FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Ondansetron orally disintegrating tablets are indicated for the prevention of nausea and vomiting associated with:
- highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m2
- initial and repeat courses of moderately emetogenic cancer chemotherapy
- radiotherapy in patients receiving either total body irradiation, single high-dose fraction to the abdomen, or daily fractions to the abdomen
Ondansetron orally disintegrating tablets also indicated for the prevention of postoperative nausea and/or vomiting.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The recommended dosage regimens for adult and pediatric patients are described in Table 1 and Table 2, respectively.
Corresponding doses of ondansetron tablets, ondansetron orally disintegrating tablets and ondansetron oral solution may be used interchangeably.
| Indication
| Dosage Regimen
|
| Highly Emetogenic Cancer Chemotherapy
| A single 24 mg dose administered 30 minutes before the start of single-day highly emetogenic chemotherapy, including cisplatin greater than or equal to 50 mg/m2. |
| Moderately Emetogenic Cancer Chemotherapy
| 8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose. Then administer 8 mg twice a day (every 12 hours) for 1 to 2 days after completion of chemotherapy. |
| Radiotherapy
| For total body irradiation: 8 mg administered 1 to 2 hours before each fraction of radiotherapy each day. For single high-dose fraction radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for 1 to 2 days after completion of radiotherapy. For daily fractionated radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for each day radiotherapy is given. |
| Postoperative
| 16 mg administered 1 hour before induction of anesthesia. |
| Indication
| Dosage Regimen
|
| Moderately Emetogenic Cancer Chemotherapy
| 12 to 17 years of age: 8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose. Then administer 8 mg twice a day (every 12 hours) for 1 to 2 days after completion of chemotherapy. 4 to 11 years of age: 4 mg administered 30 minutes before the start of chemotherapy, with a subsequent 4 mg dose 4 and 8 hours after the first dose. Then administer 4 mg three times a day for 1 to 2 days after completion of chemotherapy. |
2.2 Dosage in Hepatic Impairment
In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), do not exceed a total daily dose of 8 mg [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.3 Administration Instructions for Ondansetron Orally Disintegrating Tablets
Do not attempt to push ondansetron orally disintegrating tablets through the foil backing. With dry hands, remove the tablet from the bottle or PEEL BACK the foil backing of 1 blister and GENTLY remove the tablet. IMMEDIATELY place the ondansetron orally disintegrating tablet on top of the tongue where it will dissolve in seconds, then swallow with saliva. Administration with liquid is not necessary.
3 DOSAGE FORMS AND STRENGTHS
Ondansetron Orally Disintegrating Tablets USP, 4 mg are white to off-white, round tablets debossed with ‘5’ on one side and ‘E’ on the other side with an embossed circular edge.
Ondansetron Orally Disintegrating Tablets USP, 8 mg are white to off-white, round tablets debossed with ‘7’ on one side and ‘E’ on the other side with an embossed circular edge.
4 CONTRAINDICATIONS
Ondansetron orally disintegrating tablets are contraindicated in patients:
- known to have hypersensitivity (e.g., anaphylaxis) to ondansetron or any of the components of the formulation [see Adverse Reactions (6.2)]
- receiving concomitant apomorphine due to the risk of profound hypotension and loss of consciousness
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis and bronchospasm, have been reported in patients who have exhibited hypersensitivity to other selective 5-HT3 receptor antagonists. If hypersensitivity reactions occur, discontinue use of ondansetron; treat promptly per standard of care and monitor until signs and symptoms resolve [see Contraindications (4)].
5.2 QT Prolongation
Electrocardiogram (ECG) changes, including QT interval prolongation have been seen in patients receiving ondansetron. In addition, postmarketing cases of Torsade de Pointes have been reported in patients using ondansetron. Avoid ondansetron in patients with congenital long QT syndrome. ECG monitoring is recommended in patients with electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia), congestive heart failure, bradyarrhythmias, or patients taking other medicinal products that lead to QT prolongation [see Clinical Pharmacology (12.2)].
5.3 Serotonin Syndrome
The development of serotonin syndrome has been reported with 5-HT3 receptor antagonists alone. Most reports have been associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors, mirtazapine, fentanyl, lithium, tramadol, and intravenous methylene blue). Some of the reported cases were fatal. Serotonin syndrome occurring with overdose of ondansetron alone has also been reported. The majority of reports of serotonin syndrome related to 5-HT3 receptor antagonist use occurred in a post-anesthesia care unit or an infusion center.
Symptoms associated with serotonin syndrome may include the following combination of signs and symptoms: mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, with or without gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome, especially with concomitant use of ondansetron and other serotonergic drugs. If symptoms of serotonin syndrome occur, discontinue ondansetron and initiate supportive treatment. Patients should be informed of the increased risk of serotonin syndrome, especially if ondansetron is used concomitantly with other serotonergic drugs [see Drug Interactions (7.1), Overdosage (10)].
