FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Bepotastine Besilate Ophthalmic Solution, 1.5% is a histamine H1 receptor antagonist indicated for the treatment of itching associated with signs and symptoms of allergic conjunctivitis.
2 DOSAGE AND ADMINISTRATION
Instill one drop of bepotastine besilate ophthalmic solution into the affected eye(s) twice a day.
Remove contact lenses prior to instillation of bepotastine besilate ophthalmic solution.
4 CONTRAINDICATIONS
Bepotastine besilate ophthalmic solution is contraindicated in patients with a history of hypersensitivity reactions to bepotastine or any of the other ingredients [see Adverse Reactions (6.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Contamination of Tip and Solution
To minimize contaminating the dropper tip and solution, advise the patient not to touch the eyelids or surrounding areas with the dropper tip of the bottle and to keep the bottle tightly closed when not in use.
5.2 Contact Lens Wear
Bepotastine besilate ophthalmic solution should not be used to treat contact lens-related irritation.
Bepotastine besilate ophthalmic solution should not be instilled while wearing contact lenses. Patient should remove contact lenses prior to instillation of bepotastine besilate ophthalmic solution, because benzalkonium chloride may be absorbed by soft contact lenses. Lenses may be reinserted after 10 minutes following administration of bepotastine besilate ophthalmic solution.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most common reported adverse reaction occurring in approximately 25% of subjects was a mild taste following instillation. Other adverse reactions occurring in 2 to 5% of subjects were eye irritation, headache, and nasopharyngitis.
6.2 Post-Marketing Experience
Hypersensitivity reactions have been reported rarely during the post-marketing use of bepotastine besilate ophthalmic solution. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The hypersensitivity reactions may include itching, body rash, and swelling of lips, tongue and/or throat.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available human data for the use of bepotastine besilate during pregnancy to inform any drug-associated risks.
Oral administration of bepotastine besilate to pregnant rats or rabbits during organogenesis or during the pre/postnatal period did not produce adverse embryofetal or offspring effects at clinically relevant systemic exposures. Maternal toxicity was observed in the rabbits at the lowest dose administered, 20 mg/kg/day (215 times the maximum recommended human ophthalmic dose, RHOD, on a mg/m2 basis) [see Data].
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4%, and of miscarriage is 15 to 20%, of clinically recognized pregnancies.
Data
Animal Data
In embryofetal development studies, oral administration of bepotastine besilate to pregnant rabbits throughout organogenesis did not produce teratogenic effects at maternal doses up to 500 mg/kg/day (approximately 5400 times the maximum RHOD, on a mg/m2 basis). A maternal no observed adverse effect level (NOAEL) was not identified in this study due to spontaneous abortion observed at the lowest dose tested, 20 mg/kg/day (approximately 215 times higher than the maximum RHOD, on a mg/m2 basis). Oral administration of bepotastine besilate to pregnant rats throughout organogenesis produced skeletal anomalies at 1000 mg/kg/day (5400 times higher than the maximum RHOD, on a mg/m2 basis), a dose that also produced maternal toxicity and lethality. No teratogenic effects were observed in rats at maternal doses up to 200 mg/kg/day (corresponding to an estimated blood plasma concentration 3300 times higher than that anticipated in humans at the maximum RHOD). A maternal NOAEL was observed at 10 mg/kg/day (54 times higher than the maximum RHOD, on a mg/m2 basis). Following a single 3 mg/kg oral dose in rats (16 times higher than the maximum RHOD, on a mg/m2 basis), the concentration of radio-labeled bepotastine besilate was similar in fetal liver and maternal blood plasma. The concentration in other fetal tissues was one-third to one-tenth the concentration in maternal blood plasma.
In a pre/postnatal development study, oral administration of bepotastine besilate to rats during the perinatal and lactation periods produced an increase in stillbirths and decreased growth and development in offspring at a maternal dose of 1000 mg/kg/day (5400 times higher than the maximum RHOD, on a mg/m2 basis). There were no observed adverse effects on offspring of rats treated with 100 mg/kg/day (540 times higher than the maximum RHOD, on a mg/m2 basis). Effects on parturition and maternal lethality were observed at 100 mg/kg/day and 1000 mg/kg/day, respectively. A maternal NOAEL was observed at 10 mg/kg/day (54 times higher than the maximum RHOD, on a mg/m2 basis).
