USE IN PREGNANCY
When used in pregnancy during the second and third trimester, ACE inhibitors can cause injury and even death to the developing fetus. When pregnancy is detected, captopril should be discontinued as soon as possible. See WARNINGS: Fetal/Neonatal Morbidity and Mortality.
DESCRIPTION
Captopril is a specific competitive inhibitor of angiotensin I-converting enzyme (ACE), the enzyme responsible for the conversion of angiotensin I to angiotensin II.
Captopril is designated chemically as 1-[(2S)-3-mercapto-2-methylpropionyl]-L-proline and has the following structural formula:
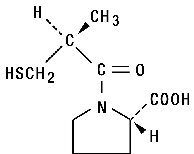
MW=217.29 C9H15NO3 S
Captopril is a white to off-white crystalline powder that may have a slight sulfurous odor; it is soluble in water (approx. 160 mg/mL), methanol, and ethanol and sparingly soluble in chloroform and ethyl acetate.
Each tablet, for oral administration, contains 12.5 mg, 25 mg, 50 mg, or 100 mg of captopril. In addition, each tablet contains the following inactive ingredients: anhydrous lactose, colloidal silicon dioxide, microcrystalline cellulose, sodium starch glycolate, starch, and stearic acid.
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of captopril has not yet been fully elucidated. Its beneficial effects in hypertension and heart failure appear to result primarily from suppression of the renin-angiotensin-aldosterone system. However, there is no consistent correlation between renin levels and response to the drug. Renin, an enzyme synthesized by the kidneys, is released into the circulation where it acts on a plasma globulin substrate to produce angiotensin I, a relatively inactive decapeptide. Angiotensin I is then converted by angiotensin converting enzyme (ACE) to angiotensin II, a potent endogenous vasoconstrictor substance. Angiotensin II also stimulates aldosterone secretion from the adrenal cortex, thereby contributing to sodium and fluid retention.
Captopril prevents the conversion of angiotensin I to angiotensin II by inhibition of ACE, a peptidyldipeptide carboxy hydrolase. This inhibition has been demonstrated in both healthy human subjects and in animals by showing that the elevation of blood pressure caused by exogenously administered angiotensin I was attenuated or abolished by captopril. In animal studies, captopril did not alter the pressor responses to a number of other agents, including angiotensin II and norepinephrine, indicating specificity of action.
ACE is identical to “bradykininase,” and captopril may also interfere with the degradation of the vasodepressor peptide, bradykinin. Increased concentrations of bradykinin or prostaglandin E2 may also have a role in the therapeutic effect of captopril.
Inhibition of ACE results in decreased plasma angiotensin II and increased plasma renin activity (PRA), the latter resulting from loss of negative feedback on renin release caused by reduction in angiotensin II. The reduction of angiotensin II leads to decreased aldosterone secretion, and, as a result, small increases in serum potassium may occur along with sodium and fluid loss.
The antihypertensive effects persist for a longer period of time than does demonstrable inhibition of circulating ACE. It is not known whether the ACE present in vascular endothelium is inhibited longer than the ACE in circulating blood.
Pharmacokinetics
After oral administration of therapeutic doses of captopril, rapid absorption occurs with peak blood levels at about one hour. The presence of food in the gastrointestinal tract reduces absorption by about 30 to 40 percent; captopril therefore should be given one hour before meals. Based on carbon-14 labeling, average minimal absorption is approximately 75 percent. In a 24-hour period, over 95 percent of the absorbed dose is eliminated in the urine; 40 to 50 percent is unchanged drug; most of the remainder is the disulfide dimer of captopril and captopril-cysteine disulfide.
Approximately 25 to 30 percent of the circulating drug is bound to plasma proteins. The apparent elimination half-life for total radioactivity in blood is probably less than 3 hours. An accurate determination of half-life of unchanged captopril is not, at present, possible, but it is probably less than 2 hours. In patients with renal impairment, however, retention of captopril occurs (see DOSAGE AND ADMINISTRATION).
Pharmacodynamics
Administration of captopril results in a reduction of peripheral arterial resistance in hypertensive patients with either no change, or an increase in cardiac output. There is an increase in renal blood flow following administration of captopril and glomerular filtration rate is usually unchanged.
Reductions of blood pressure are usually maximal 60 to 90 minutes after oral administration of an individual dose of captopril. The duration of effect is dose related. The reduction in blood pressure may be progressive, so to achieve maximal therapeutic effects, several weeks of therapy may be required. The blood pressure lowering effects of captopril and thiazide-type diuretics are additive. In contrast, captopril and beta-blockers have a less than additive effect.
Blood pressure is lowered to about the same extent in both standing and supine positions. Orthostatic effects and tachycardia are infrequent but may occur in volume-depleted patients. Abrupt withdrawal of captopril has not been associated with a rapid increase in blood pressure.
In patients with heart failure, significantly decreased peripheral (systemic vascular) resistance and blood pressure (afterload), reduced pulmonary capillary wedge pressure (preload) and pulmonary vascular resistance, increased cardiac output, and increased exercise tolerance time (ETT) have been demonstrated. These hemodynamic and clinical effects occur after the first dose and appear to persist for the duration of therapy. Placebo controlled studies of 12 week duration in patients who did not respond adequately to diuretics and digitalis show no tolerance to beneficial effects on ETT; open studies, with exposure up to 18 months in some cases, also indicate that ETT benefit is maintained. Clinical improvement has been observed in some patients where acute hemodynamic effects were minimal.