5.4 Myocardial Ischemia
Myocardial ischemia has been reported in patients treated with ondansetron. In some cases, predominantly during intravenous administration, the symptoms appeared immediately after administration but resolved with prompt treatment. Coronary artery spasm appears to be the most common underlying cause. Therefore, monitor or advise patients for signs or symptoms of myocardial ischemia after oral administration of ondansetron [see Adverse Reactions (6.2)].
5.5 Masking of Progressive Ileus and Gastric Distension
The use of ondansetron in patients following abdominal surgery or in patients with chemotherapy-induced nausea and vomiting may mask a progressive ileus and/or gastric distension. Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction.
Ondansetron is not a drug that stimulates gastric or intestinal peristalsis. It should not be used instead of nasogastric suction.
5.6 Phenylketonuria
Phenylketonuric patients should be informed that ondansetron orally disintegrating tablets contain phenylalanine (a component of aspartame). Each 4 mg orally disintegrating tablet contains 1.68 mg phenylalanine and 8 mg orally disintegrating tablet contains 3.37 mg phenylalanine.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- QT Prolongation [see Warnings and Precautions (5.2)]
- Serotonin Syndrome [see Warnings and Precautions (5.3)]
- Myocardial Ischemia [see Warnings and Precautions (5.4)]
- Masking of Progressive Ileus and Gastric Distension [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following adverse reactions have been reported in clinical trials of patients treated with ondansetron, the active ingredient of ondansetron orally disintegrating tablets. A causal relationship to therapy with ondansetron was unclear in many cases.
Prevention of Chemotherapy-Induced Nausea and Vomiting
The most common adverse reactions reported in greater than or equal to 4% of 300 adults receiving a single 24 mg dose of ondansetron orally in 2 trials for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy (cisplatin greater than or equal to 50 mg/m2) were: headache (11%) and diarrhea (4%).
The most common adverse reactions reported in 4 trials in adults for the prevention of nausea and vomiting associated with moderately emetogenic chemotherapy (primarily cyclophosphamide-based regimens) are shown in Table 3.
| a Reported in greater than or equal to 5% of patients treated with ondansetron orally disintegrating tablets and at a rate that exceeded placebo. | ||
| Adverse Reaction
| Ondansetron Orally
Disintegrating Tablets 8 mg Twice Daily (n = 242) | Placebo
(n = 262) |
| Headache | 58 (24%) | 34 (13%) |
| Malaise/Fatigue | 32 (13%) | 6 (2%) |
| Constipation | 22 (9%) | 1 (< 1%) |
| Diarrhea | 15 (6%) | 10 (4%) |
Less Common Adverse Reactions
Central Nervous System: Extrapyramidal reactions (less than 1% of patients).
Hepatic: Aspartate transaminase (AST) and/or alanine transaminase (ALT) values exceeded twice the upper limit of normal in approximately 1% to 2% of 723 patients receiving ondansetron and cyclophosphamide-based chemotherapy in U.S. clinical trials. The increases were transient and did not appear to be related to dose or duration of therapy. On repeat exposure, similar transient elevations in transaminase values occurred in some courses, but symptomatic hepatic disease did not occur. The role of cancer chemotherapy in these biochemical changes is unclear.
Liver failure and death has been reported in cancer patients receiving concurrent medications, including potentially hepatotoxic cytotoxic chemotherapy and antibiotics. The etiology of the liver failure is unclear.
Integumentary: Rash (approximately 1% of patients).
Other (less than 2%): Anaphylaxis, bronchospasm, tachycardia, angina, hypokalemia, electrocardiographic alterations, vascular occlusive events, and grand mal seizures. Except for bronchospasm and anaphylaxis, the relationship to ondansetron is unclear.
Prevention of Radiation-Induced Nausea and Vomiting
The most common adverse reactions (greater than or equal to 2%) reported in patients receiving ondansetron and concurrent radiotherapy were similar to those reported in patients receiving ondansetron and concurrent chemotherapy and were headache, constipation, and diarrhea.
Prevention of Postoperative Nausea and/or Vomiting
The most common adverse reactions reported in adults in trial(s) of prevention of postoperative nausea and vomiting are shown in Table 4. In these trial(s), patients were receiving multiple concomitant perioperative and postoperative medications in both treatment groups.
| a Reported in greater than or equal to 5% of patients treated with ondansetron orally disintegrating tablets and at a rate that exceeded placebo. | ||
| Adverse Reaction
| Ondansetron Orally
Disintegrating Tablets 16 mg as a Single Dose (n = 550) | Placebo (n = 531) |
| Headache | 49 (9%) | 27 (5%) |
| Hypoxia | 49 (9%) | 35 (7%) |
| Pyrexia | 45 (8%) | 34 (6%) |
| Dizziness | 36 (7%) | 34 (6%) |
| Gynecological disorder | 36 (7%) | 33 (6%) |
| Anxiety/Agitation | 33 (6%) | 29 (5%) |
| Urinary retention | 28 (5%) | 18 (3%) |
| Pruritus | 27 (5%) | 20 (4%) |
In a crossover study with 25 subjects, headache was reported in 6 subjects administered ondansetron orally disintegrating tablets with water (24%) as compared with 2 subjects administered ondansetron orally disintegrating tablets without water (8%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ondansetron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular
Arrhythmias (including ventricular and supraventricular tachycardia, premature ventricular contractions, and atrial fibrillation), bradycardia, electrocardiographic alterations (including second-degree heart block, QT/QTc interval prolongation, and ST segment depression), palpitations, and syncope. Rarely and predominantly with intravenous ondansetron, transient ECG changes, including QT interval prolongation have been reported.
