FULL PRESCRIBING INFORMATION
1. INDICATIONS AND USAGE
SILENOR is indicated for the treatment of insomnia characterized by difficulty with sleep maintenance. The clinical trials performed in support of efficacy were up to 3 months in duration .
2. DOSAGE AND ADMINISTRATION
The dose of SILENOR should be individualized.
2.1. Dosing in Adults
The recommended dose of SILENOR for adults is 6 mg once daily. A 3 mg once daily dose may be appropriate for some patients, if clinically indicated.
2.2. Dosing in the Elderly
The recommended starting dose of SILENOR in elderly patients (≥ 65 years old) is 3 mg once daily. The daily dose can be increased to 6 mg, if clinically indicated.
2.3. Administration
SILENOR should be taken within 30 minutes of bedtime.
To minimize the potential for next day effects, SILENOR should not be taken within 3 hours of a meal [see Clinical Pharmacology (12.3)].
The total SILENOR dose should not exceed 6 mg per day.
3. DOSAGE FORMS AND STRENGTHS
SILENOR is an immediate-release, oval-shaped, tablet for oral administration available in strengths of 3 mg and 6 mg. The tablets are blue (3 mg) or green (6 mg) and are debossed with 3 or 6, respectively, on one side and SP on the other. SILENOR tablets are not scored.
4. CONTRAINDICATIONS
4.1. Hypersensitivity
SILENOR is contraindicated in individuals who have shown hypersensitivity to doxepin HCl, any of its inactive ingredients, or other dibenzoxepines.
4.2. Co-administration with Monoamine Oxidase Inhibitors (MAOIs)
Serious side effects and even death have been reported following the concomitant use of certain drugs with MAO inhibitors. Do not administer SILENOR if patient is currently on MAOIs or has used MAOIs within the past two weeks. The exact length of time may vary depending on the particular MAOI dosage and duration of treatment.
5. WARNINGS AND PRECAUTIONS
5.1. Need to Evaluate for Comorbid Diagnoses
Because sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Exacerbation of insomnia or the emergence of new cognitive or behavioral abnormalities may be the consequence of an unrecognized psychiatric or physical disorder. Such findings have emerged during the course of treatment with hypnotic drugs.
5.2. Abnormal Thinking and Behavioral Changes
Complex behaviors such as "sleep-driving" (i.e., driving while not fully awake after ingestion of a hypnotic, with amnesia for the event) have been reported with hypnotics. These events can occur in hypnotic-naive as well as in hypnotic-experienced persons. Although behaviors such as "sleep-driving" may occur with hypnotics alone at therapeutic doses, the use of alcohol and other CNS depressants with hypnotics appears to increase the risk of such behaviors, as does the use of hypnotics at doses exceeding the maximum recommended dose. Due to the risk to the patient and the community, discontinuation of SILENOR should be strongly considered for patients who report a "sleep-driving" episode. Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a hypnotic. As with "sleep-driving", patients usually do not remember these events. Amnesia, anxiety and other neuro-psychiatric symptoms may occur unpredictably.
5.3. Suicide Risk and Worsening of Depression
In primarily depressed patients, worsening of depression, including suicidal thoughts and actions (including completed suicides), has been reported in association with the use of hypnotics.
Doxepin, the active ingredient in SILENOR, is an antidepressant at doses 10- to 100-fold higher than in SILENOR. Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Risk from the lower dose of doxepin in SILENOR can not be excluded.
It can rarely be determined with certainty whether a particular instance of the abnormal behaviors listed above is drug induced, spontaneous in origin, or a result of an underlying psychiatric or physical disorder. Nonetheless, the emergence of any new behavioral sign or symptom of concern requires careful and immediate evaluation.
5.4. CNS Depressant Effects
After taking SILENOR, patients should confine their activities to those necessary to prepare for bed. Patients should avoid engaging in hazardous activities, such as operating a motor vehicle or heavy machinery, at night after taking SILENOR, and should be cautioned about potential impairment in the performance of such activities that may occur the day following ingestion.
When taken with SILENOR, the sedative effects of alcoholic beverages, sedating antihistamines, and other CNS depressants may be potentiated [see Warnings and Precautions (5.2) and Drug Interactions (7.3, 7.4)]. Patients should not consume alcohol with SILENOR [see Warnings and Precautions (5.2) and Drug Interactions (7.3)]. Patients should be cautioned about potential additive effects of SILENOR used in combination with CNS depressants or sedating antihistamines [see Warnings and Precautions (5.2) and Drug Interactions (7.4)].
6. ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of labeling:
- Abnormal thinking and behavioral changes [see Warnings and Precautions (5.2)].
- Suicide risk and worsening of depression [see Warnings and Precautions (5.3)].
- CNS Depressant effects [see Warnings and Precautions (5.4)].
6.1. Clinical Trials Experience
The pre-marketing development program for SILENOR included doxepin HCl exposures in 1017 subjects (580 insomnia patients and 437 healthy subjects) from 12 studies conducted in the United States. 863 of these subjects (580 insomnia patients and 283 healthy subjects) participated in six randomized, placebo-controlled efficacy studies with SILENOR doses of 1 mg, 3 mg, and 6 mg for up to 3-months in duration.
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. However, data from the SILENOR studies provide the physician with a basis for estimating the relative contributions of drug and non-drug factors to adverse reaction incidence rates in the populations studied.
Associated with Discontinuation of Treatment
The percentage of subjects discontinuing Phase 1, 2, and 3 trials for an adverse reaction was 0.6% in the placebo group compared to 0.4%, 1.0%, and 0.7% in the SILENOR 1 mg, 3 mg, and 6 mg groups, respectively. No reaction that resulted in discontinuation occurred at a rate greater than 0.5%.
Adverse Reactions Observed at an Incidence of ≥ 2% in Controlled Trials
Table 1 shows the incidence of treatment-emergent adverse reactions from three long-term (28 to 85 days) placebo-controlled studies of SILENOR in adult (N=221) and elderly (N=494) subjects with chronic insomnia.
Reactions reported by Investigators were classified using a modified MedDRA dictionary of preferred terms for purposes of establishing incidence. The table includes only reactions that occurred in 2% or more of subjects who received SILENOR 3 mg or 6 mg in which the incidence in subjects treated with SILENOR was greater than the incidence in placebo-treated subjects.
| System Organ Class Preferred Term* | Placebo (N=278) | SILENOR 3 mg (N=157) | SILENOR 6 mg (N=203) |
|---|---|---|---|
|
|||
| Nervous System Disorders | |||
| Somnolence/Sedation | 4 | 6 | 9 |
| Infections and Infestations | |||
| Upper Respiratory Tract Infection/Nasopharyngitis | 2 | 4 | 2 |
| Gastroenteritis | 0 | 2 | 0 |
| Gastrointestinal Disorders | |||
| Nausea | 1 | 2 | 2 |
| Vascular Disorders | |||
| Hypertension | 0 | 3 | < 1 |
The most common treatment-emergent adverse reaction in the placebo and each of the SILENOR dose groups was somnolence/sedation.
6.2. Studies Pertinent to Safety Concerns for Sleep-promoting Drugs
Residual Pharmacological Effect in Insomnia Trials
Five randomized, placebo-controlled studies in adults and the elderly assessed next-day psychomotor function within 1 hour of awakening utilizing the digit-symbol substitution test (DSST), symbol copying test (SCT), and visual analog scale (VAS) for sleepiness, following night time administration of SILENOR.
In a one-night, double-blind study conducted in 565 healthy adult subjects experiencing transient insomnia, SILENOR 6 mg showed modest negative changes in SCT and VAS.
In a 35-day, double-blind, placebo-controlled, parallel group study of SILENOR 3 and 6 mg in 221 adults with chronic insomnia, small decreases in the DSST and SCT occurred in the 6 mg group.
In a 3-month, double-blind, placebo-controlled, parallel group study in 240 elderly subjects with chronic insomnia, SILENOR 1 mg and 3 mg was comparable to placebo on DSST, SCT, and VAS.
6.3. Other Reactions Observed During the Pre-marketing Evaluation of SILENOR
SILENOR was administered to 1017 subjects in clinical trials in the United States. Treatment-emergent adverse reactions recorded by clinical investigators were standardized using a modified MedDRA dictionary of preferred terms. The following is a list of MedDRA terms that reflect treatment-emergent adverse reactions reported by subjects treated with SILENOR.
Adverse reactions are further categorized by body system and listed in order of decreasing frequency according to the following definitions: Frequent adverse reactions are those that occurred on one or more occasions in at least 1/100 subjects; Infrequent adverse reactions are those that occurred in fewer than 1/100 subjects and more than 1/1000 subjects. Rare adverse reactions are those that occurred in fewer than 1/1000 subjects. Adverse reactions that are listed in Table 1 are not included in the following listing of frequent, infrequent, and rare AEs.
Blood and Lymphatic System Disorders: Infrequent: anemia; Rare: thrombocythemia.
Cardiac Disorders: Rare: atrioventricular block, palpitations, tachycardia, ventricular extrasystoles.