8.2 Lactation
Risk Summary
There are no data on the presence of bepotastine besilate in human milk, the effects on the breastfed infant or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for bepotastine besilate, and any potential adverse effects on the breastfed infant from bepotastine besilate.
Animal Data
Following a single 3 mg/kg oral dose (16 times the maximum RHOD, on a mg/m2 basis) of radiolabeled bepotastine besilate to nursing rats 11 days after delivery, the maximum concentration of radioactivity in milk was 0.40 mcg-eq/mL 1 hour after administration; at 48 hours after administration, the radioactivity concentration was below detection limits. The milk radioactivity concentration was higher than the maternal blood plasma radioactivity concentration at each time of measurement. It is not known whether bepotastine besilate would be present in maternal milk following topical ocular administration.
8.4 Pediatric Use
Safety and efficacy of bepotastine besilate ophthalmic solution, 1.5% have not been established in pediatric patients under 2 years of age. Efficacy in pediatric patients under 10 years of age was extrapolated from clinical trials conducted in pediatric patients greater than 10 years of age and from adults.
11 DESCRIPTION
Bepotastine Besilate Ophthalmic Solution, 1.5% is a sterile, topically administered drug for ophthalmic use. Each mL of Bepotastine Besilate Ophthalmic Solution contains 15 mg bepotastine besilate. Bepotastine besilate is designated chemically as (+) -4-[[(S)-p-chloro-alpha -2-pyridylbenzyl]oxy]-1-piperidine butyric acid monobenzenesulfonate. The chemical structure for bepotastine besilate is:
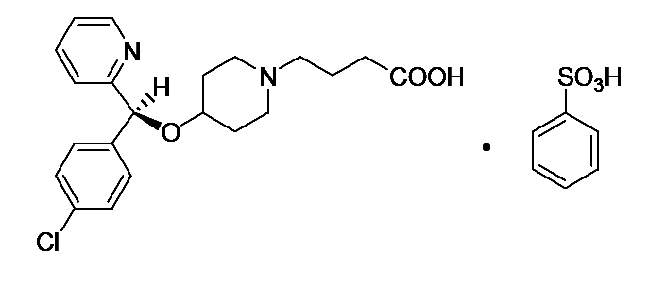
Bepotastine besilate is a white or creamish white crystalline powder. The molecular weight of bepotastine besilate is 547.06 daltons. Bepotastine Besilate Ophthalmic Solution is supplied as a sterile, aqueous 1.5% solution, with a pH of 6.8.
The osmolality of Bepotastine Besilate Ophthalmic Solution, 1.5% is approximately 290 mOsm/kg.
Each mL of Bepotastine Besilate Ophthalmic Solution, 1.5% contains:
Active: bepotastine besilate 15 mg (equivalent to 10.7 mg bepotastine)
Preservative: benzalkonium chloride 0.005%
Inactives: sodium chloride, sodium hydroxide to adjust pH, sodium phosphate monobasic dihydrate and water for injection.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bepotastine is a topically active, direct H1-receptor antagonist and an inhibitor of the release of histamine from mast cells.
12.3 Pharmacokinetics
Absorption: The extent of systemic exposure to bepotastine following topical ophthalmic administration of bepotastine besilate 1% and 1.5% ophthalmic solutions was evaluated in 12 healthy adults. Following one drop of 1% or 1.5% bepotastine besilate ophthalmic solution to both eyes four times daily (QID) for seven days, bepotastine plasma concentrations peaked at approximately one to two hours post-instillation. Maximum plasma concentrations for the 1% and 1.5% strengths were 5.1 ± 2.5 ng/mL and 7.3 ± 1.9 ng/mL, respectively. Plasma concentrations at 24 hours post-instillation were below the quantifiable limit (2 ng/mL) in 11/12 subjects in the two dose groups.