Myocardial ischemia was reported predominately with intravenous administration [see Warnings and Precautions (5.4)].
General
Flushing: Rare cases of hypersensitivity reactions, sometimes severe (e.g., anaphylactic reactions, angioedema, bronchospasm, shortness of breath, hypotension, laryngeal edema, stridor) have also been reported.
Laryngospasm, shock, and cardiopulmonary arrest have occurred during allergic reactions in patients receiving injectable ondansetron.
Hepatobiliary
Liver enzyme abnormalities.
Lower Respiratory
Hiccups.
Neurology
Oculogyric crisis, appearing alone, as well as with other dystonic reactions.
Skin
Urticaria, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
Eye Disorders
Cases of transient blindness, predominantly during intravenous administration, have been reported. These cases of transient blindness were reported to resolve within a few minutes up to 48 hours.
7 DRUG INTERACTIONS
7.1 Serotonergic Drugs
Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been described following the concomitant use of 5-HT3 receptor antagonists and other serotonergic drugs, including SSRIs and SNRIs. Monitor for the emergence of serotonin syndrome. If symptoms occur, discontinue ondansetron and initiate supportive treatment [see Warnings and Precautions (5.3)].
7.2 Drugs Affecting Cytochrome P-450 Enzymes
Ondansetron does not itself appear to induce or inhibit the cytochrome P-450 drug-metabolizing enzyme system of the liver [see Clinical Pharmacology (12.3)]. Because ondansetron is metabolized by hepatic cytochrome P-450 drug-metabolizing enzymes (CYP3A4, CYP2D6, CYP1A2), inducers or inhibitors of these enzymes may change the clearance and, hence, the half-life of ondansetron. In patients treated with potent inducers of CYP3A4 (i.e., phenytoin, carbamazepine, and rifampin), the clearance of ondansetron was significantly increased and ondansetron blood concentrations were decreased. However, on the basis of available data, no dosage adjustment for ondansetron is recommended for patients on these drugs [see Clinical Pharmacology (12.3)].
7.3 Tramadol
Although no pharmacokinetic drug interaction between ondansetron and tramadol has been observed, data from 2 small trials indicate that when used together, ondansetron may increase patient-controlled administration of tramadol. Monitor patients to ensure adequate pain control when ondansetron is administered with tramadol.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Published epidemiological studies on the association between ondansetron use and major birth defects have reported inconsistent findings and have important methodological limitations that preclude conclusions about the safety of ondansetron use in pregnancy (see Data). Available postmarketing data have not identified a drug-associated risk of miscarriage or adverse maternal outcomes. Reproductive studies in rats and rabbits did not show evidence of harm to the fetus when ondansetron was administered during organogenesis at approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, based on body surface area (BSA), respectively (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriages, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Available data on ondansetron use in pregnant women from several published epidemiological studies preclude an assessment of a drug-associated risk of adverse fetal outcomes due to important methodological limitations, including the uncertainty of whether women who filled a prescription actually took the medication, the concomitant use of other medications or treatments, recall bias, and other unadjusted confounders.
Ondansetron exposure in utero has not been associated with overall major congenital malformations in aggregate analyses. One large retrospective cohort study examined 1970 women who received a prescription for ondansetron during pregnancy and reported no association between ondansetron exposure and major congenital malformations, miscarriage, stillbirth, preterm delivery, infants of low birth weight, or infants small for gestational age.
Two large retrospective cohort studies and one case-control study have assessed ondansetron exposure in the first trimester and risk of cardiovascular defects with inconsistent findings. Relative risks (RR) ranged from 0.97 (95% CI 0.86 to 1.10) to 1.62 (95% CI 1.04, 2.54). A subset analysis in one of the cohort studies observed that ondansetron was specifically associated with cardiac septal defects (RR 2.05, 95% CI 1.19, 3.28); however, this association was not confirmed in other studies.
Several studies have assessed ondansetron and the risk of oral clefts with inconsistent findings. A retrospective cohort study of 1.8 million pregnancies in the U.S. Medicaid Database showed an increased risk of oral clefts among 88,467 pregnancies in which oral ondansetron was prescribed in the first trimester (RR 1.24, 95% CI 1.03, 1.48), but no such association was reported with intravenous ondansetron in 23,866 pregnancies (RR 0.95, 95% CI 0.63, 1.43). In the subgroup of women who received both forms of administration, the RR was 1.07 (95% CI 0.59, 1.93). Two case-control studies, using data from birth defects surveillance programs, reported conflicting associations between maternal use of ondansetron and isolated cleft palate (OR 1.6 [95% CI 1.1, 2.3] and 0.5 [95% CI 0.3, 1.0]). It is unknown whether ondansetron exposure in utero in the cases of cleft palate occurred during the time of palate formation (the palate is formed between the 6th and 9th weeks of pregnancy).