Ear and Labyrinth Disorders: Rare: ear pain, hypoacusis, motion sickness, tinnitus, tympanic membrane perforation.
Eye Disorders: Infrequent: eye redness, vision blurred; Rare: blepharospasm, diplopia, eye pain, lacrimation decreased.
Gastrointestinal Disorders: Infrequent: abdominal pain, dry mouth, gastroesophageal reflux disease, vomiting; Rare: dyspepsia, constipation, gingival recession, haematochezia, lip blister.
General Disorders and Administration Site Conditions: Infrequent: asthenia, chest pain, fatigue; Rare: chills, gait abnormal, edema peripheral.
Hepatobiliary Disorders: Rare: hyperbilirubinemia.
Immune System Disorders: Rare: hypersensitivity.
Infections and Infestations: Infrequent: bronchitis, fungal infection, laryngitis, sinusitis, tooth infection, urinary tract infection, viral infection; Rare: cellulitis staphylococcal, eye infection, folliculitis, gastroenteritis viral, herpes zoster, infective tenosynovitis, influenza, lower respiratory tract infection, onychomycosis, pharyngitis, pneumonia.
Injury, Poisoning and Procedural Complications: Infrequent: back injury, fall, joint sprain; Rare: bone fracture, skin laceration.
Investigations: Infrequent: blood glucose increased; Rare: alanine aminotransferase increased, blood pressure decreased, blood pressure increased, electrocardiogram ST-T segment abnormal, electrocardiogram QRS complex abnormal, heart rate decreased, neutrophil count decreased, QRS axis abnormal, transaminases increased.
Metabolism and Nutrition Disorders: Infrequent: anorexia, decreased appetite, hyperkalemia, hypermagnesemia, increased appetite; Rare: hypokalemia.
Musculoskeletal and Connective Tissue Disorders: Infrequent: arthralgia, back pain, myalgia, neck pain, pain in extremity; Rare: joint range of motion decreased, muscle cramp, sensation of heaviness.
Neoplasms Benign, Malignant and Unspecified (Including Cysts and Polyps): Rare: lung adenocarcinoma stage I, malignant melanoma.
Nervous System Disorders: Frequent: dizziness; Infrequent: dysgeusia, lethargy, parasthesia, syncope; Rare: ageusia, ataxia, cerebrovascular accident, disturbance in attention, migraine, sleep paralysis, syncope vasovagal, tremor.
Psychiatric Disorders: Infrequent: abnormal dreams, adjustment disorder, anxiety, depression; Rare: confusional state, elevated mood, insomnia, libido decreased, nightmare.
Reproductive System and Breast Disorders: Rare: breast cyst, dysmenorrhea.
Renal and Urinary Disorders: Rare: dysuria, enuresis, hemoglobinuria, nocturia.
Respiratory, Thoracic and Mediastinal Disorders: Infrequent: nasal congestion, pharyngolaryngeal pain, sinus congestion, wheezing; Rare: cough, crackles lung, nasopharyngeal disorder, rhinorrhea, dyspnea.
Skin and Subcutaneous Tissue Disorders: Infrequent: skin irritation; Rare: cold sweat, dermatitis, erythema, hyperhidrosis, pruritis, rash, rosacea.
Surgical and Medical Procedures: Rare: arthrodesis.
Vascular Disorders: Infrequent: pallor; Rare: blood pressure inadequately controlled, hematoma, hot flush.
In addition, the reactions below have been reported for other tricyclics and may be idiosyncratic (not related to dose).
Allergic: photosensitization, skin rash.
Hematologic: agranulocytosis, eosinophilia, leukopenia, purpura, thrombocytopenia.
7. DRUG INTERACTIONS
7.1. Cytochrome P450 Isozymes
SILENOR is primarily metabolized by hepatic cytochrome P450 isozymes CYP2C19 and CYP2D6, and to a lesser extent, by CYP1A2 and CYP2C9. Inhibitors of these isozymes may increase the exposure of doxepin. SILENOR is not an inhibitor of any CYP isozymes at therapeutically relevant concentrations. The ability of SILENOR to induce CYP isozymes is not known.
7.2. Cimetidine
SILENOR exposure is doubled with concomitant administration of cimetidine, a nonspecific inhibitor of CYP isozymes. A maximum dose of 3 mg is recommended in adults and elderly when cimetidine is co-administered with SILENOR [see Clinical Pharmacology (12.3)]
7.3. Alcohol
When taken with SILENOR, the sedative effects of alcohol may be potentiated [see Warnings and Precautions (5.2, 5.4)].
