To reduce the development of drug-resistant bacteria and maintain the effectiveness of Maxaquin and other antibacterial drugs, Maxaquin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
DESCRIPTION
Maxaquin (lomefloxacin HCl) is a synthetic broad-spectrum antimicrobial agent for oral administration. Lomefloxacin HCl, a difluoroquinolone, is the monohydrochloride salt of (±)-1-ethyl-6,8-difluoro-1,4-dihydro-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid. Its empirical formula is C17H19F2N3O3•HCl, and its structural formula is:
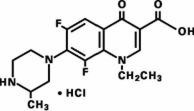
Lomefloxacin HCl is a white to pale yellow powder with a molecular weight of 387.8. It is slightly soluble in water and practically insoluble in alcohol. Lomefloxacin HCl is stable to heat and moisture but is sensitive to light in dilute aqueous solution.
Maxaquin is available as a film-coated tablet formulation containing 400 mg of lomefloxacin base, present as the hydrochloride salt. The base content of the hydrochloride salt is 90.6%. The inactive ingredients are carboxymethylcellulose calcium, hydroxypropyl cellulose, hypromellose, lactose, magnesium stearate, polyethylene glycol, polyoxyl 40 stearate, and titanium dioxide.
CLINICAL PHARMACOLOGY
Pharmacokinetics in healthy volunteers
In 6 fasting healthy male volunteers, approximately 95% to 98% of a single oral dose of lomefloxacin was absorbed. Absorption was rapid following single doses of 200 and 400 mg (Tmax 0.8 to 1.4 hours). Mean plasma concentration increased proportionally between 100 and 400 mg as shown below:
| Dose (mg) | Mean Peak Plasma Concentration (µg/mL) | Area Under Curve (AUC) (µg∙h/mL) |
|---|---|---|
| 100 | 0.8 | 5.6 |
| 200 | 1.4 | 10.9 |
| 400 | 3.2 | 26.1 |
In 6 healthy male volunteers administered 400 mg of lomefloxacin on an empty stomach qd for 7 days, the following mean pharmacokinetic parameter values were obtained:
| Cmax | 2.8 µg/mL |
| Cmin | 0.27 µg/mL |
| AUC0–24 h | 25.9 µg∙h/mL |
| Tmax | 1.5 h |
| t1/2 | 7.75 h |
The elimination half-life in 8 subjects with normal renal function was approximately 8 hours. At 24 hours postdose, subjects with normal renal function receiving single doses of 200 or 400 mg had mean plasma lomefloxacin concentrations of 0.10 and 0.24 µg/mL, respectively. Steady-state concentrations were achieved within 48 hours of initiating therapy with one-a-day dosing. There was no drug accumulation with single-daily dosing in patients with normal renal function.
Approximately 65% of an orally administered dose was excreted in the urine as unchanged drug in patients with normal renal function. Following a 400-mg dose of lomefloxacin administered qd for 7 days, the mean urine concentration 4 hours postdose was in excess of 300 µg/mL. The mean urine concentration exceeded 35 µg/mL for at least 24 hours after dosing.
Following a single 400-mg dose, the solubility of lomefloxacin in urine usually exceeded its peak urinary concentration 2- to 6-fold. In this study, urine pH affected the solubility of lomefloxacin with solubilities ranging from 7.8 mg/mL at pH 5.2, to 2.4 mg/mL at pH 6.5, and 3.03 mg/mL at pH 8.12.
The urinary excretion of lomefloxacin was virtually complete within 72 hours after cessation of dosing, with approximately 65% of the dose being recovered as parent drug and 9% as its glucuronide metabolite. The mean renal clearance was 145 mL/min in subjects with normal renal function (GFR = 120 mL/min). This may indicate tubular secretion.
Food effect
When lomefloxacin and food were administered concomitantly, the rate of drug absorption was delayed (Tmax increased to 2 hours [delayed by 41%], Cmax decreased by 18%), and the extent of absorption (AUC) was decreased by 12%.
Pharmacokinetics in the geriatric population
In 16 healthy elderly volunteers (61 to 76 years of age) with normal renal function for their age, the half-life of lomefloxacin (mean of 8 hours) and its peak plasma concentration (mean of 4.2 µg/mL) following a single 400-mg dose were similar to those in 8 younger subjects dosed with a single 400-mg dose. Thus, drug absorption appears unaffected in the elderly. Plasma clearance was, however, reduced in this elderly population by approximately 25%, and the AUC was increased by approximately 33%. This slower elimination most likely reflects the decreased renal function normally observed in the geriatric population.