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of ondansetron up to 15 mg/kg/day and 30 mg/kg/day, respectively, during the period of organogenesis. With the exception of a slight decrease in maternal body weight gain in the rabbits, there were no significant effects of ondansetron on the maternal animals or the development of the offspring. At doses of 15 mg/kg/day in rats and 30 mg/kg/day in rabbits, the maternal exposure margin was approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, respectively, based on BSA.
In a pre- and postnatal developmental toxicity study, pregnant rats received oral doses of ondansetron up to 15 mg/kg/day from Day 17 of pregnancy to litter Day 21. With the exception of a slight reduction in maternal body weight gain, there were no effects upon the pregnant rats and the pre- and postnatal development of their offspring, including reproductive performance of the mated F1 generation. At a dose of 15 mg/kg/day in rats, the maternal exposure margin was approximately 6 times the maximum recommended human oral dose of 24 mg/day, based on BSA.
8.2 Lactation
Risk Summary
It is not known whether ondansetron is present in human milk. There are no data on the effects of ondansetron on the breastfed infant or the effects on milk production. However, it has been demonstrated that ondansetron is present in the milk of rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ondansetron and any potential adverse effects on the breastfed infant from ondansetron or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of orally administered ondansetron have been established in pediatric patients 4 years and older for the prevention of nausea and vomiting associated with moderately emetogenic cancer chemotherapy. Use of ondansetron in these age-groups is supported by evidence from adequate and well- controlled studies of ondansetron in adults with additional data from 3 open-label, uncontrolled, non-U.S. trials in 182 pediatric patients aged 4 to 18 years with cancer who were given a variety of cisplatin or noncisplatin regimens [see Dosage and Administration (2.2), Clinical Studies (14.1)].
Additional information on the use of ondansetron in pediatric patients may be found in ondansetron Injection prescribing information.
The safety and effectiveness of orally administered ondansetron have not been established in pediatric patients for:
- prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy
- prevention of nausea and vomiting associated with radiotherapy
- prevention of postoperative nausea and/or vomiting
8.5 Geriatric Use
Of the total number of subjects enrolled in cancer chemotherapy-induced and postoperative nausea and vomiting in U.S.- and foreign-controlled clinical trials, for which there were subgroup analyses, 938 (19%) were aged 65 years and older.
No overall differences in safety or effectiveness were observed between subjects 65 years of age and older and younger subjects. A reduction in clearance and increase in elimination half-life were seen in patients older than 75 years compared with younger subjects [see Clinical Pharmacology (12.3)]. There were an insufficient number of patients older than 75 years of age and older in the clinical trials to permit safety or efficacy conclusions in this age group. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment is needed in elderly patients.
8.6 Hepatic Impairment
No dosage adjustment is needed in patients with mild or moderate hepatic impairment.
In patients with severe hepatic impairment, clearance is reduced and the apparent volume of distribution is increased, resulting in a significant increase in the half-life of ondansetron. Therefore, do not exceed a total daily dose of 8 mg in patients with severe hepatic impairment (Child-Pugh score of 10 or greater) [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is recommended for patients with any degree of renal impairment (mild, moderate, or severe). There is no experience beyond first-day administration of ondansetron [see Clinical Pharmacology (12.3)].
9 DRUG ABUSE AND DEPENDENCE
Animal studies have shown that ondansetron is not discriminated as a benzodiazepine nor does it substitute for benzodiazepines in direct addiction studies.
10 OVERDOSAGE
There is no specific antidote for ondansetron overdose. Patients should be managed with appropriate supportive therapy.
In addition to the adverse reactions listed above, the following adverse reactions have been described in the setting of ondansetron overdose: “Sudden blindness” (amaurosis) of 2 to 3 minutes’ duration plus severe constipation occurred in one patient that was administered 72 mg of ondansetron intravenously as a single dose. Hypotension (and faintness) occurred in a patient that took 48 mg of ondansetron tablets. Following infusion of 32 mg over only a 4-minute period, a vasovagal episode with transient second-degree heart block was observed. In all instances, the adverse reactions resolved completely.
Pediatric cases consistent with serotonin syndrome have been reported after inadvertent oral overdoses of ondansetron (exceeding estimated ingestion of 5 mg per kg) in young children. Reported symptoms included somnolence, agitation, tachycardia, tachypnea, hypertension, flushing, mydriasis, diaphoresis, myoclonic movements, horizontal nystagmus, hyperreflexia, and seizure. Patients required supportive care, including intubation in some cases, with complete recovery without sequelae within 1 to 2 days.
11 DESCRIPTION
The active ingredient in ondansetron orally disintegrating tablets, USP is ondansetron base, the racemic form of ondansetron and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (±) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one. It has the following structural formula:
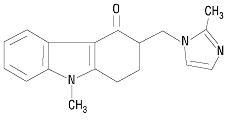
The molecular formula is C18H19N3O representing a molecular weight of 293.4 g/mol. Ondansetron is a white to off-white powder.
