FULL PRESCRIBING INFORMATION
WARNING: HEMATALOGIC TOXICITY, IMPAIRMENT OF FERTILITY, TERATOGENICITY, and CARCINOGENICITY
- Hematologic Toxicity: Granulocytopenia, anemia, thrombocytopenia, and pancytopenia have been reported in patients treated with ganciclovir [see Warnings and Precautions (5.1)].
- Impairment of Fertility: Based on animal data, GANCICLOVIR INJECTION may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females [see Warnings and Precautions (5.3)].
- Fetal Toxicity: Based on animal data, GANCICLOVIR INJECTION has the potential to cause birth defects in humans [see Warnings and Precautions (5.4)].
- Mutagenesis and Carcinogenesis: Based on animal data, GANCICLOVIR INJECTION has the potential to cause cancer in humans [see Warnings and Precautions (5.5)].
1 INDICATIONS AND USAGE
1.1 Treatment of CMV Retinitis
GANCICLOVIR INJECTION is indicated for the treatment of cytomegalovirus (CMV) retinitis in immunocompromised adult patients, including patients with acquired immunodeficiency syndrome (AIDS) [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing and Administration Information
- Do not administer GANCICLOVIR INJECTION by rapid or bolus intravenous injection which may increase toxicity as a result of excessive plasma levels.
- The recommended dosage and infusion rate for GANCICLOVIR INJECTION should not be exceeded.
- Administration of GANCICLOVIR INJECTION should be accompanied by adequate hydration.
- GANCICLOVIR INJECTION should only be infused into veins with adequate blood flow to permit rapid dilution and distribution.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
2.2 Testing Before and During Treatment
- Females of reproductive potential should undergo pregnancy testing before initiation of GANCICLOVIR INJECTION [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
- Complete blood counts with differential and platelet counts should be performed frequently, especially in patients in whom ganciclovir or other nucleoside analogues have previously resulted in cytopenias, or in whom absolute neutrophil counts are less than 1000 cells/μL at the beginning of treatment [see Warnings and Precautions (5.1)].
- All patients should be monitored for renal function before and during treatment with GANCICLOVIR INJECTION and dosage should be adjusted as needed [see Dosage and Administration (2.5), Warnings and Precautions (5.2)].
Patients with CMV retinitis should have frequent ophthalmological examinations during treatment with GANCICLOVIR INJECTION to monitor disease status and for other retinal abnormalities [see Adverse Reactions (6.1)].
2.3 Recommended Dosage for Treatment of CMV Retinitis in Adult Patients with Normal Renal Function
Induction Dosage: The recommended initial dosage of GANCICLOVIR INJECTION for patients with normal renal function is 5 mg/kg (given intravenously at a constant rate over 1 hour) every 12 hours for 14 to 21 days.
Maintenance Dosage: Following induction treatment, the recommended maintenance dosage of GANCICLOVIR INJECTION is 5 mg/kg (given intravenously at a constant rate over 1 hour) once daily for 7 days per week, or 6 mg/kg once daily for 5 days per week.
2.4 Recommended Dosage for Prevention of CMV Disease in Adult Transplant Recipients with Normal Renal Function
Induction Dosage:The recommended initial dosage of GANCICLOVIR INJECTION for patients with normal renal function is 5 mg/kg (given intravenously at a constant rate over 1 hour) every 12 hours for 7 to 14 days.
Maintenance Dosage: Following induction, the recommended maintenance dosage of GANCICLOVIR INJECTIONS is 5 mg/kg (given intravenously at a constant rate over 1 hour) once daily for 7 days per week, or 6 mg/kg once daily for 5 days per week until 100 to 120 days post-transplantation.
2.5 Recommended Dosage in Adult Patients with Renal Impairment
For patients with renal impairment, refer to Table 1 for recommended doses of GANCICLOVIR INJECTION for induction and maintenance dosage for treatment of CMV retinitis and prevention of CMV disease in transplant patients. Monitor serum creatinine or creatinine clearance during treatment to allow for dosage adjustments in patients with impaired renal function.
| Creatinine Clearance* (mL/min) | GANCICLOVIR INJECTION Induction Dose (mg/kg) | Dosing Interval (hours) for Induction | GANCICLOVIR INJECTION Maintenance Dose (mg/kg) | Dosing Interval (hours) for Maintenance |
|---|---|---|---|---|
| Greater than or equal to 70 | 5 | 12 | 5 | 24 |
| 50-69 | 2.5 | 12 | 2.5 | 24 |
| 25-49 | 2.5 | 24 | 1.25 | 24 |
| 10-24 | 1.25 | 24 | 0.625 | 24 |
| Less than 10 | 1.25 | 3 times per week, following hemodialysis | 0.625 | 3 times per week, following hemodialysis |
* Creatinine clearance can be related to serum creatinine by the formulas given below:
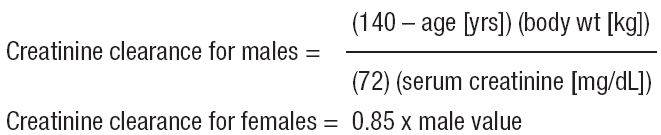
Dosing for patients undergoing hemodialysis should not exceed 1.25 mg per kg 3 times per week,
following each hemodialysis session. GANCICLOVIR INJECTION should be given shortly after completion of the hemodialysis session, since hemodialysis has been shown to reduce plasma levels by approximately 50% [see Clinical Pharmacology (12.3)].
2.6 Handling and Disposal
Because ganciclovir shares some of the properties of antitumor agents (i.e., carcinogenicity and mutagenicity), procedures for proper handling and disposal for cytotoxic drugs should be considered1[see How Supplied/Storage and Handling (16)]. The premix flexible plastic container bag contains no preservative; therefore, any unused portion should be discarded after each use.
3 DOSAGE FORMS AND STRENGTHS
Injection: 500 mg of ganciclovir in 250 mL (2 mg per mL) sterile, unpreserved, colorless solution in a single-dose bag for intravenous use.
4 CONTRAINDICATIONS
GANCICLOVIR INJECTION is contraindicated in patients who have experienced a clinically significant hypersensitivity reaction (e.g., anaphylaxis) to ganciclovir, valganciclovir or acyclovir.