Pharmacokinetics in renally impaired patients
In 8 patients with creatinine clearance (ClCr) between 10 and 40 mL/min/1.73 m2, the mean AUC after a single 400-mg dose of lomefloxacin increased 335% over the AUC demonstrated in patients with a ClCr> 80 mL/min/1.73 m2. Also, in these patients, the mean t1/2 increased to 21 hours. In 8 patients with ClCr< 10 mL/min/1.73 m2, the mean AUC after a single 400-mg dose of lomefloxacin increased 700% over the AUC demonstrated in patients with a ClCr> 80 mL/min/1.73 m2. In these patients with ClCr< 10 mL/min/1.73 m2, the mean t1/2 increased to 45 hours. The plasma clearance of lomefloxacin was closely correlated with creatinine clearance, ranging from 31 mL/min/1.73 m2 when creatinine clearance was zero to 271 mL/min/1.73 m2 at a normal creatinine clearance of 110 mL/min/1.73 m2. Peak lomefloxacin concentrations were not affected by the degree of renal function when single doses of lomefloxacin were administered. Adjustment of dosage schedules for patients with such decreases in renal function is warranted. (See Dosage and Administration.)
Pharmacokinetics in patients with cirrhosis
In 12 patients with histologically confirmed cirrhosis, no significant changes in rate or extent of lomefloxacin exposure (Cmax, Tmax, t1/2, or AUC) were observed when they were administered 400 mg of lomefloxacin as a single dose. No data are available in cirrhotic patients treated with multiple doses of lomefloxacin. Cirrhosis does not appear to reduce the nonrenal clearance of lomefloxacin. There does not appear to be a need for a dosage reduction in cirrhotic patients, provided adequate renal function is present.
Metabolism and pharmacodynamics of lomefloxacin
Lomefloxacin is minimally metabolized although 5 metabolites have been identified in human urine. The glucuronide metabolite is found in the highest concentration and accounts for approximately 9% of the administered dose. The other 4 metabolites together account for < 0.5% of the dose.
Approximately 10% of an oral dose was recovered as unchanged drug in the feces.
Serum protein binding of lomefloxacin is approximately 10%.
The following are mean tissue- or fluid-to-plasma ratios of lomefloxacin following oral administration. Studies have not been conducted to assess the penetration of lomefloxacin into human cerebrospinal fluid.
| Tissue or Body Fluid | Mean Tissue- or Fluid-to-Plasma Ratio |
|---|---|
| Bronchial mucosa | 2.1 |
| Bronchial secretions | 0.6 |
| Prostatic tissue | 2.0 |
| Sputum | 1.3 |
| Urine | 140.0 |
In two studies including 74 healthy volunteers, the minimal dose of UVA light needed to cause erythema (MED-UVA) was inversely proportional to plasma lomefloxacin concentration. The MED-UVA values (16 hours and 12 hours postdose) were significantly higher than the MED-UVA values 2 hours postdose at steady state. Increasing the interval between lomefloxacin dosing and exposure to UVA light increased the amount of light energy needed for photoreaction. In a study of 27 healthy volunteers, the steady state AUC values and Cmin values were equivalent whether the drug was administered in the morning or in the evening.
Microbiology
Lomefloxacin is a bactericidal agent with in vitro activity against a wide range of gram-negative and gram-positive organisms. The bactericidal action of lomefloxacin results from interference with the activity of the bacterial enzyme DNA gyrase, which is needed for the transcription and replication of bacterial DNA. The minimum bactericidal concentration (MBC) generally does not exceed the minimum inhibitory concentration (MIC) by more than a factor of 2, except for staphylococci, which usually have MBCs 2 to 4 times the MIC.
Lomefloxacin has been shown to be active against most strains of the following organisms both in vitro and in clinical infections: (See Indications and Usage.)
Gram-positive aerobes
- Staphylococcus saprophyticus
Gram-negative aerobes
- Citrobacter diversus
- Enterobacter cloacae
- Escherichia coli
- Haemophilus influenzae
- Klebsiella pneumoniae
- Moraxella catarrhalis
- Proteus mirabilis
- Pseudomonas aeruginosa (urinary tract only—See Indications and Usage and Warnings)
The following in vitro data are available; however, their clinical significance is unknown.
Lomefloxacin exhibits in vitro MICs of 2 µg/mL or less against most strains of the following organisms; however, the safety and effectiveness of lomefloxacin in treating clinical infections due to these organisms have not been established in adequate and well-controlled trials:
Gram-positive aerobes
- Staphylococcus aureus (including methicillin-resistant strains)
- Staphylococcus epidermidis (including methicillin-resistant strains)
Gram-negative aerobes
- Aeromonas hydrophila
- Citrobacter freundii
- Enterobacter aerogenes
- Enterobacter agglomerans
- Haemophilus parainfluenzae
- Hafnia alvei
- Klebsiella oxytoca
- Klebsiella ozaenae
- Morganella morganii
- Proteus vulgaris
- Providencia alcalifaciens
- Providencia rettgeri
- Serratia liquefaciens
- Serratia marcescens
Other organisms
- Legionella pneumophila
Beta-lactamase production should have no effect on the in vitro activity of lomefloxacin.
Most group A, B, D, and G streptococci, Streptococcus pneumoniae, Pseudomonas cepacia, Ureaplasma urealyticum, Mycoplasma hominis, and anaerobic bacteria are resistant to lomefloxacin.
