduction in blood pressure that is excessive in some patients. Such an effect was observed in one study following the concomitant administration of verapamil and prazosin.
Antiarrhythmic agents
Disopyramide
Until data on possible interactions between verapamil and disopyramide phosphate are obtained, disopyramide should not be administered within 48 hours before or 24 hours after verapamil administration.
Flecainide
A study in healthy volunteers showed that the concomitant administration of flecainide and verapamil may have additive effects on myocardial contractility, AV conduction, and repolarization. Concomitant therapy with flecainide and verapamil may result in additive negative inotropic effect and prolongation of atrioventricular conduction.
Quinidine
In a small number of patients with hypertrophic cardiomyopathy (IHSS), concomitant use of verapamil and quinidine resulted in significant hypotension. Until further data are obtained, combined therapy of verapamil and quinidine in patients with hypertrophic cardiomyopathy should probably be avoided.
The electrophysiologic effects of quinidine and verapamil on AV conduction were studied in 8 patients. Verapamil significantly counteracted the effects of quinidine on AV conduction. There has been a report of increased quinidine levels during verapamil therapy.
Other agents
Alcohol
Verapamil has been found to inhibit ethanol elimination significantly, resulting in elevated blood ethanol concentrations that may prolong the intoxicating effects of alcohol (see CLINICAL PHARMACOLOGY, Pharmacokinetics and metabolism).
Nitrates
Verapamil has been given concomitantly with short- and long-acting nitrates without any undesirable drug interactions. The pharmacologic profile of both drugs and the clinical experience suggest beneficial interactions.
Cimetidine
The interaction between cimetidine and chronically administered verapamil has not been studied. Variable results on clearance have been obtained in acute studies of healthy volunteers; clearance of verapamil was either reduced or unchanged.
Lithium
Increased sensitivity to the effects of lithium (neurotoxicity) has been reported during concomitant verapamil-lithium therapy; lithium levels have been observed sometimes to increase, sometimes to decrease, and sometimes to be unchanged. Patients receiving both drugs must be monitored carefully.
Carbamazepine
Verapamil therapy may increase carbamazepine concentrations during combined therapy. This may produce carbamazepine side effects such as diplopia, headache, ataxia, or dizziness.
Inhalation anesthetics
Animal experiments have shown that inhalation anesthetics depress cardiovascular activity by decreasing the inward movement of calcium ions. When used concomitantly, inhalation anesthetics and calcium antagonists, such as verapamil, should each be titrated carefully to avoid excessive cardiovascular depression.
Neuromuscular blocking agents
Clinical data and animal studies suggest that verapamil may potentiate the activity of neuromuscular blocking agents (curare-like and depolarizing). It may be necessary to decrease the dose of verapamil and/or the dose of the neuromuscular blocking agent when the drugs are used concomitantly.
Telithromycin
Hypotension and bradyarrhythmias have been observed in patients receiving concurrent telithromycin, an antibiotic in the ketolide class.
Clonidine
Sinus bradycardia resulting in hospitalization and pacemaker insertion has been reported in association with the use of clonidine concurrently with verapamil. Monitor heart rate in patients receiving concomitant verapamil and clonidine.
Mammalian target of rapamycin (mTOR) inhibitors
In a study of 25 healthy volunteers with co-administration of verapamil with sirolimus, whole blood sirolimus Cmax and AUC were increased 130% and 120%, respectively. Plasma S-(-) verapamil Cmax and AUC were both increased 50%. Co-administration of verapamil with everolimus in 16 healthy volunteers increased the Cmax and AUC of everolimus by 130% and 250%, respectively. With concomitant use of mTOR inhibitors (e.g., sirolimus, temsirolimus, and everolimus) and verapamil, consider appropriate dose reductions of both medications.
Carcinogenesis, mutagenesis, impairment of fertility
An 18-month toxicity study in rats, at a low multiple (6-fold) of the maximum recommended human dose, and not the maximum tolerated dose, did not suggest a tumorigenic potential. There was no evidence of a carcinogenic potential of verapamil administered in the diet of rats for two years at doses of 10, 35, and 120 mg/kg/day or approximately 1, 3.5, and 12 times, respectively, the maximum recommended human daily dose (480 mg/day or 9.6 mg/kg/day).
Verapamil was not mutagenic in the Ames test in 5 test strains at 3 mg per plate with or without metabolic activation.