The Survival and Ventricular Enlargement (SAVE) study was a multicenter, randomized, double-blind, placebo-controlled trial conducted in 2,231 patients (age 21-79 years) who survived the acute phase of myocardial infarction and did not have active ischemia. Patients had left ventricular dysfunction (LVD), defined as a resting left ventricular ejection fraction ≤40%, but at the time of randomization were not sufficiently symptomatic to require ACE inhibitor therapy for heart failure. About half of the patients had had symptoms of heart failure in the past. Patients were given a test dose of 6.25 mg oral captopril and were randomized within 3-16 days post-infarction to receive either captopril or placebo in addition to conventional therapy. Captopril was initiated at 6.25 mg or 12.5 mg t.i.d. and after two weeks titrated to a target maintenance dose of 50 mg tid. About 80% of patients were receiving the target dose at the end of the study. Patients were followed for a minimum of two years and for up to five years, with an average follow-up of 3.5 years.
Baseline blood pressure was 113/70 mmHg and 112/70 mmHg for the placebo and captopril groups, respectively. Blood pressure increased slightly in both treatment groups during the study and was somewhat lower in the captopril group (119/74 vs. 125/77 mmHg at 1 yr).
Therapy with captopril improved long-term survival and clinical outcomes compared to placebo. The risk reduction for all cause mortality was 19% (P=0.02) and for cardiovascular death was 21% (P=0.014). Captopril treated subjects had 22% (P=0.034) fewer first hospitalizations for heart failure. Compared to placebo, 22% fewer patients receiving captopril developed symptoms of overt heart failure. There was no significant difference between groups in total hospitalizations for all cause (2056 placebo; 2036 captopril).
Captopril was well tolerated in the presence of other therapies such as aspirin, beta blockers, nitrates, vasodilators, calcium antagonists and diuretics.
In a multicenter, double-blind, placebo controlled trial, 409 patients, age 18-49 of either gender, with or without hypertension, with type I (juvenile type, onset before age 30) insulin-dependent diabetes mellitus, retinopathy, proteinuria ≥500 mg per day and serum creatinine ≤2.5 mg/dL, were randomized to placebo or captopril (25 mg t.i.d.) and followed for up to 4.8 years (median 3 years). To achieve blood pressure control, additional antihypertensive agents (diuretics, beta blockers, centrally acting agents or vasodilators) were added as needed for patients in both groups.
The captopril group had a 51% reduction in risk of doubling of serum creatinine (P<0.01) and a 51% reduction in risk for the combined endpoint of end-stage renal disease (dialysis or transplantation) or death (P<0.01). Captopril treatment resulted in a 30% reduction in urine protein excretion within the first 3 months (P<0.05), which was maintained throughout the trial. The captopril group had somewhat better blood pressure control than the placebo group, but the effect of captopril on renal function were greater than would be expected from the group differences in blood pressure reduction alone. Captopril was well tolerated in this patient population.
In two multicenter, double-blind, placebo controlled studies, a total of 235 normotensive patients with insulin-dependent diabetes mellitus, retinopathy and microalbuminuria (20-200 mcg/min) were randomized to placebo or captopril (50 mg b.i.d.) and followed for up to 2 years. Captopril delayed the progression to overt nephropathy (proteinuria ≥ 500 mg/day) in both studies (risk reduction 67% to 76%; P<0.05). Captopril also reduced the albumin excretion rate. However, the long term clinical benefit of reducing the progression from microalbuminuria to proteinuria has not been established.
Studies in rats and cats indicate that captopril does not cross the blood-brain barrier to any significant extent.
INDICATIONS AND USAGE
Hypertension
Captopril tablets are indicated for the treatment of hypertension.
In using captopril, consideration should be given to the risk of neutropenia/agranulocytosis (see WARNINGS).
Captopril may be used as initial therapy for patients with normal renal function, in whom the risk is relatively low. In patients with impaired renal function, particularly those with collagen vascular disease, captopril should be reserved for hypertensives who have either developed unacceptable side effects on other drugs, or have failed to respond satisfactorily to drug combinations.
Captopril is effective alone and in combination with other antihypertensive agents, especially thiazide-type diuretics. The blood pressure lowering effects of captopril and thiazides are approximately additive.
Heart Failure
Captopril tablets are indicated in the treatment of congestive heart failure usually in combination with diuretics and digitalis. The beneficial effect of captopril in heart failure does not require the presence of digitalis, however, most controlled clinical trial experience with captopril has been in patients receiving digitalis, as well as diuretic treatment.
Left Ventricular Dysfunction After Myocardial Infarction
Captopril tablets are indicated to improve survival following myocardial infarction in clinically stable patients with left ventricular dysfunction manifested as an ejection fraction ≤40% and to reduce the incidence of overt heart failure and subsequent hospitalizations for congestive heart failure in these patients.
