DAUNORUBICIN HYDROCHLORIDE- daunorubicin hydrochloride injection, solution
Teva Parenteral Medicines, Inc.
----------
DAUNORUBICIN
Hydrochloride Injection
WARNINGS
- Daunorubicin hydrochloride injection must be given into a rapidly flowing intravenous infusion. It must never be given by the intramuscular or subcutaneous route. Severe local tissue necrosis will occur if there is extravasation during administration.
- Myocardial toxicity manifested in its most severe form by potentially fatal congestive heart failure may occur either during therapy or months to years after termination of therapy. The incidence of myocardial toxicity increases after a total cumulative dose exceeding 400 to 550 mg/m2 in adults, 300 mg/m2 in children more than 2 years of age, or 10 mg/kg in children less than 2 years of age.
- Severe myelosuppression occurs when used in therapeutic doses; this may lead to infection or hemorrhage.
- It is recommended that daunorubicin hydrochloride be administered only by physicians who are experienced in leukemia chemotherapy and in facilities with laboratory and supportive resources adequate to monitor drug tolerance and protect and maintain a patient compromised by drug toxicity. The physician and institution must be capable of responding rapidly and completely to severe hemorrhagic conditions and/or overwhelming infection.
- Dosage should be reduced in patients with impaired hepatic or renal function.
DESCRIPTION
Daunorubicin Hydrochloride Injection consists of the hydrochloride salt of an anthracycline cytotoxic antibiotic produced by a strain of Streptomyces coeruleorubidus. It is provided as a deep red sterile liquid in vials for intravenous administration only. Each mL contains daunorubicin hydrochloride, USP equivalent to 5 mg of daunorubicin, 9 mg sodium chloride, hydrochloric acid (to adjust pH), and water for injection, q.s. It has the following structural formula which may be described with the chemical name of (1S,3S)-3-Acetyl-1,2,3,4,6,11-hexahydro-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1-naphthacenyl 3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranoside hydrochloride.
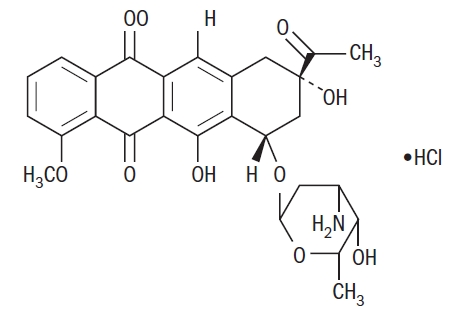
C27H29NO10•HCl M.W. 563.99
It is a hygroscopic crystalline powder. The pH of a 5 mg/mL aqueous solution is 3 to 4.
CLINICAL PHARMACOLOGY
Mechanism of Action
Daunorubicin has antimitotic and cytotoxic activity through a number of proposed mechanisms of action. Daunorubicin forms complexes with DNA by intercalation between base pairs. It inhibits topoisomerase II activity by stabilizing the DNA-topoisomerase II complex, preventing the religation portion of the ligation-religation reaction that topoisomerase II catalyzes. Single strand and double strand DNA breaks result.
Daunorubicin hydrochloride may also inhibit polymerase activity, affect regulation of gene expression, and produce free radical damage to DNA.
Daunorubicin hydrochloride possesses an antitumor effect against a wide spectrum of animal tumors, either grafted or spontaneous.
Pharmacokinetics
General
Following intravenous injection of daunorubicin hydrochloride, plasma levels of daunorubicin decline rapidly, indicating rapid tissue uptake and concentration. Thereafter, plasma levels decline slowly with a half-life of 45 minutes in the initial phase and 18.5 hours in the terminal phase. By 1 hour after drug administration, the predominant plasma species is daunorubicinol, an active metabolite, which disappears with a half-life of 26.7 hours.
Distribution
Daunorubicin hydrochloride is rapidly and widely distributed in tissues, with highest levels in the spleen, kidneys, liver, lungs, and heart. The drug binds to many cellular components, particularly nucleic acids. There is no evidence that daunorubicin crosses the blood-brain barrier, but the drug apparently crosses the placenta.