7.4. CNS Depressants and Sedating Antihistamines
When taken with SILENOR, the sedative effects of sedating antihistamines and CNS depressants may be potentiated [see Warnings and Precautions (5.2, 5.4)].
8. USE IN SPECIFIC POPULATIONS
8.1. Pregnancy
Risk Summary
Available data from published epidemiologic studies and postmarketing reports have not established an increased risk of major birth defects or miscarriage (see Data). There are risks of poor neonatal adaptation with exposure to tricyclic antidepressants (TCAs), including doxepin, during pregnancy (see Clinical Considerations). In animal reproduction studies, oral administration of doxepin to rats and rabbits during the period of organogenesis caused adverse developmental effects at doses 65 and 23 times the maximum recommended human dose (MRHD) of 6 mg/day based on AUC, respectively. Oral administration of doxepin to pregnant rats during pregnancy and lactation resulted in decreased pup survival and a delay in pup growth at doses 60 times the MRHD based on AUC (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of major birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal adverse reactions
Neonates exposed to TCAs, including doxepin, late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hyperreflexia, tremor, jitteriness, irritability and constant crying. These findings are consistent with either direct toxic effects of TCAs or possibly a drug discontinuation syndrome. Monitor neonates who were exposed to SILENOR in the third trimester of pregnancy for poor neonatal adaptation syndrome.
Data
Human Data
Published epidemiologic studies of pregnant women exposed to TCAs, including doxepin, have not established an association with major birth defects, miscarriage or adverse maternal outcomes. Methodological limitations of these observational studies include small sample size and lack of adequate controls.
Animal Data
When doxepin (30, 100, and 150 mg/kg/day) was administered orally to pregnant rats during the period of organogenesis, developmental toxicity (increased incidences of fetal structural abnormalities consisting of non-ossified bones in the skull and sternum and decreased fetal body weights) and maternal toxicity were noted at ≥100 mg/kg/day, which produced plasma exposures (AUCs) of doxepin and nordoxepin (the primary metabolite in humans) approximately 65 and 53 times, respectively, the plasma AUCs at the MRHD. The plasma exposures at the no-effect dose for embryo-fetal developmental toxicity in rats (30 mg/kg/day) are approximately 6 and 5 times the plasma AUCs for doxepin and nordoxepin, respectively, at the MRHD. When doxepin (10, 30, and 60 mg/kg/day) was administered orally to pregnant rabbits during the period of organogenesis, fetal body weights were reduced at the highest dose in the absence of maternal toxicity, which produced plasma AUCs of doxepin and nordoxepin approximately 23 and 56 times, respectively, the plasma AUCs at the MRHD. The plasma exposures at the no-effect dose for developmental effects (30 mg/kg/day) are approximately 8 and 25 times the plasma AUCs for doxepin and nordoxepin, respectively, at the MRHD. Oral administration of doxepin (10, 30, and 100 mg/kg/day) to rats throughout pregnancy and lactation resulted in decreased pup survival and transient growth delay at the highest dose, which produced plasma AUCs of doxepin and nordoxepin approximately 60 and 39 times, respectively, the plasma AUCs at the MRHD. The plasma exposures at the no-effect dose for adverse effects on pre- and postnatal development in rats (30 mg/kg/day) are approximately 2 and 1 times the plasma AUCs for doxepin and nordoxepin, respectively, at the MRHD.
8.2. Lactation
Risk Summary
Data from the published literature report the presence of doxepin and nordoxepin in human milk. There are reports of excess sedation, respiratory depression, poor sucking and swallowing, and hypotonia in breastfed infants exposed to doxepin. There are no data on the effects of doxepin on milk production. Because of the potential for serious adverse reactions, including excess sedation and respiratory depression in a breastfed infant, clinicians should advise patients that breastfeeding is not recommended during treatment with SILENOR.
8.3. Females and Males of Reproductive Potential
Infertility
Based on results from animal fertility studies conducted in rats, doxepin may reduce fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)]. It is unknown if the effects are reversible.
8.4. Pediatric Use
The safety and effectiveness of SILENOR in pediatric patients have not been evaluated.
8.5. Geriatric Use
A total of 362 subjects who were ≥ 65 years and 86 subjects who were ≥ 75 years received SILENOR in controlled clinical studies. No overall differences in safety or effectiveness were observed between these subjects and younger adult subjects. Greater sensitivity of some older individuals cannot be ruled out.
Sleep-promoting drugs may cause confusion and over-sedation in the elderly. A starting dose of 3 mg is recommended in this population and evaluation prior to considering dose escalation is recommended [see Dosage and Administration (2.2)].