Diabetic Nephropathy
Captopril is indicated for the treatment of diabetic nephropathy (proteinuria >500 mg/day) in patients with type I insulin-dependent diabetes mellitus and retinopathy. Captopril decreases the rate of progression of renal insufficiency and development of serious adverse clinical outcomes (death or need for renal transplantation or dialysis).
In considering use of captopril, it should be noted that in controlled trials ACE inhibitors have an effect on blood pressure that is less in black patients than in non-blacks. In addition, ACE inhibitors (for which adequate data are available) cause a higher rate of angioedema in black than in non-black patients (see WARNINGS: Head and Neck Angioedema and Intestinal Angioedema).
CONTRAINDICATIONS
Captopril tablets are contraindicated in patients who are hypersensitive to this product or any other angiotensin-converting enzyme inhibitor (e.g., a patient who has experienced angioedema during therapy with any other ACE inhibitor).
WARNINGS
Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors (including captopril) may be subject to a variety of adverse reactions, some of them serious.
Head and Neck Angioedema
Angioedema involving the extremities, face, lips, mucous membranes, tongue, glottis or larynx has been seen in patients treated with ACE inhibitors, including captopril. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Emergency therapy, including but not necessarily limited to, subcutaneous administration of a 1:1000 solution of epinephrine should be promptly instituted.
Swelling confined to the face, mucous membranes of the mouth, lips and extremities has usually resolved with discontinuation of captopril; some cases required medical therapy. (See PRECAUTIONS: Information for Patients and ADVERSE REACTIONS.)
Intestinal Angioedema
Intestinal angioedema has been reported in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor. Intestinal angioedema should be included in the differential diagnosis of patients on ACE inhibitors presenting with abdominal pain.
Anaphylactoid reactions during desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions were avoided when ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Anaphylactoid reactions during membrane exposure
Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
Neutropenia/Agranulocytosis
Neutropenia (<1000/mm3) with myeloid hypoplasia has resulted from use of captopril. About half of the neutropenic patients developed systemic or oral cavity infections or other features of the syndrome of agranulocytosis.
The risk of neutropenia is dependent on the clinical status of the patient:
In clinical trials in patients with hypertension who have normal renal function (serum creatinine less than 1.6 mg/dL and no collagen vascular diseases), neutropenia has been seen in one patient out of over 8600 exposed.
In patients with some degree of renal failure (serum creatinine at least 1.6 mg/dL) but no collagen vascular disease, the risk of neutropenia in clinical trials was about 1 per 500, a frequency over 15 times that for uncomplicated hypertension. Daily doses of captopril were relatively high in these patients, particularly in view of their diminished renal function. In foreign marketing experience in patients with renal failure, use of allopurinol concomitantly with captopril has been associated with neutropenia but this association has not appeared in U.S. reports.
In patients with collagen vascular diseases (e.g., systemic lupus erythematosus, scleroderma) and impaired renal function, neutropenia occurred in 3.7 percent of patients in clinical trials.
While none of the over 750 patients in formal clinical trials of heart failure developed neutropenia, it has occurred during the subsequent clinical experience. About half of the reported cases had serum creatinine ≥1.6 mg/dL and more than 75 percent were in patients also receiving procainamide. In heart failure, it appears that the same risk factors for neutropenia are present.
The neutropenia has usually been detected within three months after captopril was started. Bone marrow examinations in patients with neutropenia consistently showed myeloid hypoplasia, frequently accompanied by erythroid hypoplasia and decreased numbers of megakaryocytes (e.g., hypoplastic bone marrow and pancytopenia); anemia and thrombocytopenia were sometimes seen.
In general, neutrophils returned to normal in about two weeks after captopril was discontinued, and serious infections were limited to clinically complex patients. About 13 percent of the cases of neutropenia have ended fatally, but almost all fatalities were in patients with serious illness, having collagen vascular disease, renal failure, heart failure or immunosuppressant therapy, or a combination of these complicating factors.
Evaluation of the hypertensive or heart failure patients should always include assessment of renal function.
If captopril is used in patients with impaired renal function, white blood cell and differential counts should be evaluated prior to starting treatment and at approximately two-week intervals for about three months, then periodically.
In patients with collagen vascular disease or who are exposed to other drugs known to affect the white cells or immune response, particularly when there is impaired renal function, captopril should be used only after an assessment of benefit and risk, and then with caution.
All patients treated with captopril should be told to report any signs of infection (e.g., sore throat, fever). If infection is suspected, white cell counts should be performed without delay.
Since discontinuation of captopril and other drugs has generally led to prompt return of the white count to normal, upon confirmation of neutropenia (neutrophil count <1000/mm3) the physician should withdraw captopril and closely follow the patient’s course.
Proteinuria
Total urinary proteins greater than 1 g per day were seen in about 0.7 percent of patients receiving captopril. About 90 percent of affected patients had evidence of prior renal disease or received relatively high doses of captopril (in excess of 150 mg/day), or both. The nephrotic syndrome occurred in about one-fifth of proteinuric patients. In most cases, proteinuria subsided or cleared within six months whether or not captopril was continued. Parameters of renal function, such as BUN and creatinine, were seldom altered in the patients with proteinuria.