5 WARNINGS AND PRECAUTIONS
5.1 Hematologic Toxicity
Granulocytopenia (neutropenia), anemia,thrombocytopenia, and pancytopenia have been observed in patients treated with ganciclovir. The frequency andseverity of these events vary widely in different patient populations [see Adverse Reactions (6.1)].GANCICLOVIR INJECTION is not recommended if the absolute neutrophil count is less than 500 cells/μL, hemoglobin is less than 8 g/dL, or the platelet count is less than 25,000 cells/μL. GANCICLOVIR INJECTION should also be used with caution in patients with pre-existing cytopenias and in patients receiving myelosuppressive drugs or irradiation. Neutropenia usually occurs during the first or second week of treatment but may occur at any time during treatment. Cell counts usually begin to recover within 3 to 7 days after discontinuing drug. Colony-stimulating factors have been shown to increase neutrophil and white blood cell counts in patients receiving ganciclovir for treatment of CMV retinitis.
Due to the frequency of neutropenia, anemia and thrombocytopenia in patients receiving GANCICLOVIR INJECTION, complete blood counts with differential and platelet counts should be performed frequently, especially in patients in whom ganciclovir or other nucleoside analogues have previously resulted in leukopenia, or in whom neutrophil counts are less than 1000 cells/µL at the beginning of treatment [see Dosage and Administration (2.2)].
5.2 Impairment of Renal Function
Increased serum creatinine levels have been reported in elderly patients and in transplant patients receiving concomitant nephrotoxic medications (i.e., cyclosporine and amphotericin B). Monitoring renal function during therapy with GANCICLOVIR INJECTION is essential, especially for elderly patients and those patients receiving concomitant agents that may cause nephrotoxicity [see Dosage and Administration (2.5), Use in Specific Populations (8.5)].
5.3 Impairment of Fertility
Based on animal data, GANCICLOVIR INJECTION at the recommended human dose (RHD) may cause temporary or permanent inhibition of spermatogenesis in males, and may cause suppression of fertility in females. Advise patients that fertility may be impaired with use of GANCICLOVIR INJECTION [see Use in Specific Population (8.1), Nonclinical Toxicology (13.1)].
5.4 Fetal Toxicity
GANCICLOVIR INJECTION may cause fetal toxicity when administered to pregnant women based on findings in animal studies. Systemic exposure of ganciclovir in animals at approximately 2 times the RHD caused fetal growth retardation, embryolethality, teratogenicity, and/or maternal toxicity. Teratogenic changes in animals included cleft palate, anophthalmia/microphthalmia, aplastic organs (kidney and pancreas), hydrocephaly and brachygnathia. Women of childbearing potential should be advised to use effective contraception during treatment and for at least 30 days following treatment with GANCICLOVIR INJECTION. Similarly, men should be advised to practice barrier contraception during and for at least 90 days following treatment with GANCICLOVIR INJECTION [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Hematologic Toxicity [see Warnings and Precautions (5.1)]
- Impairment of Renal Function [see Warnings and Precautions (5.2)]
- Impairment of Fertility [see Warnings and Precautions (5.3)]
- Fetal Toxicity [see Warnings and Precautions (5.4)]
- Mutagenesis and Carcinogenesis [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Patients with CMV Retinitis
Three controlled, randomized, phase 3 trials comparing intravenous ganciclovir and ganciclovir capsules for maintenance treatment of CMV retinitis have been completed. During these trials, 9% of subjects were prematurely discontinued because of adverse reactions. Selected adverse reactions and laboratory abnormalities reported during the conduct of these controlled trials are summarized in Table 2 and Table 3, respectively [see Clinical Studies (14)].
| Adverse Event | Maintenance Treatment Studies |
|
|---|---|---|
| Intravenous Ganciclovir (n=179) | Ganciclovir Capsules (n=326) |
|
| Fever | 48% | 38% |
| Diarrhea |
44% | 41% |
| Leukopenia | 41% | 29% |
| Anemia | 25% | 19% |
| Total catheter events: | 22% | 6% |
| Catheter infection | 9% | 4% |
| Catheter sepsis | 8% | 1% |
| Other catheter related events | 5% | 1% |
| Sepsis | 15% | 4% |
| Anorexia | 14% | 15% |
| Vomiting | 13% | 13% |
| Infection | 13% | 9% |
| Sweating | 12% | 11% |
| Chills | 10% | 7% |
| Neuropathy | 9% | 8% |
| Thrombocytopenia | 6% | 6% |
| Pruritus | 5% | 6% |
Retinal Detachment
Retinal detachment has been observed in subjects with CMV retinitis both before and after initiation of therapy with ganciclovir. Its relationship to therapy with ganciclovir is unknown. Retinal detachment occurred in 11% of patients treated with intravenous ganciclovir and in 8% of patients treated with ganciclovir capsules.
| Laboratory Abnormalities | CMV Retinitis Treatment* | |
|---|---|---|
| Intravenous Ganciclovir† 5mg/kg/day (N=175) % | Ganciclovir Capsules‡ 3000mg/day (N=320) % |
|
|
Neutropenia with Absolute Neutrophil Count (ANC) per µL: | ||
| <500 |
25% | 18% |
| 500 - <749 | 14% | 17% |
| 750 - <1000 | 16% | 19% |
| Anemia with Hemoglobin (g/dL): | ||
| <6.5 | 5% | 2% |
| 6.5 - <8.0 | 16% | 10% |
| 8.0 - <9.5 | 26% | 25% |
| Serum Creatinine (mg/dL): | ||
| ≥2.5 | 2% | 1% |
| ≥1.5 - <2.5 | 14% | 12% |
* Pooled data from treatment studies ICM 1653, ICM 1774, and AVI 034
† Mean time on therapy = 103 days, including allowed re-induction treatment periods
‡ Mean time on therapy = 91 days, including allowed re-induction treatment periods
Adverse Reactions in Transplant Recipients
There have been three controlled clinical trials of intravenous ganciclovir for the prevention of CMV disease in transplant recipients. Selected laboratory abnormalities are summarized in Tables 4 and 5 below. Table 4 shows the frequency of neutropenia and thrombocytopenia and Table 5 shows the frequency of elevated serum creatinine values observed in these trials [see Clinical Studies (14)].