Each 4 mg ondansetron orally disintegrating tablet, USP for oral administration contains 4 mg ondansetron base. Each 8 mg ondansetron orally disintegrating tablet, USP for oral administration contains 8 mg ondansetron base. Each ondansetron orally disintegrating tablet, USP also contains the inactive ingredients mannitol, crospovidone, lactose monohydrate, microcrystalline cellulose, aspartame, strawberry guarana flavor, colloidal silicon dioxide, and magnesium stearate. The strawberry guarana flavor contains maltodextrin, propylene glycol, artificial flavors, and acetic acid. Ondansetron orally disintegrating tablets, USP are orally administered formulation of ondansetron which disintegrates on the tongue and does not require water to aid dissolution or swallowing.
Meets USP Disintegration Test 2.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ondansetron is a selective 5-HT3 receptor antagonist. While its mechanism of action has not been fully characterized, ondansetron is not a dopamine-receptor antagonist. Serotonin receptors of the 5-HT3 type are present both peripherally on vagal nerve terminals and centrally in the chemoreceptor trigger zone of the area postrema. It is not certain whether ondansetron’s antiemetic action is mediated centrally, peripherally, or in both sites. However, cytotoxic chemotherapy appears to be associated with release of serotonin from the enterochromaffin cells of the small intestine. In humans, urinary 5-hydroxyindoleacetic acid (5-HIAA) excretion increases after cisplatin administration in parallel with the onset of emesis. The released serotonin may stimulate the vagal afferents through the 5-HT3 receptors and initiate the vomiting reflex.
12.2 Pharmacodynamics
In healthy subjects, single intravenous doses of 0.15 mg/kg of ondansetron had no effect on esophageal motility, gastric motility, lower esophageal sphincter pressure, or small intestinal transit time. Multiday administration of ondansetron has been shown to slow colonic transit in healthy subjects. Ondansetron has no effect on plasma-prolactin concentrations.
Cardiac Electrophysiology
QTc interval prolongation was studied in a double-blind, single-intravenous dose, placebo- and positive- controlled, crossover trial in 58 healthy subjects. The maximum mean (95% upper confidence bound) difference in QTcF from placebo after baseline correction was 19.5 (21.8) milliseconds and 5.6 (7.4) milliseconds after 15-minute intravenous infusions of 32 mg and 8 mg of ondansetron injection, respectively. A significant exposure- response relationship was identified between ondansetron concentration and ΔΔQTcF. Using the established exposure-response relationship, 24 mg infused intravenously over 15 minutes had a mean predicted (95% upper prediction interval) ΔΔQTcF of 14.0 (16.3) milliseconds. In contrast, 16 mg infused intravenously over 15 minutes using the same model had a mean predicted (95% upper prediction interval) ΔΔQTcF of 9.1 (11.2) milliseconds. In this study, the 8 mg dose infused over 15 minutes did not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Absorption
Ondansetron is absorbed from the gastrointestinal tract and undergoes some first-pass metabolism. Mean bioavailability in healthy subjects, following administration of a single 8 mg tablet, is approximately 56%.
Ondansetron systemic exposure does not increase proportionately to dose. The area under curve (AUC) from a 16 mg tablet was 24% greater than predicted from an 8 mg tablet dose. This may reflect some reduction of first-pass metabolism at higher oral doses.
Food Effects: Bioavailability is also slightly enhanced by the presence of food.
Distribution
Plasma protein binding of ondansetron as measured in vitro was 70% to 76% over the concentration range of 10 to 500 ng/mL. Circulating drug also distributes into erythrocytes.
Elimination
Metabolism and Excretion: Ondansetron is extensively metabolized in humans, with approximately 5% of a radiolabeled dose recovered as the parent compound from the urine. The metabolites are observed in the urine. The primary metabolic pathway is hydroxylation on the indole ring followed by subsequent glucuronide or sulfate conjugation.
In vitro metabolism studies have shown that ondansetron is a substrate for human hepatic cytochrome P-450 enzymes, including CYP1A2, CYP2D6, and CYP3A4. In terms of overall ondansetron turnover, CYP3A4 played the predominant role. Because of the multiplicity of metabolic enzymes capable of metabolizing ondansetron, it is likely that inhibition or loss of one enzyme (e.g., CYP2D6 genetic deficiency) will be compensated by others and may result in little change in overall rates of ondansetron elimination.
Although some nonconjugated metabolites have pharmacologic activity, these are not found in plasma at concentrations likely to significantly contribute to the biological activity of ondansetron.
Specific Populations
Age: Geriatric Population: A reduction in clearance and increase in elimination half-life are seen in patients older than 75 years compared to younger subjects [see Use in Specific Populations (8.5)].