Metabolism and Elimination
Daunorubicin hydrochloride is extensively metabolized in the liver and other tissues, mainly by cytoplasmic aldo-keto reductases, producing daunorubicinol, the major metabolite which has antineoplastic activity. Approximately 40% of the drug in the plasma is present as daunorubicinol within 30 minutes and 60% in 4 hours after a dose of daunorubicin. Further metabolism via reduction cleavage of the glycosidic bond, 4-O demethylation, and conjugation with both sulfate and glucuronide have been demonstrated. Simple glycosidic cleavage of daunorubicin or daunorubicinol is not a significant metabolic pathway in man. Twenty-five percent of an administered dose of daunorubicin hydrochloride is eliminated in an active form by urinary excretion and an estimated 40% by biliary excretion.
Special Populations
Pediatric Patients
Although appropriate studies with daunorubicin hydrochloride have not been performed in the pediatric population, cardiotoxicity may be more frequent and occur at lower cumulative doses in children.
Geriatric Patients
Although appropriate studies with daunorubicin hydrochloride have not been performed in the geriatric population, cardiotoxicity may be more frequent in the elderly. Caution should also be used in patients who have inadequate bone marrow reserves due to old age. In addition, elderly patients are more likely to have age-related renal function impairment, which may require reduction of dosage in patients receiving daunorubicin hydrochloride.
Renal and Hepatic Impairment
Doses of daunorubicin hydrochloride should be reduced in patients with hepatic and renal impairment. Patients with serum bilirubin concentrations of 1.2 to 3 mg/dL should receive 75% of the usual daily dose and patients with serum bilirubin concentrations greater than 3 mg/dL should receive 50% of the usual daily dose. Patients with serum creatinine concentrations of greater than 3 mg/dL should receive 50% of the usual daily dose (see WARNINGS, Evaluation of Hepatic and Renal Functionsection).
Clinical Studies
In the treatment of adult acute nonlymphocytic leukemia, daunorubicin hydrochloride, used as a single agent, has produced complete remission rates of 40 to 50%, and in combination with cytarabine, has produced complete remission rates of 53 to 65%.
The addition of daunorubicin hydrochloride to the two-drug induction regimen of vincristine-prednisone in the treatment of childhood acute lymphocytic leukemia does not increase the rate of complete remission. In children receiving identical CNS prophylaxis and maintenance therapy (without consolidation), there is prolongation of complete remission duration (statistically significant, p < 0.02) in those children induced with the three drug (daunorubicin-vincristine-prednisone) regimen as compared to two drugs. There is no evidence of any impact of daunorubicin hydrochloride on the duration of complete remission when a consolidation (intensification) phase is employed as part of a total treatment program.
In adult acute lymphocytic leukemia, in contrast to childhood acute lymphocytic leukemia, daunorubicin hydrochloride during induction significantly increases the rate of complete remission, but not remission duration, compared to that obtained with vincristine, prednisone, and L-asparaginase alone. The use of daunorubicin hydrochloride in combination with vincristine, prednisone, and L-asparaginase has produced complete remission rates of 83% in contrast to a 47% remission in patients not receiving daunorubicin hydrochloride.
INDICATIONS AND USAGE
Daunorubicin hydrochloride injection in combination with other approved anticancer drugs is indicated for remission induction in acute nonlymphocytic leukemia (myelogenous, monocytic, erythroid) of adults and for remission induction in acute lymphocytic leukemia of children and adults.
CONTRAINDICATIONS
Daunorubicin hydrochloride is contraindicated in patients who have shown a hypersensitivity to it.
WARNINGS
Bone Marrow
Daunorubicin hydrochloride is a potent bone marrow suppressant. Suppression will occur in all patients given a therapeutic dose of this drug. Therapy with daunorubicin hydrochloride should not be started in patients with preexisting drug-induced bone marrow suppression unless the benefit from such treatment warrants the risk. Persistent, severe myelosuppression may result in superinfection or hemorrhage.