| Laboratory Abnormalities | Heart Allograft* | Bone Marrow Allograft† | ||
|---|---|---|---|---|
| Intravenous Ganciclovir (N=76) | Placebo (n=73) | Intravenous Ganciclovir (n=57) | Control (n=55) |
|
| Neutropenia | ||||
|
Absolute Neutrophil Count (ANC) per µL: | ||||
| <500 |
4% | 3% | 12% | 6% |
| <500-1000 | 3% | 8% | 29% | 17% |
|
TOTAL ANC ≤1000/µL | 7% | 11% | 41% | 23% |
| Thrombocytopenia | ||||
| Platelet count per µL: | ||||
| <25,000 | 3% | 1% | 32% | 28% |
| 25,000-50,000 | 5% | 3% | 25% | 37% |
| TOTAL Platelet Count ≤50,000/µL | 8% | 4% | 57% | 65% |
| Serum Creatinine Levels (mg/dL) | Heart Allograft ICM 1496 | Bone Marrow Allograft ICM 1570 | Bone Marrow Allograft ICM 1689 |
|||
|---|---|---|---|---|---|---|
| Intravenous
Ganciclovir (N=76) | Placebo (n=73) | Intravenous Ganciclovir (n=20) | Placebo (n=73) | Intravenous Ganciclovir (n=37) | Placebo (n=35) |
|
|
≥2.5 | 18% | 14% | 20% | 0% | 0% | 0% |
| ≥1.5 - <2.5 | 58% | 69% | 50% | 35% | 43% | 44% |
Other Adverse Reactions in Clinical Trials in Patients with CMV Retinitis and in Transplant Recipients
Other adverse reactions with intravenous ganciclovir or ganciclovir capsules in controlled clinical studies in either subjects with AIDS or transplant recipients are listed below [see Clinical Studies (14)]. All these events occurred in at least 3 subjects.
Ear and labyrinth disorders: tinnitus
Eye disorders: abnormal vision, vitreous disorder
Gastrointestinal disorders: enlarged abdomen, aphthous stomatitis, constipation, dyspepsia, eructation, gastrointestinal perforation, pancreatitis, dry mouth
General disorders and administration site conditions: asthenia, injection site inflammation, edema, malaise, multiple organ failure, pain, chest pain
Blood and lymphatic system disorders: pancytopenia
Infections and infestations: sepsis
Investigations: abnormal liver function test, decreased creatinine clearance
Metabolism and nutritional disorders: weight loss
Musculoskeletal and connective tissue disorders: arthralgia, leg cramps, myalgia, myasthenia
Renal and urinary disorders: kidney failure, abnormal kidney function, urinary frequency
Respiratory, thoracic and mediastinal disorders: increased cough, dyspnea
Nervous system disorders: confusion, dizziness, headache, insomnia, seizures, somnolence, abnormal thinking, tremor, taste perversion
Psychiatric disorders: abnormal dreams, anxiety, depression
Skin and subcutaneous disorders: alopecia, dry skin
Vascular disorders: hypertension, phlebitis, vasodilation
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of intravenous ganciclovir. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: hemolytic anemia
Cardiac disorders: cardiac arrest, cardiac conduction abnormality, torsades de pointes, ventricular tachycardia
Congenital, familial and genetic disorders: congenital anomaly
Eye disorders: cataracts, dry eyes
Gastrointestinal disorders: intestinal ulceration
Hepatobiliary disorders: hepatic failure, cholestasis, cholelithiasis, hepatitis
Immune system disorders: anaphylactic reaction, allergic reaction, vasculitis
Investigations: increased blood triglycerides, increased blood antidiuretic hormone
Metabolism and nutrition disorders: acidosis, hypercalcemia, hyponatremia
Musculoskeletal and connective tissue disorders: arthritis, rhabdomyolysis
Nervous system disorders: dysesthesia, dysphasia, encephalopathy, extrapyramidal disorder, facial palsy, intracranial hypertension, loss of memory, loss of smell, myelopathy, oculomotor nerve paralysis, stroke
Renal and urinary disorders: hemolytic uremic syndrome, renal tubular disorder
Reproductive system and breast disorders: infertility, testicular hypotrophy
Respiratory, thoracic and mediastinal disorders: bronchospasm, pulmonary fibrosis
Psychiatric disorders: hallucinations, irritability
Skin and subcutaneous tissue disorders: exfoliative dermatitis, Stevens-Johnson syndrome
Vascular disorders: peripheral ischemia
7 DRUG INTERACTIONS
Established and other potentially significant drug interactions conducted with ganciclovir are listed in Table 6 [see Clinical Pharmacology (12.3)].
| Name of the Concomitant Drug | Change in the Concentration of Ganciclovir or Concomitant Drug | Clinical Comment |
|---|---|---|
| Didanosine | ↑ Didanosine |
Patients should be closely monitored for didanosine toxicity. |
| Zidovudine | ↓ Ganciclovir ↑ Zidovudine | Dose reduction or interruption may be needed because both zidovudine and ganciclovir have the potential to cause neutropenia and anemia. Monitor with frequent tests of white blood cell counts with differential and hemoglobin levels. |
| Probenecid | ↑ Ganciclovir | GANCICLOVIR INJECTION dose may need to be reduced. Monitor for evidence of ganciclovir toxicity. |
| Imipenem-cilastatin | Unknown |
Coadministration with imipenem-cilastatin is not recommended because generalized seizures have been reported in patients who received ganciclovir and imipenem-cilastatin. |
| Cyclosporine or amphotericin B | Unknown | Monitor renal function when GANCICLOVIR INJECTION is co-administered with cyclosporine or amphotericin B because of potential increase in serum creatinine [see Warnings and Precautions (5.2)]. |
| Dapsone, pentamidine, flucytosine, vincristine, vinblastine, adriamycin, amphotericin B, trimethoprim/sulfamethoxazole combinations or other nucleoside analogues | Unknown | Co-administration with GANCICLOVIR INJECTION should be considered only if the potential benefits are judged to outweigh the risks because of potential additive toxicity. |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Inanimal studies, ganciclovir caused maternal and fetal toxicity and embryo-fetal mortality in pregnant mice and rabbits as well as teratogenicity in rabbits at exposures two times the exposure at the recommended human dose (RHD) [see Data]. Although placental transfer of ganciclovir has been shown to occur based on ex vivo experiments with human placenta and on at least one case report in a pregnant woman, no adequate human data are available to establish whether GANCICLOVIR INJECTION poses a risk to pregnancy outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in the clinically recognized pregnancies is 2-4% and 15−20%, respectively.