Sex: Gender differences were shown in the disposition of ondansetron given as a single dose. The extent and rate of absorption are greater in women than men. Slower clearance in women, a smaller apparent volume of distribution (adjusted for weight), and higher absolute bioavailability resulted in higher plasma ondansetron concentrations. These higher plasma concentrations may in part be explained by differences in body weight between men and women. It is not known whether these sex-related differences were clinically important. More detailed pharmacokinetic information is contained in Tables 5 and 6.
| Age-group (years) Sex (M/F)
| Mean
Weight (kg) | N
| Peak Plasma
Concentration (ng/mL) | Time of
Peak Plasma Concentration (h) | Mean
Elimination Half-life (h) | Systemic
Plasma Clearance L/h/kg | Absolute
Bioavailability |
| 18 to 40 M F | 69.0 62.7 | 6 5 | 26.2 42.7 | 2.0 1.7 | 3.1 3.5 | 0.403 0.354 | 0.483 0.663 |
| 61 to 74 M F | 77.5 60.2 | 6 6 | 24.1 52.4 | 2.1 1.9 | 4.1 4.9 | 0.384 0.255 | 0.585 0.643 |
| ≥ 75 M F | 78.0 67.6 | 5 6 | 37.0 46.1 | 2.2 2.1 | 4.5 6.2 | 0.277 0.249 | 0.619 0.747 |
| Age-group (years)
Sex (M/F) |
Mean Weight (kg) | N
| Peak Plasma Concentration (ng/mL)
| Time of Peak Plasma Concentration
(h) | Mean Elimination Half-life
(h) |
| 18 to 43 M F | 84.1 71.8 | 8 8 | 125.8 194.4 | 1.9 1.6 | 4.7 5.8 |
Renal Impairment: Renal impairment is not expected to significantly influence the total clearance of ondansetron as renal clearance represents only 5% of the overall clearance. However, the mean plasma clearance of ondansetron was reduced by about 50% in patients with severe renal impairment (creatinine clearance less than 30 mL/min). The reduction in clearance was variable and not consistent with an increase in half-life [see Use in Specific Populations (8.7)].
Hepatic Impairment: In patients with mild-to-moderate hepatic impairment, clearance is reduced 2-fold and mean half-life is increased to 11.6 hours compared with 5.7 hours in healthy subjects. In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), clearance is reduced 2-fold to 3-fold and apparent volume of distribution is increased with a resultant increase in half-life to 20 hours [see Dosage and Administration (2.2), Use in Specific Populations (8.6)].
Drug Interaction Studies
CYP 3A4 Inducers: Ondansetron elimination may be affected by cytochrome P-450 inducers. In a pharmacokinetic trial of 16 epileptic patients maintained chronically on CYP3A4 inducers, carbamazepine, or phenytoin, a reduction in AUC, Cmax, and t½ of ondansetron was observed. This resulted in a significant increase in the clearance of ondansetron. However, this increase is not thought to be clinically relevant [see Drug Interactions (7.2)].
Chemotherapeutic Agents: Carmustine, etoposide, and cisplatin do not affect the pharmacokinetics of ondansetron [see Drug Interactions (7.4)].
Antacids: Concomitant administration of antacids does not alter the absorption of ondansetron.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic effects were not seen in 2-year studies in rats and mice with oral ondansetron doses up to 10 mg/kg per day and 30 mg/kg per day, respectively (approximately 4 and 6 times the maximum recommended human oral dose of 24 mg per day, based on BSA).
Ondansetron was not mutagenic in standard tests for mutagenicity.
Oral administration of ondansetron up to 15 mg/kg per day (approximately 6 times the maximum recommended human oral dose of 24 mg per day, based on BSA) did not affect fertility or general reproductive performance of male and female rats.
14 CLINICAL STUDIES
14.1 Prevention of Chemotherapy-Induced Nausea and Vomiting
Highly Emetogenic Chemotherapy
In 2 randomized, double-blind, monotherapy trials, a single 24 mg oral dose of ondansetron was superior to a relevant historical placebo control in the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m2. Steroid administration was excluded from these clinical trials. More than 90% of patients receiving a cisplatin dose greater than or equal to 50 mg/m2 in the historical-placebo comparator, experienced vomiting in the absence of antiemetic therapy.
The first trial compared oral doses of ondansetron 24 mg as a single dose, 8 mg every 8 hours for 2 doses, and 32 mg as a single dose in 357 adult cancer patients receiving chemotherapy regimens containing cisplatin greater than or equal to 50 mg/m2. The first or single dose was administered 30 minutes prior to chemotherapy. A total of 66% of patients in the ondansetron 24 mg once-a-day group, 55% in the ondansetron 8 mg twice-a-day group, and 55% in the ondansetron 32 mg once-a-day group, completed the 24-hour trial period with 0 emetic episodes and no rescue antiemetic medications, the primary endpoint of efficacy. Each of the 3 treatment groups was shown to be statistically significantly superior to a historical placebo control.
In the same trial, 56% of patients receiving a single 24 mg oral dose of ondansetron experienced no nausea during the 24-hour trial period, compared with 36% of patients in the oral ondansetron 8 mg twice-a-day group (P = 0.001) and 50% in the oral ondansetron 32 mg once-a-day group. Dosage regimens of ondansetron 8 mg twice daily and 32 mg once daily are not recommended for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy [see Dosage and Administration (2.1)].
In a second trial, efficacy of a single 24 mg oral dose of ondansetron for the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m2, was confirmed.