8.6. Use in Patients with Hepatic Impairment
Patients with hepatic impairment may display higher doxepin concentrations than healthy individuals. Initiate SILENOR treatment with 3 mg in patients with hepatic impairment and monitor closely for adverse daytime effects. [see Clinical Pharmacology (12.3)]
8.7. Use in Patients with Sleep Apnea
SILENOR has not been studied in patients with obstructive sleep apnea. Since hypnotics have the capacity to depress respiratory drive, precautions should be taken if SILENOR is prescribed to patients with compromised respiratory function. In patients with severe sleep apnea, SILENOR is ordinarily not recommended for use.
9. DRUG ABUSE AND DEPENDENCE
10. OVERDOSAGE
Doxepin is routinely administered for indications other than insomnia at doses 10- to 50-fold higher than the highest recommended dose of SILENOR.
The signs and symptoms associated with doxepin use at doses several-fold higher than the maximum recommended dose (Excessive dose) of SILENOR for the treatment of insomnia are described [see Overdosage (10.1)], as are signs and symptoms associated with higher multiples of the maximum recommended dose (Critical overdose) [see Overdosage (10.2)].
10.1. Signs and Symptoms of Excessive Doses
The following adverse effects have been associated with use of doxepin at doses higher than 6 mg.
Anticholinergic Effects: constipation and urinary retention.
Central Nervous System: disorientation, hallucinations, numbness, paresthesias, extrapyramidal symptoms, seizures, tardive dyskinesia.
Cardiovascular: hypotension.
Gastrointestinal: aphthous stomatitis, indigestion.
Endocrine: raised libido, testicular swelling, gynecomastia in males, enlargement of breasts and galactorrhea in the female, raising or lowering of blood sugar levels, and syndrome of inappropriate antidiuretic hormone secretion.
Other: tinnitus, weight gain, sweating, flushing, jaundice, alopecia, exacerbation of asthma, and hyperpyrexia (in association with chlorpromazine).
10.2. Signs and Symptoms of Critical Overdose
Manifestations of doxepin critical overdose include: cardiac dysrhythmias, severe hypotension, convulsions, and CNS depression including coma. Electrocardiogram changes, particularly in QRS axis or width, are clinically significant indicators of tricyclic compound toxicity. Other signs of overdose may include, but are not limited to: confusion, disturbed concentration, transient visual hallucinations, dilated pupils, agitation, hyperactive reflexes, stupor, drowsiness, muscle rigidity, vomiting, hypothermia, hyperpyrexia.
10.3 Recommended Management
As management of overdose is complex and changing, it is recommended that the physician contact a poison control center for current information on treatment. In addition, the possibility of a multiple drug ingestion should be considered.
If an overdose is suspected, an ECG should be obtained and cardiac monitoring should be initiated immediately. The patient's airway should be protected, an intravenous line should be established, and gastric decontamination should be initiated. A minimum of six hours of observation with cardiac monitoring and observation for signs of CNS or respiratory depression, hypotension, cardiac dysrhythmias and/or conduction blocks, and seizures is strongly advised. If signs of toxicity occur at any time during this period, extended monitoring is recommended. There are case reports of patients succumbing to fatal dysrhythmias late after overdose; these patients had clinical evidence of significant poisoning prior to death and most received inadequate gastrointestinal decontamination. Monitoring of plasma drug levels should not guide management of the patient.
Gastrointestinal Decontamination
All patients suspected of overdose should receive gastrointestinal decontamination. This should include large volume gastric lavage followed by administration of activated charcoal. If consciousness is impaired, the airway should be secured prior to lavage. Emesis is contraindicated.
Cardiovascular
A maximal limb-lead QRS duration of ≥0.10 seconds may be the best indication of the severity of an overdose. Serum alkalinization, using intravenous sodium bicarbonate should be used to maintain the serum pH in the range of 7.45 to 7.55 for patients with dysrhythmias and/or QRS widening. If the pH response is inadequate, hyperventilation may also be used. Concomitant use of hyperventilation and sodium bicarbonate should be done with extreme caution, with frequent pH monitoring. A pH >7.60 or a pCO2 <20 mm Hg is undesirable. Dysrhythmias unresponsive to sodium bicarbonate therapy/hyperventilation may respond to lidocaine or phenytoin. Type 1A and 1C antiarrhythmics are generally contraindicated (e.g., quinidine, disopyramide, and procainamide).