Studies in female rats at daily dietary doses up to 5.5 times (55 mg/kg/day) the maximum recommended human dose did not show impaired fertility. Effects on male fertility have not been determined.
Pregnancy
Reproduction studies have been performed in rabbits and rats at oral doses up to 1.5 (15 mg/kg/day) and 6 (60 mg/kg/day) times the human oral daily dose, respectively, and have revealed no evidence of teratogenicity. In the rat, however, this multiple of the human dose was embryocidal and retarded fetal growth and development, probably because of adverse maternal effects reflected in reduced weight gains of the dams. This oral dose has also been shown to cause hypotension in rats. There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed. Verapamil crosses the placental barrier and can be detected in umbilical vein blood at delivery.
Labor and delivery
It is not known whether the use of verapamil during labor or delivery has immediate or delayed adverse effects on the fetus, or whether it prolongs the duration of labor or increases the need for forceps delivery or other obstetric intervention. Such adverse experiences have not been reported in the literature, despite a long history of use of verapamil in Europe in the treatment of cardiac side effects of beta-adrenergic agonist agents used to treat premature labor.
Nursing mothers
Verapamil is excreted in human milk. Because of the potential for adverse reactions in nursing infants from verapamil, nursing should be discontinued while verapamil is administered.
Pediatric use
Safety and efficacy of verapamil hydrochloride extended-release tablets in pediatric patients below the age of 18 years have not been established.
Animal pharmacology and/or animal toxicology
In chronic animal toxicology studies, verapamil caused lenticular and/or suture line changes at 30 mg/kg/day or greater, and frank cataracts at 62.5 mg/kg/day or greater in the beagle dog but not in the rat. Development of cataracts due to verapamil has not been reported in man.
ADVERSE REACTIONS
Serious adverse reactions are uncommon when verapamil therapy is initiated with upward dose titration within the recommended single and total daily dose. See WARNINGS for discussion of heart failure, hypotension, elevated liver enzymes, AV block, and rapid ventricular response. Reversible (upon discontinuation of verapamil) non-obstructive, paralytic ileus has been infrequently reported in association with the use of verapamil. The following reactions to orally administered verapamil occurred at rates greater than 1.0% or occurred at lower rates but appeared clearly drug-related in clinical trials in 4,954 patients:
| Constipation | 7.3% | Dyspnea | 1.4% |
| Dizziness | 3.3% | Bradycardia | |
| Nausea | 2.7% | (HR <50/min) | 1.4% |
| Hypotension | 2.5% | AV block | |
| Headache | 2.2% | (total 1°, 2°, 3°) | 1.2% |
| Edema | 1.9% | (2° and 3°) | 0.8% |
| CHF, Pulmonary edema | 1.8% | Rash | 1.2% |
| Fatigue | 1.7% | Flushing | 0.6% |
Elevated liver enzymes (see WARNINGS)
In clinical trials related to the control of ventricular response in digitalized patients who had atrial fibrillation or flutter, ventricular rates below 50/min at rest occurred in 15% of patients and asymptomatic hypotension occurred in 5% of patients.
The following reactions, reported in 1% or less of patients, occurred under conditions (open trials, marketing experience) where a causal relationship is uncertain; they are listed to alert the physician to a possible relationship:
Cardiovascular: angina pectoris, atrioventricular dissociation, chest pain, claudication, myocardial infarction, palpitations, purpura (vasculitis), syncope.
Digestive system: diarrhea, dry mouth, gastrointestinal distress, gingival hyperplasia.
Hemic and lymphatic: ecchymosis or bruising.
Nervous system: cerebrovascular accident, confusion, equilibrium disorders, insomnia, muscle cramps, paresthesia, psychotic symptoms, shakiness, somnolence.
Skin: arthralgia and rash, exanthema, hair loss, hyperkeratosis, macules, sweating, urticaria, Stevens-Johnson syndrome, erythema multiforme.
Special senses: blurred vision, tinnitus.
Urogenital: gynecomastia, galactorrhea/hyperprolactinemia, increased urination, spotty menstruation, impotence.