Hypotension
Excessive hypotension was rarely seen in hypertensive patients but is a possible consequence of captopril use in salt/volume depleted persons (such as those treated vigorously with diuretics), patients with heart failure or those patients undergoing renal dialysis. (See PRECAUTIONS: Drug Interactions.)
In heart failure, where the blood pressure was either normal or low, transient decreases in mean blood pressure greater than 20 percent were recorded in about half of the patients. This transient hypotension is more likely to occur after any of the first several doses and is usually well tolerated, producing either no symptoms or brief mild lightheadedness, although in rare instances it has been associated with arrhythmia or conduction defects. Hypotension was the reason for discontinuation of drug in 3.6 percent of patients with heart failure.
BECAUSE OF THE POTENTIAL FALL IN BLOOD PRESSURE IN THESE PATIENTS, THERAPY SHOULD BE STARTED UNDER VERY CLOSE MEDICAL SUPERVISION. A starting dose of 6.25 or 12.5 mg tid may minimize the hypotensive effect. Patients should be followed closely for the first two weeks of treatment and whenever the dose of captopril and/or diuretic is increased. In patients with heart failure, reducing the dose of diuretic, if feasible, may minimize the fall in blood pressure.
Hypotension is not per se a reason to discontinue captopril. Some decrease of systemic blood pressure is a common and desirable observation upon initiation of captopril treatment in heart failure. The magnitude of the decrease is greatest early in the course of treatment; this effect stabilizes within a week or two, and generally returns to pretreatment levels, without a decrease in therapeutic efficacy, within two months.
Fetal/Neonatal Morbidity and Mortality
ACE inhibitors can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature. When pregnancy is detected, ACE inhibitors should be discontinued as soon as possible.
The use of ACE inhibitors during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to the ACE-inhibitor exposure.
These adverse effects do not appear to have resulted from intrauterine ACE-inhibitor exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to ACE inhibitors only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should make every effort to discontinue the use of captopril as soon as possible.
Rarely (probably less often than once in every thousand pregnancies), no alternative to ACE inhibitors will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses and serial ultrasound examinations should be performed to assess the intraamniotic environment.
If oligohydramnios is observed, captopril should be discontinued unless it is considered life-saving for the mother. Contraction stress testing (CST), a non-stress test (NST), or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Infants with histories of in utero exposure to ACE inhibitors should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function. While captopril may be removed from the adult circulation by hemodialysis, there is inadequate data concerning the effectiveness of hemodialysis for removing it from the circulation of neonates or children. Peritoneal dialysis is not effective for removing captopril; there is no information concerning exchange transfusion for removing captopril from the general circulation.
When captopril was given to rabbits at doses about 0.8 to 70 times (on a mg/kg basis) the maximum recommended human dose, low incidences of craniofacial malformations were seen. No teratogenic effects of captopril were seen in studies of pregnant rats and hamsters. On a mg/kg basis, the doses used were up to 150 times (in hamsters) and 625 times (in rats) the maximum recommended human dose.
Hepatic Failure
Rarely, ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and (sometimes) death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice or marked elevations of hepatic enzymes should discontinue the ACE inhibitor and receive appropriate medical follow-up.
PRECAUTIONS
Hypertension
Some patients with renal disease, particularly those with severe renal artery stenosis, have developed increases in BUN and serum creatinine after reduction of blood pressure with captopril. Captopril dosage reduction and/or discontinuation of diuretic may be required. For some of these patients, it may not be possible to normalize blood pressure and maintain adequate renal perfusion.
Heart Failure
About 20 percent of patients develop stable elevations of BUN and serum creatinine greater than 20 percent above normal or baseline upon long-term treatment with captopril. Less than 5 percent of patients, generally those with severe preexisting renal disease, required discontinuation of treatment due to progressively increasing creatinine; subsequent improvement probably depends upon the severity of the underlying renal disease.
See CLINICAL PHARMACOLOGY, DOSAGE AND ADMINISTRATION, ADVERSE REACTIONS: Altered Laboratory Findings.
Hyperkalemia
Elevations in serum potassium have been observed in some patients treated with ACE inhibitors, including captopril. When treated with ACE inhibitors, patients at risk for the development of hyperkalemia include those with: renal insufficiency; diabetes mellitus; and those using concomitant potassium-sparing diuretics, potassium supplements or potassium-containing salt substitutes; or other drugs associated with increases in serum potassium. In a trial of type I diabetic patients with proteinuria, the incidence of withdrawal of treatment with captopril for hyperkalemia was 2% (4/207). In two trials of normotensive type I diabetic patients with microalbuminuria, no captopril group subjects had hyperkalemia (0/116). (See PRECAUTIONS: Information for Patients and Drug Interactions; ADVERSE REACTIONS: Altered Laboratory Findings.)
Cough
Presumably due to the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. ACE inhibitor-induced cough should be considered in the differential diagnosis of cough.
Valvular Stenosis
There is concern, on theoretical grounds, that patients with aortic stenosis might be at particular risk of decreased coronary perfusion when treated with vasodilators because they do not develop as much afterload reduction as others.
Surgery/Anesthesia
In patients undergoing major surgery or during anesthesia with agents that produce hypotension, captopril will block angiotensin II formation secondary to compensatory renin release. If hypotension occurs and is considered to be due to this mechanism, it can be corrected by volume expansion.