In rare instances, hemoperfusion may be beneficial in acute refractory cardiovascular instability in patients with acute toxicity. However, hemodialysis, peritoneal dialysis, exchange transfusions, and forced diuresis generally have been reported as ineffective in treatment of tricyclic compound poisoning.
Central Nervous System
In patients with central nervous system depression, early intubation is advised because of the potential for abrupt deterioration. Seizures should be controlled with benzodiazepines, or, if these are ineffective, other anticonvulsants (e.g., phenobarbital or phenytoin). Physostigmine is not recommended except to treat life-threatening symptoms that have been unresponsive to other therapies, and then only in consultation with a poison control center.
11. DESCRIPTION
SILENOR (doxepin) is available in 3 mg and 6 mg strength tablets for oral administration. Each tablet contains 3.39 mg or 6.78 mg doxepin hydrochloride, equivalent to 3 mg and 6mg of doxepin, respectively.
Chemically, doxepin hydrochloride is an (E) and (Z) geometric, isomeric mixture of 1 propanamine, 3-dibenz[b,e]oxepin-11(6H)ylidene-N,N-dimethyl-hydrochloride. It has the following structure:
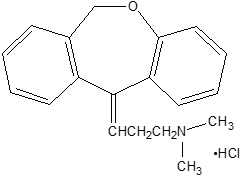
Doxepin hydrochloride is a white crystalline powder, with a slight amine-like odor, that is readily soluble in water. It has a molecular weight of 315.84 and molecular formula of C19 H21 NO∙HCl.
Each SILENOR tablet includes the following inactive ingredients: microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate. The 3 mg tablet also contains FD&C Blue No.1. The 6 mg tablet also contains D&C Yellow No. 10 and FD&C Blue No. 1.
12. CLINICAL PHARMACOLOGY
12.1. Mechanism of Action
The mechanism of action of doxepin in sleep maintenance is unclear; however, doxepin's effect could be mediated through antagonism of the H1 receptor.
12.3. Pharmacokinetics
Absorption
The median time to peak concentrations (Tmax) of doxepin occurred at 3.5 hours postdose after oral administration of a 6 mg dose to fasted healthy subjects. Peak plasma concentrations (Cmax) of SILENOR increased in approximately a dose-proportional manner for 3 mg and 6 mg doses. The AUC was increased by 41% and Cmax by 15% when 6 mg SILENOR was administered with a high fat meal. Additionally, compared to the fasted state, Tmax was delayed by approximately 3 hours. Therefore, for faster onset and to minimize the potential for next day effects, it is recommended that SILENOR not be taken within 3 hours of a meal [see Dosage and Administration (2.3)].
Distribution
SILENOR is widely distributed throughout the body tissues. The mean apparent volume of distribution following a single 6 mg oral dose of SILENOR to healthy subjects was 11,930 liters. SILENOR is approximately 80% bound to plasma proteins.
Metabolism
Following oral administration, SILENOR is extensively metabolized by oxidation and demethylation. The primary metabolite is N-desmethyldoxepin (nordoxepin).
The primary metabolite undergoes further biotransformation to glucuronide conjugates.
In vitro studies have shown that CYP2C19 and CYP2D6 are the major enzymes involved in doxepin metabolism, and that CYP1A2 and CYP2C9 are involved to a lesser extent.
Doxepin appears not to have inhibitory effects on human CYP enzymes at therapeutic concentrations. The potential of doxepin to induce metabolizing enzymes is not known. Doxepin is not a Pgp substrate.
Excretion
Doxepin is excreted in the urine mainly in the form of glucuronide conjugates.
Less than 3% of a doxepin dose is excreted in the urine as parent compound or nordoxepin. The apparent terminal half-life (t ½) of doxepin was 15.3 hours and for nordoxepin was 31 hours.
Drug Interactions
Since doxepin is metabolized by CYP2C19 and CYP2D6, inhibitors of these CYP isozymes may increase the exposure of doxepin.
Cimetidine
The effect of cimetidine, a non-specific inhibitor of CYP1A2, 2C19, 2D6, and 3A4, on SILENOR plasma concentrations was evaluated in healthy subjects. When cimetidine 300 mg BID was co-administered with a single dose of SILENOR 6 mg, there was approximately a 2-fold increase in SILENOR Cmax and AUC compared to SILENOR given alone. A maximum dose of doxepin in adults and elderly should be 3 mg, when doxepin is co-administered with cimetidine.