Clinical considerations
Disease-associated maternal and/or fetal risk
Most maternal CMV infections are subclinical or they may be associated with a mononucleosis-like syndrome. However, in immunocompromised patients, CMV infections are often symptomatic and are associated with significant morbidity and mortality. The transmission of CMV to the fetus is a result of maternal viremia and transplacental infection. CMV infection can also occur perinatally from mother to infant by exposure to CMV in cervicovaginal secretions. Approximately 10% of infected newborns are symptomatic at birth. Mortality in symptomatic infants is about 10%, and approximately 50 to 90% of survivors experience significant problems, including sensorineural hearing loss, mental retardation, and other neurologic deficits. The risk and severity of congenital CMV infection appear to be higher in infants born to mothers with primary CMV infection than in those born to mothers with reactivation of CMV infection.
Data
Animal Data
Daily intravenous doses of ganciclovir were administered to pregnant mice (108 mg/kg/day) and rabbits (60 mg/kg/day), and also to female mice (90 mg/kg) prior to mating, during gestation, and during lactation. Fetal resorptions were present in at least 85% of rabbits and mice. Additional effects observed in rabbits included fetal growth retardation, embryolethality, teratogenicity, and/or maternal toxicity. Teratogenic changes included cleft palate, anophthalmia/microphthalmia, aplastic organs (kidney and pancreas), hydrocephaly and brachygnathia. In pre/postnatal development studies in mice, there were maternal/fetal toxicity and embryolethality which included fetal effects of hypoplasia of the testes and seminal vesicles in the male offspring, as well as pathologic changes in the nonglandular region of the stomach. The systemic exposure (AUC) of ganciclovir during these studies was approximately 2-times (pregnant mice and rabbits) and 1.7-times (pre/postnatal mice) the exposure in humans at the RHD [see Nonclinical Toxicology (13.1)].
8.2 Lactation
Risk Summary
No data are available regarding the presence of ganciclovir in human milk, the effects on the breastfed infant, or the effects on milk production. When ganciclovir was administered to lactating rats, ganciclovir was present in milk [see Data]. Advise nursing mothers that breastfeeding is not recommended during treatment with GANCICLOVIR INJECTION because of the potential for serious adverse reactions in nursing infants. Furthermore, the Centers for Disease Control and Prevention recommends that HIV-infected mothers not breastfeed their infants to avoid potential postnatal transmission of HIV [see Warnings and Precautions (5.1, 5.3, 5.5), Nonclinical Toxicology (13.1)].
Data
Animal Data
Ganciclovir administered intravenously (at 0.13 mg/h) to lactating rats (on lactation day 15) resulted in passive transfer into milk. The milk-to-serum ratio for ganciclovir at steady state was 1.6 ± 0.33.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Females of reproductive potential should undergo pregnancy testing before initiation of GANCICLOVIR INJECTION [see Dosage and Administration (2.2), Use in Specific Populations (8.1)].
Contraception
Females
Because of the mutagenic and teratogenic potential of ganciclovir, females of reproductive potential should be advised to use effective contraception during treatment and for at least 30 days following treatment with GANCICLOVIR INJECTION [see Warnings and Precautions (5.3, 5.4), Nonclinical Toxicology (13.1)].
Males
Because of its mutagenic potential, males should be advised to practice barrier contraception during and for at least 90 days following, treatment with GANCICLOVIR INJECTION [see Warnings and Precautions (5.3, 5.4), Nonclinical Toxicology (13.1)].
Infertility
GANCICLOVIR INJECTION at the recommended doses may cause temporary or permanent female and male infertility [see Warnings and Precautions (5.3, 5.4), Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and efficacy of GANCICLOVIR INJECTION have not been established in pediatric patients.
A total of 120 pediatric patients with serious CMV infections participated in clinical trials. Granulocytopenia and thrombocytopenia were the most common adverse reactions.
The pharmacokinetic characteristics of ganciclovir administered intravenously were studied in 27 neonates (aged 2 to 49 days) and 10 pediatric patients, aged 9 months to 12 years. In neonates, the pharmacokinetic parameters after ganciclovir intravenous doses of 4 mg/kg (n=14) and 6 mg/kg (n=13) were Cmax 5.5 ± 1.6 and 7.0 ± 1.6 μg/mL, systemic clearance 3.14 ± 1.75 and 3.56 ± 1.27 mL/min/kg, and t1/2 of 2.4 hours (harmonic mean) for both doses, respectively.
In pediatric patients 9 months to 12 years of age, the pharmacokinetic characteristics of ganciclovir were the same after single and multiple (every 12 hours) intravenous doses (5 mg/kg). The steady-state volume of distribution was 0.64 ± 0.22 L/kg, Cmax was 7.9 ± 3.9 μg/mL, systemic clearance was 4.7 ± 2.2 mL/min/kg, and t1/2 was 2.4 ± 0.7 hours.
Although the pharmacokinetics of intravenous ganciclovir in pediatric patients were similar to those observed in adults, the safety and efficacy of ganciclovir at these exposures in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of ganciclovir did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Ganciclovir is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection. In addition, renal function should be monitored and dosage adjustments should be made accordingly [see Dosage and Administration (2.5), Warnings and Precautions (5.2), Use in Specific Populations (8.6)].
8.6 Renal Impairment
Dose reduction is recommended when administering GANCICLOVIR INJECTION to patients with renal impairment [see Dosage and Administration (2.5), Warnings and Precautions (5.2), Adverse Reactions (6.1)].
Hemodialysis has been shown to reduce plasma levels of ganciclovir by approximately 50%.
10 OVERDOSAGE
Adverse reactions reported with overdosage of intravenous ganciclovir included irreversible pancytopenia, acute renal failure requiring hemodialysis, neutropenia, thrombocytopenia, hepatitis, and seizures.
Since ganciclovir is dialyzable, dialysis may be useful in reducing serum concentrations. Adequate hydration should be maintained. The use of hematopoietic growth factors should be considered in patients with cytopenias [see Dosage and Administration (2.5), Warnings and Precautions (5.1)].