Cardiac Effects
Special attention must be given to the potential cardiac toxicity of daunorubicin hydrochloride, particularly in infants and children. Preexisting heart disease and previous therapy with doxorubicin are co-factors of increased risk of daunorubicin-induced cardiac toxicity and the benefit-to-risk ratio of daunorubicin hydrochloride therapy in such patients should be weighed before starting daunorubicin hydrochloride. In adults, at total cumulative doses less than 550 mg/m2, acute congestive heart failure is seldom encountered. However, rare instances of pericarditis-myocarditis, not dose-related, have been reported.
In adults, at cumulative doses exceeding 550 mg/m2, there is an increased incidence of drug-induced congestive heart failure. Based on prior clinical experience with doxorubicin, this limit appears lower, namely 400 mg/m2, in patients who received radiation therapy that encompassed the heart.
In infants and children, there appears to be a greater susceptibility to anthracycline-induced cardiotoxicity compared to that in adults, which is more clearly dose-related. Anthracycline therapy (including daunorubicin) in pediatric patients has been reported to produce impaired left ventricular systolic performance, reduced contractility, congestive heart failure or death. These conditions may occur months to years following cessation of chemotherapy. This appears to be dose-dependent and aggravated by thoracic irradiation. Long-term periodic evaluation of cardiac function in such patients should, thus, be performed. In both children and adults, the total dose of daunorubicin hydrochloride administered should also take into account any previous or concomitant therapy with other potentially cardiotoxic agents or related compounds such as doxorubicin.
There is no absolutely reliable method of predicting the patients in whom acute congestive heart failure will develop as a result of the cardiac toxic effect of daunorubicin hydrochloride. However, certain changes in the electrocardiogram and a decrease in the systolic ejection fraction from pretreatment baseline may help to recognize those patients at greatest risk to develop congestive heart failure. On the basis of the electrocardiogram, a decrease equal to or greater than 30% in limb lead QRS voltage has been associated with a significant risk of drug-induced cardiomyopathy. Therefore, an electrocardiogram and/or determination of systolic ejection fraction should be performed before each course of daunorubicin hydrochloride. In the event that one or the other of these predictive parameters should occur, the benefit of continued therapy must be weighed against the risk of producing cardiac damage.
Early clinical diagnosis of drug-induced congestive heart failure appears to be essential for successful treatment.
Evaluation of Hepatic and Renal Function
Significant hepatic or renal impairment can enhance the toxicity of the recommended doses of daunorubicin hydrochloride; therefore, prior to administration, evaluation of hepatic function and renal function using conventional clinical laboratory tests is recommended (see DOSAGE AND ADMINISTRATIONsection).
Pregnancy
Daunorubicin hydrochloride may cause fetal harm when administered to a pregnant woman. An increased incidence of fetal abnormalities (parieto-occipital cranioschisis, umbilical hernias, or rachischisis) and abortions was reported in rabbits at doses of 0.05 mg/kg/day or approximately 1/100th of the highest recommended human dose on a body surface area basis. Rats showed an increased incidence of esophageal, cardiovascular and urogenital abnormalities as well as rib fusions at doses of 4 mg/kg/day or approximately 1/2 the human dose on a body surface area basis. Decreases in fetal birth weight and post-delivery growth rate were observed in mice. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Secondary Leukemias
There have been reports of secondary leukemias in patients exposed to topoisomerase II inhibitors when used in combination with other antineoplastic agents or radiation therapy.
Extravasation at Injection Site
Extravasation of daunorubicin hydrochloride at the site of intravenous administration can cause severe local tissue necrosis (see ADVERSE REACTIONSsection).
PRECAUTIONS
General
Therapy with daunorubicin hydrochloride requires close patient observation and frequent complete blood-count determinations. Cardiac, renal, and hepatic function should be evaluated prior to each course of treatment.
Appropriate measures must be taken to control any systemic infection before beginning therapy with daunorubicin hydrochloride.
Daunorubicin hydrochloride may transiently impart a red coloration to the urine after administration, and patients should be advised to expect this.