Sertraline
The effect of sertraline HCl, a selective serotonin reuptake inhibitor, on doxepin plasma concentrations was evaluated in a daytime study conducted with 24 healthy subjects. Following co-administration of doxepin 6 mg with sertraline 50 mg (at steady-state), the doxepin mean AUC and Cmax estimates were approximately 21% and 32% higher, respectively, than those obtained following administration of doxepin alone. Psychomotor function as measured by the digit symbol substitution test and symbol copy test performance was decreased more at 2-4 hours post dosing for the combination of sertraline and doxepin as compared to doxepin alone, but subjective measures of alertness were comparable for the two treatments.
Special Populations
Renal Impairment
The effects of renal impairment on doxepin pharmacokinetics have not been studied. Because only small amounts of doxepin and nordoxepin are eliminated in the urine, renal impairment would not be expected to result in significantly altered doxepin concentrations.
13. NONCLINICAL TOXICOLOGY
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of carcinogenic potential was observed when doxepin was administered orally to hemizygous Tg.rasH2 mice for 26 weeks at doses of 25, 50, 75, and 100 mg/kg/day.
Mutagenesis
Doxepin was negative in in vitro (bacterial reverse mutation, chromosomal aberration in human lymphocytes) and in vivo (rat micronucleus) assays.
Impairment of Fertility
When doxepin (10, 30, and 100 mg/kg/day) was orally administered to male and female rats prior to, during and after mating, adverse effects on fertility (increased copulatory interval and decreased corpora lutea, implantation, viable embryos and litter size) and sperm parameters (increased percentages of abnormal sperm and decreased sperm motility) were observed. The plasma exposures (AUC) for doxepin and nordoxepin at the no-effect dose for adverse effects on reproductive performance and fertility in rats (10 mg/kg/day) are less than those in humans at the maximum recommended human dose of 6 mg/day.
14. CLINICAL STUDIES
14.1. Controlled Clinical Trials
The efficacy of SILENOR for improving sleep maintenance was supported by six randomized, double-blind studies up to 3 months in duration that included 1,423 subjects, 18 to 93 years of age, with chronic (N=858) or transient (N=565) insomnia. SILENOR was evaluated at doses of 1 mg, 3 mg, and 6 mg relative to placebo in inpatient (sleep laboratory) and outpatient settings.
The primary efficacy measures for assessment of sleep maintenance were the objective and subjective time spent awake after sleep onset (respectively, objective Wake After Sleep Onset [WASO] and subjective WASO).
Subjects in studies of chronic insomnia were required to have at least a 3-month history of insomnia.
Chronic Insomnia
Adults
A randomized, double-blind, parallel-group study was conducted in adults (N = 221) with chronic insomnia.
SILENOR 3 mg and 6 mg was compared to placebo out to 30 days.
SILENOR 3 mg and 6 mg were superior to placebo on objective WASO. SILENOR 3 mg was superior to placebo on subjective WASO at night 1 only. SILENOR 6 mg was superior to placebo on subjective WASO at night 1, and nominally superior at some later time points out to Day 30.
Elderly
Elderly subjects with chronic insomnia were assessed in two parallel-group studies.
The first randomized, double-blind study assessed SILENOR 1 mg and 3 mg relative to placebo for 3 months in inpatient and outpatient settings in elderly subjects (N=240) with chronic insomnia. SILENOR 3 mg was superior to placebo on objective WASO.
The second randomized, double-blind study assessed SILENOR 6 mg relative to placebo for 4 weeks in an outpatient setting in elderly subjects (N=254) with chronic insomnia. On subjective WASO, SILENOR 6 mg was superior to placebo.
Transient Insomnia
Healthy adult subjects (N=565) experiencing transient insomnia during the first night in a sleep laboratory were evaluated in a randomized, double-blind, parallel-group, single-dose study of SILENOR 6 mg relative to placebo. SILENOR 6 mg was superior to placebo on objective WASO and subjective WASO.
Withdrawal Effects
Potential withdrawal effects were assessed in a 35-day double blind study of adults with chronic insomnia who were randomized to placebo, SILENOR 3 mg, or SILENOR 6 mg. There was no indication of a withdrawal syndrome after discontinuation of SILENOR treatment (3 mg or 6 mg), as measured by the Tyrer's Symptom Checklist. Discontinuation-period emergent nausea and vomiting occurred in 5% of subjects treated with 6 mg SILENOR, versus 0% in 3 mg and placebo subjects.