11 DESCRIPTION
GANCICLOVIR INJECTION, 500 mg is a sterile, unpreserved solution for intravenous administration. The appearance of the solution is clear and colorless. Each mL contains 2.0 mg of ganciclovir, 8.0 mg of sodium chloride in water for injection, and may contain sodium hydroxide, and/or hydrochloric acid, as required to adjust the pH to 7.5.
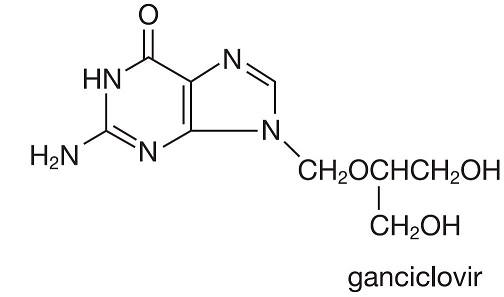
Ganciclovir, an antiviral agent, is a synthetic guanine derivative, 9-[[2-hydroxy-1-(hydroxymethyl)- ethoxy]methyl]guanine. Ganciclovir is a white to off-white crystalline powder with a molecular formula of C9H13 N5 04 and a molecular weight of 255.23. Ganciclovir is a polar hydrophilic compound with a solubility of 2.6 mg/mL in water at 25°C and an n-octanol/water partition coefficient of 0.022. The pKas for ganciclovir are 2.2 and 9.4.
The chemical structure of ganciclovir is:
The plastic container is fabricated from a multilayer film designed for medical use. The solution is in contact with the inner polypropylene layer of the container. No components of the plastic container material were found to migrate into the solution.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ganciclovir is an antiviral drug with activity against cytomegalovirus (CMV) [see Microbiology (12.4)].
12.3 Pharmacokinetics
Absorption:
At the end of a 1-hour intravenous infusion of 5 mg/kg ganciclovir, total AUC ranged between 22.1 ±3.2 (n=16) and 26.8 ± 6.1 g·hr/mL (n=16) and Cmax ranged between 8.27 ± 1.02 (n=16) and 9.0 ± 1.4 g/mL (n=16).
Distribution:
The steady-state volume of distribution of ganciclovir after intravenous administration was 0.74 ± 0.15 L/kg (n=98). Cerebrospinal fluid concentrations obtained 0.25 to 5.67 hours post-dose in 3 patients who received 2.5 mg/kg ganciclovir intravenously every 8 hours or every 12 hours ranged from 0.31 to 0.68 g/mL representing 24% to 70% of the respective plasma concentrations. Binding to plasma proteins was 1% to 2% over ganciclovir concentrations of 0.5 and 51 g/mL.
Elimination:
Renal excretion of unchanged drug by glomerular filtration and active tubular secretion is the major route of elimination of ganciclovir. In patients with normal renal function, 91.3 ± 5.0% (n=4) of intravenously administered ganciclovir was recovered unchanged in the urine. Systemic clearance of intravenously administered ganciclovir was 3.52 ± 0.80 mL/min/kg (n=98) while renal clearance was 3.20 ± 0.80 mL/min/kg (n=47), accounting for 91 ± 11% of the systemic clearance (n=47). Following intravenous administration, ganciclovir half-life was 3.5 ± 0.9 hours (n=98) with linear pharmacokinetics over the dose range of 1.6 to 5.0 mg/kg.
Specific Populations
Patients with Renal Impairment
The pharmacokinetics of ganciclovir following intravenous administration of ganciclovir were evaluated in 10 immunocompromised patients with renal impairment who received doses ranging from 1.25 to 5.0 mg/kg (Table 7). Hemodialysis reduces plasma concentrations of ganciclovir by about 50% after intravenous administration.
|
Estimated Creatinine Clearance (mL/min) |
n | Dose |
Clearance (mL/min) Mean ± SD |
Half-life (hours) Mean ± SD |
| 50-79 | 4 | 3.2-5 mg/kg | 128 ± 63 | 4.6 ± 1.4 |
| 25-49 |
3 | 3-5 mg/kg | 57 ± 8 | 4.4 ± 0.4 |
| <25 | 3 | 1.25-5 mg/kg | 30 ± 13 | 10.7 ± 5.7 |
Race (Ethnicity) and Gender
The effects of race/ethnicity and gender were studied in subjects receiving a dose regimen of 1000 mg every 8 hours. Although the numbers of Blacks (16%) and Hispanics (20%) were small, there appeared to be a trend towards a lower steady-state Cmax and AUC0-8 in these subpopulations as compared to Caucasians. No definitive conclusions regarding gender differences could be made because of the small number of females (12%); however, no differences between males and females were observed.
Drug Interactions
Didanosine
When the standard intravenous ganciclovir induction dose (5 mg/kg infused over 1 hour every 12 hours) was coadministered with didanosine at a dose of 200 mg orally every 12 hours, the steady-state didanosine AUC0-12 increased 70 ± 40% (range: 3% to 121%, n=11) and Cmax increased 49 ± 48% (range: -28% to 125%). In a separate study, when the standard intravenous ganciclovir maintenance dose (5 mg/kg infused over 1 hour every 24 hours) was coadministered with didanosine at a dose of 200 mg orally every 12 hours, didanosine AUC0-12 increased 50 ± 26% (range: 22% to 110%, n=11) and Cmax increased 36 ± 36% (range: -27% to 94%) over the first didanosine dosing interval. Ganciclovir pharmacokinetics were not affected by didanosine. In both studies, there was no significant change in the renal clearance of either drug.
Zidovudine
At an oral dose of 1000 mg of ganciclovir every 8 hours, mean steady-state ganciclovir AUC0-8
decreased 17 ± 25% (range: -52% to 23%) in the presence of zidovudine, 100 mg every 4 hours (n=12). Steady-state zidovudine AUC0-4 increased 19 ± 27% (range: -11% to 74%) in the presence of ganciclovir. No drug-drug interaction studies have been conducted with intravenous ganciclovir and zidovudine.