Laboratory Tests
Daunorubicin hydrochloride may induce hyperuricemia secondary to rapid lysis of leukemic cells. As a precaution, allopurinol administration is usually begun prior to initiating antileukemic therapy. Blood uric acid levels should be monitored and appropriate therapy initiated in the event that hyperuricemia develops.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Daunorubicin hydrochloride, when injected subcutaneously into mice, causes a fibrosarcomas to develop at the injection site. When administered to mice thrice weekly intraperitoneally, no carcinogenic effect was noted after 18 months of observation. In male rats administered daunorubicin thrice weekly for 6 months, at 1/70th the recommended human dose on a body surface area basis, peritoneal sarcomas were found at 18 months. A single IV dose of daunorubicin administered to rats at 1.6 fold the recommended human dose on a body surface area basis caused mammary adenocarcinomas to appear at 1 year. Daunorubicin was mutagenic in vitro (Ames assay, V79 hamster cell assay), and clastogenic in vitro (CCRFCEM human lymphoblasts) and in vivo (SCE assay in mouse bone marrow) tests.
In male dogs at a daily dose of 0.25 mg/kg administered intravenously, testicular atrophy was noted at autopsy. Histologic examination revealed total aplasia of the spermatocyte series in the seminiferous tubules with complete aspermatogenesis.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from daunorubicin, mothers should be advised to discontinue nursing during daunorubicin therapy.
Pediatric Use
See CLINICAL PHARMACOLOGY, Special Populations, Pediatric Patients section and WARNINGS, Cardiac Effects section.
Drug Interactions
Use of daunorubicin in a patient who has previously received doxorubicin increases the risk of cardiotoxicity. Daunorubicin hydrochloride should not be used in patients who have previously received the recommended maximum cumulative doses of doxorubicin or daunorubicin hydrochloride. Cyclophosphamide used concurrently with daunorubicin hydrochloride may also result in increased cardiotoxicity.
Dosage reduction of daunorubicin hydrochloride may be required when used concurrently with other myelosuppressive agents.
Hepatotoxic medications, such as high-dose methotrexate, may impair liver function and increase the risk of toxicity.
ADVERSE REACTIONS
Dose-limiting toxicity includes myelosuppression and cardiotoxicity (see WARNINGSsection). Other reactions include:
Cutaneous
Reversible alopecia occurs in most patients. Rash, contact dermatitis and urticaria have occurred rarely.
Gastrointestinal
Acute nausea and vomiting occur but are usually mild. Antiemetic therapy may be of some help. Mucositis may occur 3 to 7 days after administration. Diarrhea and abdominal pain have occasionally been reported.
DOSAGE AND ADMINISTRATION
Parenteral drug products should be inspected visually for particulate matter prior to administration, whenever solution and container permit.
Principles
In order to eradicate the leukemic cells and induce a complete remission, a profound suppression of the bone marrow is usually required. Evaluation of both the peripheral blood and bone marrow is mandatory in the formulation of appropriate treatment plans.
It is recommended that the dosage of daunorubicin hydrochloride be reduced in instances of hepatic or renal impairment. For example, using serum bilirubin and serum creatinine as indicators of liver and kidney function, the following dose modifications are recommended:
| Serum Bilirubin | Serum Creatinine | Dose Reduction |
|---|---|---|
| 1.2 to 3 mg% | — | 25% |
| > 3 mg% | — | 50% |
| — | > 3 mg% | 50% |
Representative Dose Schedules and Combination for the Approved Indication of Remission Induction in Adult Acute Nonlymphocytic Leukemia
In Combination
For patients under age 60, daunorubicin hydrochloride 45 mg/m2/day IV on days 1, 2, and 3 of the first course and on days 1, 2 of subsequent courses AND cytosine arabinoside 100 mg/m2/day IV infusion daily for 7 days for the first course and for 5 days for subsequent courses.
For patients 60 years of age and above, daunorubicin hydrochloride 30 mg/m2/day IV on days 1, 2, and 3 of the first course and on days 1, 2 of subsequent courses AND cytosine arabinoside 100 mg/m2/day IV infusion daily for 7 days for the first course and for 5 days for subsequent courses. This daunorubicin hydrochloride dose-reduction is based on a single study and may not be appropriate if optimal supportive care is available.