Hemodialysis
Recent clinical observations have shown an association of hypersensitivity-like (anaphylactoid) reactions during hemodialysis with high-flux dialysis membranes (e.g., AN69) in patients receiving ACE inhibitors. In these patients, consideration should be given to using a different type of dialysis membrane of a different class medication. (See WARNINGS: Anaphylactoid reactions during membrane exposure.)
Information for Patients
Patients should be advised to immediately report to their physician any signs or symptoms suggesting angioedema (e.g., swelling of face, eyes, lips, tongue, larynx and extremities; difficulty in swallowing or breathing; hoarseness) and to discontinue therapy. (See WARNINGS: Head and Heck Angioedema and Intestinal Angioedema.)
Patients should be told to report promptly any indication of infection (e.g., sore throat, fever), which may be a sign of neutropenia, or of progressive edema which might be related to proteinuria and nephrotic syndrome.
All patients should be cautioned that excessive perspiration and dehydration may lead to an excessive fall in blood pressure because of reduction in fluid volume. The causes of volume depletion such as vomiting or diarrhea may also lead to a fall in blood pressure; patients should be advised to consult with the physician.
Patients should be advised not to use potassium-sparing diuretics, potassium supplements or potassium-containing salt substitutes without consulting their physician. (See PRECAUTIONS: General and Drug Interactions; ADVERSE REACTIONS.)
Patients should be warned against interruption or discontinuation of medication unless instructed by the physician.
Heart failure patients on captopril therapy should be cautioned against rapid increases in physical activity.
Patients should be informed that captopril should be taken one hour before meals (see DOSAGE AND ADMINISTRATION).
Pregnancy
Female patients of childbearing age should be told about the consequences of second- and third-trimester exposure to ACE-inhibitors, and they should also be told that these consequences do not appear to have resulted from intrauterine ACE-inhibitor exposure that has been limited to the first trimester. These patients should be asked to report pregnancies to their physicians as soon as possible.
Hypotension-Patients on Diuretic Therapy
Patients on diuretics and especially those in whom diuretic therapy was recently instituted, as well as those on severe dietary salt restriction or dialysis, may occasionally experience a precipitous reduction of blood pressure usually within the first hour after receiving the initial dose of captopril.
The possibility of hypotensive effects with captopril can be minimized by either discontinuing the diuretic or increasing the salt intake approximately one week prior to initiation of treatment with captopril or initiating therapy with small doses (6.25 or 12.5 mg). Alternatively, provide medical supervision for at least one hour after the initial dose. If hypotension occurs, the patient should be placed in a supine position and, if necessary, receive an intravenous infusion of normal saline. This transient hypotensive response is not a contraindication to further doses which can be given without difficulty once the blood pressure has increased after volume expansion.
Agents Having Vasodilator Activity
Data on the effect of concomitant use of vasodilators in patients receiving captopril for heart failure are not available; therefore, nitroglycerin or other nitrates (as used for management of angina) or other drugs having vasodilator activity should, if possible, be discontinued before starting captopril. If resumed during captopril therapy, such agents should be administered cautiously, and perhaps at lower dosage.
Agents Causing Renin Release
Captopril’s effect will be augmented by antihypertensive agents that cause renin release. For example, diuretics (e.g., thiazides) may activate the renin-angiotensin-aldosterone system.
Agents Affecting Sympathetic Activity
The sympathetic nervous system may be especially important in supporting blood pressure in patients receiving captopril alone or with diuretics. Therefore, agents affecting sympathetic activity (e.g., ganglionic blocking agents or adrenergic neuron blocking agents) should be used with caution. Beta-adrenergic blocking drugs add some further anti-hypertensive effect to captopril, but the overall response is less than additive.
Agents Increasing Serum Potassium
Since captopril decreases aldosterone production, elevation of serum potassium may occur. Potassium-sparing diuretics such as spironolactone, triamterene, or amiloride, or potassium supplements should be given only for documented hypokalemia, and then with caution, since they may lead to a significant increase of serum potassium. Salt substitutes containing potassium should also be used with caution.
Inhibitors of Endogenous Prostaglandin Synthesis
It has been reported that indomethacin may reduce the antihypertensive effect of captopril, especially in cases of low renin hypertension. Other nonsteroidal anti-inflammatory agents (e.g., aspirin) may also have this effect.
Lithium
Increased serum lithium levels and symptoms of lithium toxicity have been reported in patients receiving concomitant lithium and ACE inhibitor therapy. These drugs should be coadministered with caution and frequent monitoring of serum lithium levels is recommended. If a diuretic is also used, it may increase the risk of lithium toxicity.
Cardiac Glycosides
In a study of young healthy male subjects no evidence of a direct pharmacokinetic captopril-digoxin interaction could be found.
Loop Diuretics
Furosemide administered concurrently with captopril does not alter the pharmacokinetics of captopril in renally impaired hypertensive patients.
Allopurinol
In a study of healthy male volunteers no significant pharmacokinetic interaction occurred when captopril and allopurinol were administered concomitantly for 6 days.
Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE inhibitor therapy including captopril.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year studies with doses of 50 to 1350 mg/kg/day in mice and rats failed to show any evidence of carcinogenic potential. The high dose in these studies is 150 times the maximum recommended human dose of 450 mg, assuming a 50-kg subject. On a body-surface-area basis, the high doses for mice and rats are 13 and 26 times the maximum recommended human dose, respectively.
Studies in rats have revealed no impairment of fertility.
Animal Toxicology
Chronic oral toxicity studies were conducted in rats (2 years), dogs (47 weeks; 1 year), mice (2 years), and monkeys (1 year). Significant drug related toxicity included effects on hematopoiesis, renal toxicity, erosion/ulceration of the stomach, and variation of retinal blood vessels.
Reductions in hemoglobin and/or hematocrit values were seen in mice, rats, and monkeys at doses 50 to 150 times the maximum recommended human dose (MRHD) of 450 mg, assuming a 50-kg subject. On a body-surface-area-basis, these doses are 5 to 25 times maximum recommended dose (MRHD). Anemia, leukopenia, thrombocytopenia, and bone marrow suppression occurred in dogs at doses 8 to 30 times MRHD on a body-weight basis (4 to 15 times MRHD on a surface-area basis). The reductions in hemoglobin and hematocrit values in rats and mice were only significant at 1 year and returned to normal with continued dosing by the end of the study. Marked anemia was seen at all dose levels (8 to 30 times MRHD) in dogs, whereas moderate to marked leukopenia was noted only at 15 and 30 times MRHD and thrombocytopenia at 30 times MRHD. The anemia could be reversed upon discontinuation of dosing. Bone marrow suppression occurred to a varying degree, being associated only with dogs that died or were sacrificed in a moribund condition in the 1 year study. However, in the 47-week study at a dose 30 times MRHD, bone marrow suppression was found to be reversible upon continued drug administration.
Captopril caused hyperplasia of the juxtaglomerular apparatus of the kidneys in mice and rats at doses 7 to 200 times MRHD on a body-weight basis (0.6 to 35 times MRHD on a surface-area basis); in monkeys at 20 to 60 times MRHD on a body-weight basis (7 to 20 times MRHD on a surface-area basis); and in dogs at 30 times MRHD on a body-weight basis (15 times MRHD on a surface-area basis).
Gastric erosion/ulcerations were increased in incidence in male rats at 20 to 200 times MRHD on a body-weight basis (3.5 and 35 times MRHD on a surface-area basis); in dogs at 30 times MRHD on a body-weight basis (15 times on MRHD on a surface-area basis); and in monkeys at 65 times MRHD on a body-weight basis (20 times MRHD on a surface-area basis). Rabbits developed gastric and intestinal ulcers when given oral doses approximately 30 times MRHD on a body-weight basis (10 times MRHD on surface-area basis) for only 5 to 7 days.
In the two-year rat study, irreversible and progressive variations in the caliber of retinal vessels (focal sacculations and constrictions) occurred at all dose levels (7 to 200 times MRHD) on a body-weight basis; 1 to 35 times MRHD on a surface-area basis in a dose-related fashion. The effect was first observed in the 88th week of dosing, with a progressively increased incidence thereafter, even after cessation of dosing.
Nursing Mothers
Concentrations of captopril in human milk are approximately one percent of those in maternal blood. Because of the potential for serious adverse reactions in nursing infants from captopril, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of captopril to the mother. (See PRECAUTIONS: Pediatric Use.)
Pediatric Use
Safety and effectiveness in pediatric patients have not been established. There is limited experience reported in the literature with the use of captopril in the pediatric population; dosage, on a weight basis, was generally reported to be comparable to or less than that used in adults.
Infants, especially newborns, may be more susceptible to the adverse hemodynamic effects of captopril. Excessive, prolonged and unpredictable decreases in blood pressure and associated complications, including oliguria and seizures, have been reported.
Captopril should be used in pediatric patients only if other measures for controlling blood pressure have not been effective.
ADVERSE REACTIONS
Reported incidences are based on clinical trials involving approximately 7000 patients.
Renal: About one of 100 patients developed proteinuria (see WARNINGS).
Each of the following has been reported in approximately 1 to 2 of 1000 patients and are of uncertain relationship to drug use: renal insufficiency, renal failure, nephrotic syndrome, polyuria, oliguria, and urinary frequency.
Hematologic: Neutropenia/agranulocytosis has occurred (see WARNINGS). Cases of anemia, thrombocytopenia, and pancytopenia have been reported.
Dermatologic: Rash, often with pruritus, and sometimes with fever, arthralgia, and eosinophilia, occurred in about 4 to 7 (depending on renal status and dose) of 100 patients, usually during the first four weeks of therapy. It is usually maculopapular, and rarely urticarial. The rash is usually mild and disappears within a few days of dosage reduction, short-term treatment with an antihistaminic agent, and/or discontinuing therapy; remission may occur even if captopril is continued. Pruritus, without rash, occurs in about 2 of 100 patients. Between 7 and 10 patients with skin rash have shown an eosinophilia and/or positive ANA titers. A reversible associated pemphigoid-like lesion, and photosensitivity, have also been reported.
Flushing or pallor has been reported in 2 to 5 of 1000 patients.