Distribution: The extent of protein binding of bepotastine is approximately 55% and independent of bepotastine concentration.
Metabolism: In vitro metabolism studies with human liver microsomes demonstrated that bepotastine is minimally metabolized by CYP450 isozymes.
In vitro studies demonstrated that bepotastine besilate does not inhibit the metabolism of various cytochrome P450 substrates via inhibition of CYP3A4, CYP2C9, and CYP2C19. The effect of bepotastine besilate on the metabolism of substrates of CYP1A2, CYP2C8 and CYP2D6 was not studied. Bepotastine besilate has a low potential for drug interaction via inhibition of CYP3A4, CYP2C9, and CYP2C19.
Excretion: The main route of elimination of bepotastine besilate is urinary excretion (with approximately 75 to 90% excreted unchanged in urine).
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term dietary studies in mice and rats were conducted to evaluate the carcinogenic potential of bepotastine besilate. Bepotastine besilate did not significantly induce neoplasms in mice receiving a nominal dose of up to 200 mg/kg/day for 21 months or in rats receiving a nominal dose of up to 97 mg/kg/day for 24 months. These dose levels correspond to systemic exposures approximately 350 and 200 times higher than that achieved at the RHOD, respectively.
The no observable adverse effect level for bepotastine besilate based on nominal dose levels in carcinogenicity tests were 18.7 to 19.9 mg/kg/day in mice and 9.6 to 9.8 mg/kg/day in rats (corresponding to systemic exposures approximately 60 and 20 times higher than that anticipated in humans at RHOD, respectively).
Mutagenesis
There was no evidence of genotoxicity in the Ames test (mutagenicity), in CHO cells (chromosome aberration), in mouse hepatocytes (unscheduled DNA synthesis), or in the mouse micronucleus test.
Impairment of Fertility
Oral administration of bepotastine to male and female rats at doses up to 1000 mg/kg/day (5400 times higher than the maximum RHOD, on a mg/m2 basis) resulted in reduction in fertility index and surviving fetuses. Oral administration of bepotastine besilate produced no observed adverse effects on fertility or reproduction in rats at oral doses up to 200 mg/kg/day (corresponding to an estimated blood plasma concentration 3300 times higher than that anticipated in humans at the RHOD).
14 CLINICAL STUDIES
Clinical efficacy was evaluated in two conjunctival allergen challenge (CAC) studies (237 patients). Bepotastine besilate ophthalmic solution, 1.5% was more effective than its vehicle for relieving ocular itching induced by an ocular allergen challenge, both at a CAC 15 minutes post-dosing and a CAC 8 hours post dosing of bepotastine besilate ophthalmic solution.
The safety of bepotastine besilate ophthalmic solution was evaluated in a randomized clinical study of 861 subjects over a period of 6 weeks.
16 HOW SUPPLIED/STORAGE AND HANDLING
Bepotastine Besilate Ophthalmic Solution, 1.5% is supplied in a white opaque low density polyethylene bottle with a white translucent low density polyethylene dropper and a white opaque high density polyethylene cap in the following sizes:
5 mL (NDC 60505-6111-1)
10 mL (NDC 60505-6111-2)
STORAGE
Store at 20ºC to 25ºC (68ºF to 77ºF) [See USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
- Sterility of Dropper Tip
Advise patients not to touch dropper tip to any surface, as this may contaminate the solution and to keep the bottle tightly closed when not in use.
- Concomitant Use of Contact Lenses
Advise patients not to wear a contact lens if their eye is red and bepotastine besilate ophthalmic solution should not be used to treat contact lens-related irritation. Patients should also be advised to remove contact lenses prior to instillation of bepotastine besilate ophthalmic solution, which may be reinserted after 10 minutes following administration.
APOTEX INC.
BEPOTASTINE BESILATE OPHTHALMIC SOLUTION
1.5%
| Manufactured by: Apotex Inc. Toronto, Ontario Canada M9L 1T9 | Manufactured for: Apotex Corp. Weston, FL USA 33326 |
Revised: February 2018