Moderately Emetogenic Chemotherapy
A randomized, placebo-controlled, double-blind trial was conducted in the U.S. in 67 patients receiving a cyclophosphamide-based chemotherapy regimen containing doxorubicin. The first 8 mg dose of ondansetron was administered 30 minutes before the start of chemotherapy, with a subsequent dose 8 hours after the first dose, followed by 8 mg of ondansetron twice a day for 2 days after the completion of chemotherapy. Ondansetron orally disintegrating tablets was significantly more effective than placebo in preventing vomiting. Treatment response was based on the total number of emetic episodes over the 3-day trial period. The results of this trial are summarized in Table 7.
| a Median undefined since at least 50% of the patients were withdrawn or had more than 2 emetic episodes. b Median undefined since at least 50% of patients did not have any emetic episodes. |
|||
|
| Ondansetron Orally Disintegrating Tablets
(n = 33) | Placebo
(n = 34) | P-value
|
| Treatment response 0 Emetic episodes 1 to 2 Emetic episodes More than 2 emetic episodes/withdrawn |
20 (61%) 6 (18%) 7 (21%) |
2 (6%) 8 (24%) 24 (71%) |
< 0.001 < 0.001 |
| Median number of emetic episodes | 0.0 | Undefineda
|
|
| Median time to first emetic episode (hours) | Undefinedb
| 6.5 |
|
In a double-blind, U.S. trial in 336 patients receiving a cyclophosphamide-based chemotherapy regimen containing either methotrexate or doxorubicin, ondansetron 8 mg administered twice a day, was as effective as ondansetron 8 mg administered 3 times a day in preventing nausea and vomiting. Ondansetron 8 mg three times daily is not a recommended regimen for the treatment of moderately emetogenic chemotherapy [see Dosage and Administration (2.1)].
Treatment response was based on the total number of emetic episodes over the 3-day trial period. See Table 8 for the details of the dosage regimens studied and results of this trial.
| a The first 8 mg dose was administered 30 minutes before the start of emetogenic chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose, followed by 8 mg administered twice a day for 2 days after the completion of chemotherapy. b The first 8 mg dose was administered 30 minutes before the start of emetogenic chemotherapy, with subsequent 8 mg doses at 4 hours and 8 hours after the first dose, followed by 8 mg administered 3 times a day for 2 days after the completion of chemotherapy. c Median undefined since at least 50% of patients did not have any emetic episodes. d Visual analog scale assessment: 0 = no nausea, 100 = nausea as bad as it can be. |
||||||||||||||||
|
| Ondansetron Tablets
|
|||||||||||||||
| 8 mg Twice Dailya
(n = 165) | 8 mg Three Times a Dayb
(n = 171) |
|||||||||||||||
| Treatment response 0 Emetic episodes 1 to 2 Emetic episodes More than 2 emetic episodes/withdrawn |
101 (61%) 16 (10%) 48 (29%) |
99 (58%) 17 (10%) 55 (32%) |
||||||||||||||
| Median number of emetic episodes | 0.0 | 0.0 |
||||||||||||||
| Median time to first emetic episode (h) | Undefinedc
| Undefinedc
|
||||||||||||||
| Median nausea scores (0 to 100)d
| 6 | 6 |
||||||||||||||
Re-treatment
In single-arm trials, 148 patients receiving cyclophosphamide-based chemotherapy were re-treated with ondansetron 8 mg three times daily during subsequent chemotherapy for a total of 396 re-treatment courses. No emetic episodes occurred in 314 (79%) of the re-treatment courses, and only 1 to 2 emetic episodes occurred in 43 (11%) of the re-treatment courses.
Pediatric Trials
Three open-label, single-arm, non-U.S. trials have been performed with 182 pediatric patients aged 4 to 18 years with cancer who were given a variety of cisplatin or noncisplatin regimens. The initial dose of ondansetron injection ranged from 0.04 to 0.87 mg per kg (total dose of 2.16 mg to 12 mg) followed by the administration of oral doses of ondansetron ranging from 4 to 24 mg daily for 3 days. In these trials, 58% of the 170 evaluable patients had a complete response (no emetic episodes) on Day 1. In 2 trials, the response rates to ondansetron 4 mg three times a day in patients younger than 12 years was similar to ondansetron 8 mg three times daily in patients 12 to 18 years. Prevention of emesis in these pediatric patients was essentially the same as for adults.
14.2 Radiation-Induced Nausea and Vomiting
Total Body Irradiation
In a randomized, placebo-controlled, double-blind trial in 20 patients, 8 mg of ondansetron administered 1.5 hours before each fraction of radiotherapy for 4 days was significantly more effective than placebo in preventing vomiting induced by total body irradiation. Total body irradiation consisted of 11 fractions (120 cGy per fraction) over 4 days for a total of 1,320 cGy. Patients received 3 fractions for 3 days, then 2 fractions on Day 4.