Cardiovascular: Hypotension may occur; see WARNINGS and PRECAUTIONS: Drug Interactions for discussion of hypotension with captopril therapy.
Tachycardia, chest pain, and palpitations have each been observed in approximately 1 of 100 patients.
Angina pectoris, myocardial infarction, Raynaud’s syndrome and congestive heart failure have each occurred in 2 to 3 of 1000 patients.
Dysgeusia: Approximately 2 to 4 (depending on renal status and dose) of 100 patients developed a diminution or loss of taste perception. Taste impairment is reversible and usually self-limited (2 to 3 months) even with continued drug administration. Weight loss may be associated with the loss of taste.
Angioedema: Angioedema involving the extremities, face, lips, mucous membranes, tongue, glottis or larynx has been reported in approximately one in 1000 patients. Angioedema involving the upper airways has caused fatal airway obstruction. (See WARNINGS: Head and Neck Angioedema, Intestinal Angioedema and PRECAUTIONS: Information for Patients.)
Cough: Cough has been reported in 0.5 to 2% of patients treated with captopril in clinical trials (see PRECAUTIONS: General: Cough).
The following have been reported in about 0.5 to 2 percent of patients but did not appear at increased frequency compared to placebo or other treatments used in controlled trials; gastric irritation, abdominal pain, nausea, vomiting, diarrhea, anorexia, constipation, aphthous ulcers, peptic ulcer, dizziness, headache, malaise, fatigue, insomnia, dry mouth, dyspnea, alopecia, paresthesias.
Other clinical adverse effects reported since the drug was marketed are listed below by body system. In this setting, an incidence or causal relationship cannot be accurately determined.
Body as a Whole: Anaphylactoid reactions (see WARNINGS: Anaphylactoid and Possible Related Reactions and PRECAUTIONS: Hemodialysis).
General: Asthenia, gynecomastia.
Cardiovascular: Cardiac arrest, cerebrovascular accident/insufficiency, rhythm disturbances, orthostatic hypotension, syncope.
Dermatologic: Bullous pemphigus, erythema multiforme (including Stevens-Johnson syndrome), exfoliative dermatitis.
Gastrointestinal: Pancreatitis, glossitis, dyspepsia.
Hematologic: Anemia, including aplastic and hemolytic.
Hepatobiliary: Jaundice, hepatitis, including rare cases of necrosis, cholestasis.
Metabolic: Symptomatic hyponatremia.
Musculoskeletal: Myalgia, myasthenia.
Nervous/Psychiatric: Ataxia, confusion, depression, nervousness, somnolence.
Respiratory: Bronchospasm, eosinophilic pneumonitis, rhinitis.
Special Senses: Blurred vision.
Urogenital: Impotence.
As with other ACE inhibitors, a syndrome has been reported which may include: fever, myalgia, arthralgia, interstitial nephritis, vasculitis, rash or other dermatologic manifestations, eosinophilia and an elevated ESR.
Altered Laboratory Findings
Serum Electrolytes: Hyperkalemia: Small increases in serum potassium, especially in patients with renal impairment (see PRECAUTIONS).
Hyponatremia: Particularly in patients receiving a low sodium diet or concomitant diuretics.
BUN/Serum Creatinine: Transient elevations of BUN or serum creatinine especially in volume or salt depleted patients or those with renovascular hypertension may occur. Rapid reduction of long-standing or markedly elevated blood pressure can result in decreases in the glomerular filtration rate and, in turn, lead to increases in BUN or serum creatinine.
Hematologic: A positive ANA has been reported.
Liver Funtion Tests: Elevations of liver transaminases, alkaline phosphatase, and serum bilirubin have occurred.
OVERDOSAGE
Correction of hypotension would be of primary concern. Volume expansion with an intravenous infusion of normal saline is the treatment of choice for restoration of blood pressure.
While captopril may be removed from the adult circulation by hemodialysis, there is inadequate data concerning the effectiveness of hemodialysis for removing it from the circulation of neonates or children. Peritoneal dialysis is not effective for removing captopril; there is no information concerning exchange transfusion for removing captopril from the general circulation.
DOSAGE AND ADMINISTRATION
Captopril should be taken one hour before meals. Dosage must be individualized.
Hypertension
Initiation of therapy requires consideration of recent antihypertensive drug treatment, the extent of blood pressure elevation, salt restriction, and other clinical circumstances. If possible, discontinue the patient’s previous antihypertensive drug regimen for one week before starting captopril.
The initial dose of captopril is 25 mg b.i.d. or t.i.d. If satisfactory reduction of blood pressure has not been achieved after one or two weeks, the dose may be increased to 50 mg b.i.d. or t.i.d. Concomitant sodium restriction may be beneficial when captopril is used alone.
The dose of captopril in hypertension usually does not exceed 50 mg t.i.d. Therefore, if the blood pressure has not been satisfactorily controlled after one to two weeks at this dose, (and the patient is not already receiving a diuretic), a modest dose of thiazide-type diuretic (e.g., hydrochlorothiazide, 25 mg daily), should be added. The diuretic dose may be increased at one- to two-week intervals until its highest usual antihypertensive dose is reached.