Probenecid
At an oral dose of 1000 mg of ganciclovir every 8 hours (n=10), ganciclovir AUC0-8 increased 53 ± 91% (range: -14% to 299%) in the presence of probenecid, 500 mg every 6 hours. Renal clearance of ganciclovir decreased 22 ± 20% (range: -54% to -4%), which is consistent with an interaction involving competition for renal tubular secretion. No drug-drug interaction studies have been conducted with intravenous ganciclovir and probenecid.
Cyclosporine
In a retrospective analysis of 93 liver allograft recipients receiving ganciclovir (5 mg/kg infused over 1 hour every 12 hours) and oral cyclosporine (at therapeutic doses), there was no evidence of an effect on cyclosporine whole blood concentrations.
12.4 Microbiology
Antiviral Activity
Thciclovir is a synthetic analogue of 2'-deoxyguanosine, which inhibits replication of human CMV in cell culture and in vivo. In CMV-infected cells, ganciclovir is initially phosphorylated to ganciclovir monophosphate by the viral protein kinase, pUL97. Further phosphorylation occurs by cellular kinases to produce ganciclovir triphosphate, which is then slowly metabolized intracellularly. As the phosphorylation is largely dependent on the viral kinase, phosphorylation of ganciclovir occurs preferentially in virus-infected cells. The virustatic activity of ganciclovir is due to inhibition of the viral DNA polymerase, pUL54, by ganciclovir triphosphate.
Antiviral Activity
The median concentration of ganciclovir that inhibits CMV replication (EC50 value) in cell culture (laboratory strains or clinical isolates) has ranged from 0.08 to 13.6 micromolar (0.02 to 3.48 μg/mL). Ganciclovir inhibits mammalian cell proliferation (CC50 value) in cell culture at higher concentrations ranging from 118 to 2840 micromolar (30 to 725 μg/mL). Bone marrow-derived colony-forming cells are more sensitive [CC50 value = 0.1 to 2.7 micromolar (0.028 to 0.7 μg/mL)]. The relationship between the antiviral activity in cell culture and clinical response has not been established.
Resistance
Cell Culture: CMV isolates with reduced susceptibility to ganciclovir have been selected in cell culture. Growth of CMV strains in the presence of ganciclovir resulted in the selection of amino acid substitutions in the viral protein kinase pUL97 and the viral DNA polymerase pUL54.
In vivo: Viruses resistant to ganciclovir can arise after prolonged treatment or prophylaxis with ganciclovir by selection of substitutions in pUL97 and/or pUL54. Limited clinical data are available on the development of clinical resistance to ganciclovir and many pathways to resistance exist. The possibility of viral resistance should be considered in patients who show poor clinical response or experience persistent viral excretion during therapy.
CMV resistance to ganciclovir has been observed in individuals with AIDS and CMV retinitis who have never received ganciclovir therapy. Viral resistance has also been observed in patients receiving prolonged treatment for CMV retinitis with ganciclovir. In a controlled study of oral ganciclovir for prevention of AIDS-associated CMV disease, 364 individuals had one or more cultures performed after at least 90 days of ganciclovir treatment. Of these, 113 had at least one positive culture. The last available isolate from each subject was tested for reduced sensitivity, and 2 of 40 were found to be resistant to ganciclovir. These resistant isolates were associated with subsequent treatment failure for retinitis.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis, Mutagenesis
Ganciclovir was carcinogenic in mice at the same mean drug exposure in humans as at the RHD (5 mg/kg). At the dose of 1000 mg/kg/day (1.4 times the exposure at the RHD) there was a significant increase in the incidence of tumors of the preputial gland in males, forestomach (nonglandular mucosa) in males and females, and reproductive tissues (ovaries, uterus, mammary gland, clitoral gland and vagina) and liver in females. At the dose of 20 mg/kg/day (0.1 times the exposure at the RHD), a slightly increased incidence of tumors was noted in the preputial and harderian glands in males, forestomach in males and females, and liver in females. No carcinogenic effect was observed in mice administered ganciclovir at 1 mg/kg/day (exposure estimated as 0.01 times the RHD). Except for histiocytic sarcoma of the liver, ganciclovir-induced tumors were generally of epithelial or vascular origin. Although the preputial and clitoral glands, forestomach and harderian glands of mice do not have human counterparts, ganciclovir should be considered a potential carcinogen in humans.
Ganciclovir increased mutations in mouse lymphoma cells and DNA damage in human lymphocytes in vitro at concentrations of 50 to 500 and 250 to 2000 μg/mL, respectively. In the mouse micronucleus assay, ganciclovir was clastogenic at doses of 150 and 500 mg/kg (2.8 to 10 times the exposure at the RHD) but not at doses of 50 mg/kg (exposure approximately comparable to the RHD). Ganciclovir was not mutagenic in the Ames Salmonella assay at concentrations of 500 to 5000 μg/mL.
Impairment of Fertility
Ganciclovir caused decreased mating behavior, decreased fertility, and an increased incidence of embryolethality in female mice following doses of 90 mg/kg/day (exposures approximately 1.7 times the RHD). Ganciclovir caused decreased fertility in male mice and hypospermatogenesis in mice and dogs following daily oral or intravenous administration of doses ranging from 0.2 to 10 mg/kg. Systemic drug exposure (AUC) at the lowest dose showing toxicity in each species ranged from 0.03 to 0.1 times the exposure at the RHD.
14 CLINICAL STUDIES
14.1 Treatment of CMV Retinitis
In a retrospective, non-randomized, single-center analysis of 41 patients with AIDS and CMV retinitis diagnosed by ophthalmologic examination between August 1983 and April 1988, treatment with intravenous ganciclovir resulted in a delay in mean (median) time to first retinitis progression compared to untreated controls [105 (71) days from diagnosis vs. 35 (29) days from diagnosis]. Patients in this series received induction treatment of intravenous ganciclovir 5 mg/kg twice daily for 14 to 21 days followed by maintenance treatment with either 5 mg/kg once daily, 7 days per week or 6 mg/kg once daily, 5 days per week.
In a controlled, randomized trial conducted between February 1989 and December 1990, immediate treatment with intravenous ganciclovir was compared to delayed treatment in 42 patients with AIDS and peripheral CMV retinitis; 35 of 42 patients (13 in the immediate-treatment group and 22 in the delayed-treatment group) were included in the analysis of time to retinitis progression. Based on masked assessment of fundus photographs, the mean [95% CI] and median [95% CI] times to progression of retinitis were 66 days [39, 94] and 50 days [40, 84], respectively, in the immediate-treatment group compared to 19 days [11, 27] and 13.5 days [8, 18], respectively, in the delayed-treatment group.