Treatment of acute cardiovascular adverse reactions
The frequency of cardiovascular adverse reactions that require therapy is rare; hence, experience with their treatment is limited. Whenever severe hypotension or complete AV block occurs following oral administration of verapamil, the appropriate emergency measures should be applied immediately; eg, intravenously administered norepinephrine bitartrate, atropine sulfate, isoproterenol HCl (all in the usual doses), or calcium gluconate (10% solution). In patients with hypertrophic cardiomyopathy (IHSS), alpha-adrenergic agents (phenylephrine HCl, metaraminol bitartrate, or methoxamine HCl) should be used to maintain blood pressure, and isoproterenol and norepinephrine should be avoided. If further support is necessary, dopamine HCl or dobutamine HCl may be administered. Actual treatment and dosage should depend on the severity of the clinical situation and the judgment and experience of the treating physician.
OVERDOSAGE
Overdose with verapamil may lead to pronounced hypotension, bradycardia, and conduction system abnormalities (eg, junctional rhythm with AV dissociation and high degree AV block, including asystole). Other symptoms secondary to hypoperfusion (eg, metabolic acidosis, hyperglycemia, hyperkalemia, renal dysfunction, and convulsions) may be evident.
Treat all verapamil overdoses as serious and maintain observation for at least 48 hours (especially verapamil hydrochloride extended-release tablets), preferably under continuous hospital care. Delayed pharmacodynamic consequences may occur with the sustained-release formulation. Verapamil is known to decrease gastrointestinal transit time.
In overdose, verapamil hydrochloride extended-release tablets have occasionally been reported to form concretions within the stomach or intestines. These concretions have not been visible on plain radiographs of the abdomen, and no medical means of gastrointestinal emptying is of proven efficacy in removing them. Endoscopy might reasonably be considered in cases of massive overdose when symptoms are unusually prolonged.
Treatment of overdosage should be supportive. Beta-adrenergic stimulation or parenteral administration of calcium solutions may increase calcium ion flux across the slow channel and have been used effectively in treatment of deliberate overdosage with verapamil. Continued treatment with large doses of calcium may produce a response. In a few reported cases, overdose with calcium channel blockers that was initially refractory to atropine became more responsive to this treatment when the patients received large doses (close to 1 g/hr for more than 24 hr) of calcium chloride. Verapamil cannot be removed by hemodialysis. Clinically significant hypotensive reactions or high degree AV block should be treated with vasopressor agents or cardiac pacing, respectively. Asystole should be handled by the usual measures including cardiopulmonary resuscitation.
DOSAGE AND ADMINISTRATION
Essential hypertension
The dose of verapamil hydrochloride extended-release tablets should be individualized by titration and the drug should be administered with food. Initiate therapy with 180 mg of sustained-release verapamil HCl, verapamil hydrochloride extended-release tablets, given in the morning. Lower initial doses of 120 mg a day may be warranted in patients who may have an increased response to verapamil (eg, the elderly or small people). Upward titration should be based on therapeutic efficacy and safety evaluated weekly and approximately 24 hours after the previous dose. The antihypertensive effects of verapamil hydrochloride extended-release tablets are evident within the first week of therapy.
If adequate response is not obtained with 180 mg of verapamil hydrochloride extended-release tablets, the dose may be titrated upward in the following manner:
- a)
- 240 mg each morning,
- b)
- 180 mg each morning plus
180 mg each evening; or
240 mg each morning plus
120 mg each evening, - c)
- 240 mg every 12 hours.
When switching from immediate-release verapamil hydrochloride tablets to verapamil hydrochloride extended-release tablets, the total daily dose in milligrams may remain the same.
HOW SUPPLIED
Verapamil hydrochloride extended-release tablets, USP 240 mg are supplied as light blue, capsule shaped, bevelled edged, scored, film coated tablets debossed with C 77 on one side and plain on other side.
NDC: 71335-1394-1: 30 Tablets in a BOTTLE
NDC: 71335-1394-2: 90 Tablets in a BOTTLE
NDC: 71335-1394-3: 60 Tablets in a BOTTLE
NDC: 71335-1394-4: 10 Tablets in a BOTTLE
NDC: 71335-1394-5: 100 Tablets in a BOTTLE
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F) [See USP Controlled Room Temperature] and protect from light and moisture. Dispense in tight, light-resistant containers.
Repackaged/Relabeled by:
Bryant Ranch Prepack, Inc.
Burbank, CA 91504
NDC: 71335-1394-1: 30 Tablets in a BOTTLE
NDC: 71335-1394-2: 90 Tablets in a BOTTLE
NDC: 71335-1394-3: 60 Tablets in a BOTTLE
NDC: 71335-1394-4: 10 Tablets in a BOTTLE