The attainment of a normal-appearing bone marrow may require up to three courses of induction therapy. Evaluation of the bone marrow following recovery from the previous course of induction therapy determines whether a further course of induction treatment is required.
Representative Dose Schedule and Combination for the Approved Indication of Remission Induction in Pediatric Acute Lymphocytic Leukemia
In Combination
Daunorubicin hydrochloride 25 mg/m2 IV on day 1 every week, vincristine 1.5 mg/m2 IV on day 1 every week, prednisone 40 mg/m2 PO daily. Generally, a complete remission will be obtained within four such courses of therapy; however, if after four courses the patient is in partial remission, an additional one or, if necessary, two courses may be given in an effort to obtain a complete remission.
In children less than 2 years of age or below 0.5 m2 body surface area, it has been recommended that the daunorubicin hydrochloride dosage calculation should be based on weight (1 mg/kg) instead of body surface area.
Representative Dose Schedules and Combination for the Approved Indication of Remission Induction in Adult Acute Lymphocytic Leukemia
In Combination
Daunorubicin hydrochloride 45 mg/m2/day IV on days 1, 2, and 3 AND vincristine 2 mg IV on days 1, 8, and 15; prednisone 40 mg/m2/day PO on days 1 through 22, then tapered between days 22 to 29; L-asparaginase 500 IU/kg/day X 10 days IV on days 22 through 32.
The sterile 4 mL vial contents provide 20 mg of daunorubicin with 5 mg of daunorubicin per mL. The desired dose is withdrawn into a syringe containing 10 mL to 15 mL of 0.9% sodium chloride injection, USP and then injected into the tubing or sidearm in a rapidly flowing IV infusion of 5% dextrose injection, USP or 0.9% sodium chloride injection, USP. Daunorubicin hydrochloride should not be administered mixed with other drugs or heparin.
Storage and Handling
Store unopened vials in refrigerator, 2° to 8°C (36° to 46°F). Store prepared solution for infusion at room temperature, 20° to 25°C (68° to 77°F) for up to 24 hours. Contains no preservative. Discard unused portion. Protect from light. Retain in carton until time of use.
If daunorubicin hydrochloride contacts the skin or mucosae, the area should be washed thoroughly with soap and water. Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.1-7 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
HOW SUPPLIED
Daunorubicin Hydrochloride Injection, 5 mg (base)/mL, is available as follows:
| NDC Number | Contents | Size |
|---|---|---|
| 0703-5233-13 | 20 mg (as base) | 4 mL fill in a 6 mL Single Dose Vial |
The 20 mg base/4 mL vials are packaged in tens.
REFERENCES
- Recommendations for the Safe Handling of Parenteral Antineoplastic Drugs. NIH Publication No. 83-2621. For Sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, D.C. 20402.
- AMA Council Report. Guidelines for Handling Parenteral Antineoplastics. JAMA. 253 (11): 1590–1592, 1985.
- National Study Commission on Cytotoxic Exposure—Recommendations for Handling Cytotoxic Agents. Available from Louis P. Jeffrey, Sc.D., Chairman, National Study Commission on Cytotoxic Exposure, Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, Massachusetts 02115.
- Clinical Oncological Society of Australia: Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia 1:426–428, 1983.
- Jones RB, et al.: Safe handling of chemotherapeutic agents: A report from the Mount Sinai Medical Center, Ca – A Cancer Journal for Clinicians Sept/Oct, 258–263, 1983.
- American Society of Hospital Pharmacists technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm 47:1033–1049, 1990.
- OSHA Work Practice Guidelines for Personnel Dealing with Cyotoxic (Antineoplastic) Drugs. AM J Hosp Pharm 43:1193–1204, 1986.

| DAUNORUBICIN HYDROCHLORIDE
daunorubicin hydrochloride injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Teva Parenteral Medicines, Inc. (794362533) |