Data from trials ICM 1653, ICM 1774, and AVI034, which were performed comparing intravenous to oral ganciclovir for treatment of CMV retinitis in patients with AIDS, are shown in Table 8, and Figures 1, 2, and 3, and are discussed below.
| ICM 1653
(n=121) | ICM 1774
(n=225) | AVI 034
(n=159) |
||
| Median age (years) | 38 | 37 | 39 | |
| Range | 24-62 | 22-56 | 23-62 | |
| Sex | Males | 116 (96%) | 222 (99%) | 148 (93%) |
| Females | 5 (4%) | 3 (1%) | 10 (6%) | |
| Asian | 3 (3%) | 5 (2%) | 7 (4%) | |
| Ethnicity | Black | 11 (9%) | 9 (4%) | 3 (2%) |
| Caucasian | 98 (81%) | 186 (83%) | 140 (88%) | |
| Other | 9 (7%) | 25 (11%) | 8 (5%) | |
| Median CD4 Count | 9.5 | 7.0 | 10.0 | |
| Range | 0-141 | 0-80 | 0-320 | |
| Mean (SD) Observation Time (days) | 107.9 (43.0) | 97.6 (42.5) | 80.9 (47.0) | |
Trial ICM 1653
In this randomized, open-label, parallel group trial, conducted between March 1991 and November1992, patients with AIDS and newly diagnosed CMV retinitis received a 3-week induction course of intravenous ganciclovir, 5 mg/kg twice daily for 14 days followed by 5 mg/kg once daily for 1 additional week. Following the 21-day intravenous induction course, patients with stable CMV retinitis were randomized to receive 20 weeks of maintenance treatment with either intravenous ganciclovir, 5 mg/kg once daily, or ganciclovir capsules, 500 mg 6 times daily (3000 mg/day). The study showed that the mean [95% CI] and median [95% CI] times to progression of CMV retinitis, as assessed by masked reading of fundus photographs, were 57 days [44, 70] and 29 days [28, 43], respectively, for patients on oral therapy compared to 62 days [50, 73] and 49 days [29, 61], respectively, for patients on intravenous therapy. The difference [95% CI] in the mean time to progression between the oral and intravenous therapies (oral - IV) was -5 days [-22, 12]. See Figure 1 for comparison of the proportion of patients remaining free of progression over time.
Trial ICM 1774
In this three-arm, randomized, open-label, parallel group trial, conducted between June 1991 and August 1993, patients with AIDS and stable CMV retinitis following from 4 weeks to 4 months of treatment with intravenous ganciclovir were randomized to receive maintenance treatment with intravenous ganciclovir, 5 mg/kg once daily, ganciclovir capsules, 500 mg 6 times daily, or ganciclovir capsules, 1000 mg three times daily for 20 weeks. The study showed that the mean [95% CI] and median [95% CI] times to progression of CMV retinitis, as assessed by masked reading of fundus photographs, were 54 days [48, 60] and 42 days [31, 54], respectively, for patients on oral therapy compared to 66 days [56, 76] and 54 days [41, 69], respectively, for patients on intravenous therapy. The difference [95% CI] in the mean time to progression between the oral and intravenous therapies (oral - IV) was -12 days [-24, 0]. See Figure 2 for comparison of the proportion of patients remaining free of progression over time.
Trial AVI 034
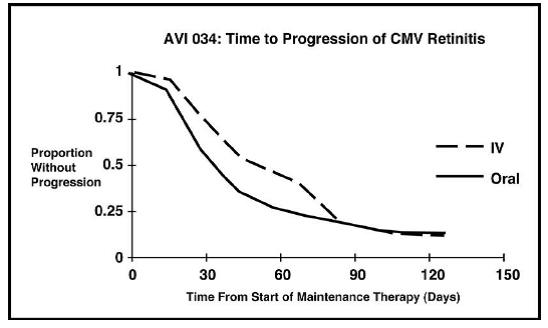
In this randomized, open-label, parallel group trial, conducted between June 1991 and February 1993, patients with AIDS and newly diagnosed (81%) or previously treated (19%) CMV retinitis who had tolerated 10 to 21 days of induction treatment with intravenous ganciclovir, 5 mg/kg twice daily, were randomized to receive 20 weeks of maintenance treatment with either ganciclovir capsules, 500 mg 6 times daily or intravenous ganciclovir, 5 mg/kg/day. The mean [95% CI] and median [95% CI] times to progression of CMV retinitis, as assessed by masked reading of fundus photographs, were 51 days [44, 57] and 41 days [31, 45], respectively, for patients on oral therapy compared to 62 days [52, 72] and 60 days [42, 83], respectively, for patients on intravenous therapy. The difference [95% CI] in the mean time to progression between the oral and intravenous therapies (oral - IV) was -11 days [-24, 1]. See Figure 3 for comparison of the proportion of patients remaining free of progression over time.
Comparison of other CMV retinitis outcomes between oral and intravenous formulations (development of bilateral retinitis, progression into Zone 1, and deterioration of visual acuity), while not definitive, showed no marked differences between treatment groups in these studies. Because of low event rates among these endpoints, these studies are underpowered to rule out significant differences in these endpoints.
Figure 1. ICM 1653: Time to Progression of CMV Retinitis
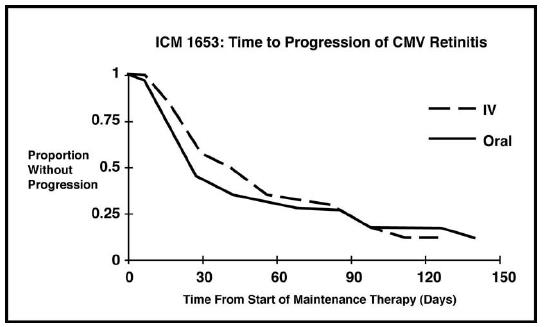
Figure 2. ICM 1774: Time to Progression of CMV Retinitis
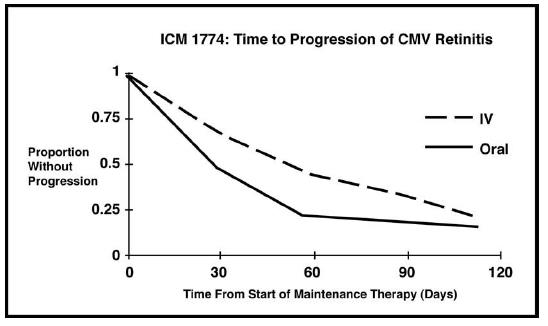
Figure 3. AVI 034: Time to Progression of CMV Retinitis
14.2 Prevention of CMV Disease in Transplant Recipients
Intravenous ganciclovir was evaluated in three randomized, controlled trials of prevention of CMV disease in organ transplant recipients.