If captopril is being started in a patient already receiving a diuretic, captopril therapy should be initiated under close medical supervision (see WARNINGS and PRECAUTIONS: Drug Interactions regarding hypotension), with dosage and titration of captopril as noted above.
If further blood pressure reduction is required, the dose of captopril may be increased to 100 mg b.i.d. or t.i.d. and then, if necessary, to 150 mg b.i.d. or t.i.d. (while continuing the diuretic). The usual dose range is 25 to 150 mg b.i.d. or t.i.d. A maximum daily dose of 450 mg captopril should not be exceeded.
For patients with severe hypertension (e.g., accelerated or malignant hypertension), when temporary discontinuation of current antihypertensive therapy is not practical or desirable, or when prompt titration to more normotensive blood pressure levels is indicated, diuretic should be continued but other current antihypertensive medication stopped and captopril dosage promptly initiated at 25 mg b.i.d. or t.i.d., under close medical supervision.
When necessitated by the patient’s clinical condition, the daily dose of captopril may be increased every 24 hours or less under continuous medical supervision until a satisfactory blood pressure response is obtained or the maximum dose of captopril is reached. In this regimen, addition of a more potent diuretic, e.g., furosemide, may also be indicated.
Beta-blockers may also be used in conjunction with captopril therapy (see PRECAUTIONS: Drug Interactions), but the effects of the two drugs are less than additive.
Heart Failure
Initiation of therapy requires consideration of recent diuretic therapy and the possibility of severe salt/volume depletion. In patients with either normal or low blood pressure, who have been vigorously treated with diuretics and who may be hyponatremic and/or hypovolemic, a starting dose of 6.25 or 12.5 mg tid may minimize the magnitude or duration of the hypotensive effect (see WARNINGS: Hypotension); for these patients, titration of the usual daily dosage can then occur within the next several days.
For most patients the usual initial daily dosage is 25 mg tid. After a dose of 50 mg tid is reached, further increases in dosage should be delayed, where possible, for at least two weeks to determine if a satisfactory response occurs. Most patients studied have had a satisfactory clinical improvement at 50 or 100 mg tid. A maximum daily dose of 450 mg of captopril should not be exceeded.
Captopril should generally be used in conjunction with a diuretic and digitalis. Captopril therapy must be initiated under very close medical supervision.
Left Ventricular Dysfunction After Myocardial Infarction
The recommended dose for long-term use in patients following a myocardial infarction is a target maintenance dose of 50 mg t.i.d.
Therapy may be initiated as early as three days following a myocardial infarction. After a single dose of 6.25 mg, captopril therapy should be initiated at 12.5 mg t.i.d. Captopril should then be increased to 25 mg t.i.d. during the next several days and to a target dose of 50 mg t.i.d. over the next several weeks as tolerated (see CLINICAL PHARMACOLOGY).
Captopril may be used in patients treated with other post-myocardial infarction therapies, e.g., thrombolytics, aspirin, beta blockers.
Diabetic Nephropathy
The recommended dose of captopril for long term use to treat diabetic nephropathy is 25 mg t.i.d.
Other antihypertensives such as diuretics, beta blockers, centrally acting agents or vasodilators may be used in conjunction with captopril if additional therapy is required to further lower blood pressure.
Dosage Adjustment in Renal Impairment
Because captopril is excreted primarily by the kidneys, excretion rates are reduced in patients with impaired renal function. These patients will take longer to reach steady-state captopril levels and will reach higher steady-state levels for a given daily dose than patients with normal renal function. Therefore, these patients may respond to smaller or less frequent doses.
Accordingly, for patients with significant renal impairment, initial daily dosage of captopril should be reduced, and smaller increments utilized for titration, which should be quite slow (one- to two-week intervals). After the desired therapeutic effect has been achieved, the dose should be slowly back-titrated to determine the minimal effective dose. When concomitant diuretic therapy is required, a loop diuretic (e.g., furosemide), rather than a thiazide diuretic, is preferred in patients with severe renal impairment. (See also WARNINGS: Anaphylactoid reactions during membrane exposure and PRECAUTIONS: Hemodialysis.)
HOW SUPPLIED
Captopril Tablets, USP are available as follows:
12.5 mg - White, Round, Scored Tablets; Debossed "W7".
25 mg - White, Round, Quadrisect Scored Tablets; Debossed "WW 172".
50 mg - White, Oblong, Bisect Scored Tablets; Debossed "WW 173".
100 mg - White, Oblong Tablets, Bisect Scored; Debossed "WW 174".
Captopril tablets may exhibit a slight sulfurous odor.
They are supplied by State of Florida DOH Central Pharmacy as follows:
| NDC | Strength | Quantity/Form | Color | Source Prod. Code |
| 53808-0217-1 | 50 mg | 30 Tablets in a Blister Pack | WHITE | 0143-1173 |
| 53808-0359-1 | 100 mg | 30 Tablets in a Blister Pack | WHITE | 0143-1174 |
Storage
Store at 20-25°C (68-77°F). [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Manufactured by:
West-ward Pharmaceutical Corp.
Eatontown, NJ 07724
And Repackaged By:
State of Florida DOH Central Pharmacy
104-2 Hamilton Park Drive
Tallahassee, FL 32304
United States