16. HOW SUPPLIED/STORAGE AND HANDLING
16.1. How Supplied
SILENOR 3 mg tablets are oval shaped, blue, identified with debossed markings of "3" on one side and "SP" on the other, and are supplied as:
| NDC 42847-103-30 | Bottle of 30 |
SILENOR 6 mg tablets are oval shaped, green, identified with debossed markings of "6" on one side and "SP" on the other, and are supplied as:
| NDC 42847-106-30 | Bottle of 30 |
17. PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Sleep-driving and Other Complex Behaviors
There have been reports of people getting out of bed after taking a hypnotic and driving their cars while not fully awake, often with no memory of the event. If a patient experiences such an episode, it should be reported to his or her doctor immediately, since "sleep-driving" can be dangerous. This behavior is more likely to occur when a hypnotic is taken with alcohol or other central nervous system depressants [see Warnings and Precautions (5.2, 5.4) and Drug Interactions (7.3, 7.4)]. Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a hypnotic. As with "sleep-driving", patients usually do not remember these events.
In addition, patients should be advised to report all concomitant medications to the prescriber. Patients should be instructed to report events such as "sleep-driving" and other complex behaviors immediately to the prescriber.
Suicide risk and Worsening of Depression
Patients, their families, and their caregivers should be encouraged to be alert to worsening of depression, including suicidal thoughts and actions. Such symptoms should be reported to the patient's prescriber or health professional.
Administration Instructions
Patients should be counseled to take SILENOR within 30 minutes of bedtime and should confine their activities to those necessary to prepare for bed. SILENOR tablets should not be taken with or immediately after a meal [see Dosage and Administration (2.3)]. Advise patients NOT to take SILENOR when drinking alcohol [see Warnings and Precautions (5.2, 5.4) and Drug Interactions (7.3)].
Pregnancy
Advise patients that SILENOR use late in pregnancy may increase the risk for neonatal complications requiring prolonged hospitalization, respiratory support or tube feeding [see Use in Specific Populations (8.1)].
Lactation
Advise patients that breastfeeding is not recommended during treatment with SILENOR [see Use in Specific Populations (8.2)].
Infertility
Inform patients that SILENOR may cause reduced fertility. It is not known whether these effects on fertility are reversible [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 12/2022 | |
| MEDICATION GUIDE
SILENOR® [si-leh-nor] (doxepin) tablets |
||
| What is the most important information I should know about SILENOR? | ||
| SILENOR can cause serious side effects including: | ||
After taking SILENOR, you may get up out of bed while not being fully awake and do an activity that you do not know you are doing. The next morning, you may not remember that you did anything during the night. You have a higher chance for doing these activities if you drink alcohol or take other medicines that make you sleepy with SILENOR. Reported activities include:
|
||
| Stop taking SILENOR and call your healthcare provider right away if you find out that you have done any of the above activities after taking SILENOR. Important:
|
||
| Take SILENOR 30 minutes before bedtime. After taking SILENOR, you should only do activities needed to get ready for bed. | ||
| What is SILENOR? | ||
| SILENOR is a prescription medicine used to treat adults who have trouble staying asleep. | ||
| It is not known if SILENOR is safe and effective in children. | ||
Do not take SILENOR if you:
|
||
Before taking SILENOR, tell your healthcare provider about all of your medical conditions, including if you:
|
||
| Tell your healthcare provider about all of the medicines you take including prescription and over-the-counter medicines, vitamins and herbal supplements. | ||
| SILENOR and other medicines may affect each other causing side effects. SILENOR may affect the way other medicines work, and other medicines may affect how SILENOR works. | ||
Especially tell your healthcare provider if you take:
|
||
| Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider and pharmacist each time you get a new medicine. | ||
How should I take SILENOR?
|
||
What should I avoid during treatment with SILENOR?
|
||
| What are the possible side effects of SILENOR? | ||
SILENOR can cause serious side effects including:
|
||
| The most common side effects of SILENOR include: | ||
|
|
|
| SILENOR may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility. | ||
| These are not all of the possible side effects of SILENOR. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
How should I store SILENOR?
|
||
| Keep SILENOR and all medicines out of the reach of children. | ||
| General Information about the safe and effective use of SILENOR. | ||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SILENOR for a condition for which it was not prescribed. Do not give SILENOR to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about SILENOR that is written for healthcare professionals. | ||
| What are the ingredients in SILENOR? | ||
| Active Ingredient: doxepin hydrochloride | ||
| Inactive Ingredients: microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate. The 3 mg tablet also contains FD&C Blue No. 1. The 6 mg tablet also contains FD&C Yellow No. 10 and FD&C Blue No. 1. | ||
| Distributed by: Currax™ Pharmaceuticals LLC, Brentwood, TN 37027 USA SIL-LC088.02 For more information, contact Currax Pharmaceuticals LLC at 1-800-793-2145. |
||