Trial ICM 1496
In a randomized, double-blind, placebo-controlled study of 149 heart transplant recipients at risk for CMV infection (CMV seropositive or a seronegative recipient of an organ from a CMV seropositive donor), there was a reduction in the overall incidence of CMV disease in patients treated with intravenous ganciclovir. Immediately post-transplant, patients received intravenous ganciclovir 5 mg/kg twice daily for 14 days followed by 6 mg/kg once daily for 5 days/week for an additional 14 days. Twelve of the 76 (16%) patients treated with intravenous ganciclovir vs. 31 of the 73 (43%) placebo-treated patients developed CMV disease during the 120-day post-transplant observation period. No significant differences in hematologic toxicities were seen between the two treatment groups [see Adverse Reactions (6.1)].
Trial ICM 1689
In a randomized, double-blind, placebo-controlled study of 72 bone marrow transplant recipients with asymptomatic CMV infection (CMV positive culture of urine, throat or blood) there was a reduction in the incidence of CMV disease in patients treated with intravenous ganciclovir following successful hematopoietic engraftment. Patients with virologic evidence of CMV infection received intravenous ganciclovir 5 mg/kg twice daily for 7 days followed by 5 mg/kg once daily through day 100 post-transplant. One of the 37 (3%) patients treated with intravenous ganciclovir vs 15 of the 35 (43%) placebo-treated patients developed CMV disease during the study. At 6 months post-transplant, there continued to be a reduction in the incidence of CMV disease in patients treated with intravenous ganciclovir. Six of 37 (16%) patients treated with intravenous ganciclovir vs 15 of the 35 (43%) placebo-treated patients developed disease through 6 months post-transplant. The overall rate of survival was higher in the group treated with intravenous ganciclovir, both at day 100 and day 180 post-transplant. Although the differences in hematologic toxicities were not statistically significant, the incidence of neutropenia was higher in the group treated with intravenous ganciclovir [seeDosage and Administration (2.4), Adverse Reactions (6.1)].
Trial ICM 1570
This was a randomized, unblinded trial which evaluated 40 allogeneic bone marrow transplant recipients at risk for CMV disease. Patients underwent bronchoscopy and bronchoalveolar lavage (BAL) on day 35 post-transplant. Patients with histologic, immunologic or virologic evidence of CMV infection in the lung were then randomized to observation or treatment with intravenous ganciclovir (5 mg/kg twice daily for 14 days followed by 5 mg/kg once daily 5 days/week until day 120). Four of 20 (20%) patients treated with intravenous ganciclovir and 14 of 20 (70%) control patients developed interstitial pneumonia. The incidence of CMV disease was lower in the group treated with intravenous ganciclovir [see Dosage and Administration (2.4)].
16 HOW SUPPLIED/STORAGE AND HANDLING
GANCICLOVIR INJECTION is supplied as a sterile, unpreserved, colorless solution in a single-dose polymeric bag containing 500 mg ganciclovir in 250 mL of solution (2 mg/mL) sealed with a Twist Off port from Technoflex, and oversealed in an aluminum pouch (NDC 51754-2500-1), in cases of 10 (NDC 51754-2500-3). Follow guidelines for handling and disposal for cytotoxic drugs.1
The premix flexible plastic container bag contains no preservative; any unused portion should be discarded [see Dosage and Administration (2.6)].
Gently shaking should redissolve any crystals that may have formed during transportation and/or storage at temperatures lower than recommended. The solution must be clear at the time of use.
Storage
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Hematologic Toxicity
Inform patients of the potential for hematologic toxicity associated with the use of GANCICLOVIR INJECTION including granulocytopenia (neutropenia), anemia and thrombocytopenia. Inform patients that their blood counts will be closely monitored while on therapy [see Warnings and Precautions (5.1)].
Impairment of Renal Function
Inform patients that ganciclovir has been associated with decreased renal function and that serum creatinine or creatinine clearance will be monitored carefully to allow for dosage adjustment in patients with renal impairment [see Dosage and Administration (2.5), Warnings and Precautions (5.2)].
Impairment of Fertility
Inform patients that ganciclovir has caused decreased fertility in animals and may cause temporary or permanent infertility in humans [see Warnings and Precautions (5.3), Use in Specific Populations (8.3)].
Pregnancy and Contraception
Inform women of childbearing potential that ganciclovir causes birth defects in animals. Advise female patients to use effective contraception during treatment and for at least 30 days following treatment with GANCICLOVIR INJECTION. Similarly, advise men to practice barrier contraception during and for at least 90 days following treatment with GANCICLOVIR INJECTION [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
Carcinogenicity
Inform patients that ganciclovir causes tumors in animals. Although there is no information from human studies, ganciclovir should be considered a potential carcinogen [see Warnings and Precautions (5.5)].
Drug Interactions
Inform patients that GANCICLOVIR INJECTION may interact with other drugs. Advise patients to report to their healthcare provider the use of any other medication [see Drug Interactions (7)].
Ophthalmological Examination in Patients with CMV Retinitis
Advise patients with CMV retinitis to have frequent ophthalmological examinations while being treated with GANCICLOVIR INJECTION to monitor disease status and for other retinal abnormalities. More frequent ophthalmological follow-up may be needed in some cases [see Dosage and Administration (2.2), Adverse Reactions (6.1)].
Lactation
Advise nursing mothers that breastfeeding is not recommended during treatment with GANCICLOVIR INJECTION because of the potential for serious adverse events in nursing infants and because HIV can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].