Single High-Dose Fraction Radiotherapy
In an active-controlled, double-blind trial in 105 patients receiving single high-dose radiotherapy (800 to 1,000 cGy) over an anterior or posterior field size of greater than or equal to 80 cm2 to the abdomen, ondansetron was significantly more effective than metoclopramide with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron (8 mg) or metoclopramide (10 mg) 1 to 2 hours before radiotherapy. If radiotherapy was given in the morning, 8 mg of ondansetron or 10 mg of metoclopramide was administered in the late afternoon and repeated again before bedtime. If radiotherapy was given in the afternoon, patients took 8 mg of ondansetron or 10 mg of metoclopramide only once before bedtime. Patients continued the doses of oral medication three times daily for 3 days.
Daily Fractionated Radiotherapy
In an active-controlled, double-blind trial in 135 patients receiving a 1- to 4-week course of fractionated radiotherapy (180 cGy doses) over a field size of greater than or equal to 100 cm2 to the abdomen, ondansetron was significantly more effective than prochlorperazine with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron (8 mg) or prochlorperazine (10 mg) 1 to 2 hours before the first daily radiotherapy fraction, with subsequent 8 mg doses approximately every 8 hours on each day of radiotherapy.
14.3 Postoperative Nausea and/or Vomiting
In 2 placebo-controlled, double-blind trials (one conducted in the U.S. and the other outside the U.S.) in 865 females undergoing inpatient surgical procedures, ondansetron 16 mg as a single dose or placebo was administered one hour before the induction of general balanced anesthesia (barbiturate, opioid, nitrous oxide, neuromuscular blockade, and supplemental isoflurane or enflurane), ondansetron was significantly more effective than placebo in preventing postoperative nausea and vomiting.
No trials have been performed in males.
16 HOW SUPPLIED/STORAGE AND HANDLING
Ondansetron Orally Disintegrating Tablets USP, 8 mg are white to off-white, round tablets debossed with ‘7’ on one side and ‘E’ on the other side with an embossed circular edge.
NDC: 71335-1731-1: 30 Tablets in a Blister Pack
NDC: 71335-1731-2: 10 Tablets in a Blister Pack
NDC: 71335-1731-3: 5 Tablets in a Blister Pack
NDC: 71335-1731-4: 4 Tablets in a Blister Pack
NDC: 71335-1731-5: 6 Tablets in a Blister Pack
NDC: 71335-1731-6: 14 Tablets in a Blister Pack
NDC: 71335-1731-7: 60 Tablets in a Blister Pack
NDC: 71335-1731-8: 12 Tablets in a Blister Pack
NDC: 71335-1731-9: 90 Tablets in a Blister Pack
NDC: 71335-1731-0: 3 Tablets in a Blister Pack
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Dispense in a tight, light-resistant container as defined in the USP.
Repackaged/Relabeled by:
Bryant Ranch Prepack, Inc.
Burbank, CA 91504
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions
Inform patients that ondansetron may cause hypersensitivity reactions, some as severe as anaphylaxis and bronchospasm. Instruct patients to immediately report any signs and symptoms of hypersensitivity reactions, including fever, chills, rash, or breathing problems to their healthcare provider [see Warnings and Precautions (5.1)].
QT Prolongation
Inform patients that ondansetron may cause serious cardiac arrhythmias, such as QT prolongation. Instruct patients to tell their healthcare provider right away if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode [see Warnings and Precautions (5.2)].
Drug Interactions
- Instruct the patient to report the use of all medications, especially apomorphine, to their healthcare provider. Concomitant use of apomorphine and ondansetron may cause a significant drop in blood pressure and loss of consciousness.
- Advise patients of the possibility of serotonin syndrome with concomitant use of ondansetron and another serotonergic agent, such as medications to treat depression and migraines. Advise patients to seek immediate medical attention if the following symptoms occur: changes in mental status, autonomic instability, neuromuscular symptoms with or without gastrointestinal symptoms [see Warnings and Precautions (5.3)].
Myocardial Ischemia
Inform patients that ondansetron may cause myocardial ischemia. Advise patients to seek immediate medical help if any symptoms suggestive of a myocardial ischemia occur, such as sudden chest pain or chest tightness [see Warnings and Precautions (5.4)].
Masking of Progressive Ileus and Gastric Distension
Inform patients following abdominal surgery or those with chemotherapy-induced nausea and vomiting that ondansetron may mask signs and symptoms of bowel obstruction. Instruct patients to immediately report any signs or symptoms consistent with a potential bowel obstruction to their healthcare provider [see Warnings and Precautions (5.5)].
Administration of Ondansetron Orally Disintegrating Tablets
Instruct patients not to remove ondansetron orally disintegrating tablets from the blister until just prior to dosing.
- Do not attempt to push ondansetron orally disintegrating tablets through the foil backing.
- With dry hands, remove the tablet from the bottle or peel back the foil backing of 1 blister and gently remove the tablet.
- Immediately place the ondansetron orally disintegrating tablet on top of the tongue where it will dissolve in seconds, then swallow with saliva.
- Administration with liquid is not necessary.
- Peelable illustrated stickers are affixed to the product carton that can be provided with the prescription to ensure proper use and handling of the product.
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad–500 032, India
Revised: 11/2021