Lomefloxacin appears slightly less active in vitro when tested at acidic pH. An increase in inoculum size has little effect on the in vitro activity of lomefloxacin. In vitro resistance to lomefloxacin develops slowly (multiple-step mutation). Rapid one-step development of resistance occurs only rarely (< 10–9) in vitro.
Cross-resistance between lomefloxacin and other quinolone-class antimicrobial agents has been reported; however, cross-resistance between lomefloxacin and members of other classes of antimicrobial agents, such as aminoglycosides, penicillins, tetracyclines, cephalosporins, or sulfonamides has not yet been reported. Lomefloxacin is active in vitro against some strains of cephalosporin- and aminoglycoside-resistant gram-negative bacteria.
Susceptibility tests
Diffusion techniques
Quantitative methods that require measurement of zone diameters give the most precise estimate of the susceptibility of bacteria to antimicrobial agents. One such standardized procedure1 that has been recommended for use with disks to test the susceptibility of organisms to lomefloxacin uses the 10-µg lomefloxacin disk. Interpretation involves correlation of the diameter obtained in the disk test with the MIC for lomefloxacin.
Reports from the laboratory giving results of the standard single-disk susceptibility test with a 10-µg lomefloxacin disk should be interpreted according to the following criteria:
| Zone Diameter (mm) | Interpretation |
|---|---|
| ≥ 22 | Susceptible (S) |
| 19–21 | Intermediate (I) |
| ≤ 18 | Resistant (R) |
A report of "susceptible" indicates that the pathogen is likely to be inhibited by generally achievable drug concentrations. A report of "intermediate" indicates that the result should be considered equivocal, and, if the organism is not fully susceptible to alternative clinically feasible drugs, the test should be repeated. This category provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "resistant" indicates that achievable drug concentrations are unlikely to be inhibitory, and other therapy should be selected.
Standardized susceptibility test procedures require the use of laboratory control organisms. The 10-µg lomefloxacin disk should give the following zone diameters:
| Organism | Zone Diameter (mm) |
|---|---|
| S aureus (ATCC 25923) | 23–29 |
| E coli (ATCC 25922) | 27–33 |
| P aeruginosa (ATCC 27853) | 22–28 |
Dilution techniques
Use a standardized dilution method2 (broth, agar, or microdilution) or equivalent with lomefloxacin powder. The MIC values obtained should be interpreted according to the following criteria:
| MIC (µg/mL) | Interpretation |
|---|---|
| ≤ 2 | Susceptible (S) |
| 4 | Intermediate (I) |
| ≥ 8 | Resistant (R) |
As with standard diffusion techniques, dilution methods require the use of laboratory control organisms. Standard lomefloxacin powder should provide the following MIC values:
| Organism | MIC (µg/mL) |
|---|---|
| S aureus (ATCC 29213) | 0.25–2.0 |
| E coli (ATCC 25922) | 0.03–0.12 |
| P aeruginosa (ATCC 27853) | 1.0–4.0 |
INDICATIONS AND USAGE
Treatment
Maxaquin (lomefloxacin HCl) film-coated tablets are indicated for the treatment of adults with mild to moderate infections caused by susceptible strains of the designated microorganisms in the conditions listed below: (See Dosage and Administration for specific dosing recommendations.)
LOWER RESPIRATORY TRACT
Acute Bacterial Exacerbation of Chronic Bronchitis caused by Haemophilus influenzae or Moraxella catarrhalis.1
NOTE: MAXAQUIN IS NOT INDICATED FOR THE EMPIRIC TREATMENT OF ACUTE BACTERIAL EXACERBATION OF CHRONIC BRONCHITIS WHEN IT IS PROBABLE THAT S PNEUMONIAE IS A CAUSATIVE PATHOGEN. S PNEUMONIAE EXHIBITS IN VITRO RESISTANCE TO LOMEFLOXACIN, AND THE SAFETY AND EFFICACY OF LOMEFLOXACIN IN THE TREATMENT OF PATIENTS WITH ACUTE BACTERIAL EXACERBATION OF CHRONIC BRONCHITIS CAUSED BY S PNEUMONIAE HAVE NOT BEEN DEMONSTRATED. IF LOMEFLOXACIN IS TO BE PRESCRIBED FOR GRAM–STAIN–GUIDED EMPIRIC THERAPY OF ACUTE BACTERIAL EXACERBATION OF CHRONIC BRONCHITIS, IT SHOULD BE USED ONLY IF SPUTUM GRAM STAIN DEMONSTRATES AN ADEQUATE QUALITY OF SPECIMEN (> 25 PMNs/LPF) AND THERE IS BOTH A PREDOMINANCE OF GRAM-NEGATIVE MICROORGANISMS AND NOT A PREDOMINANCE OF GRAM-POSITIVE MICROORGANISMS.
- 1
- Although treatment of infections due to this microorganism in this organ system demonstrated a clinically acceptable overall outcome, efficacy was studied in fewer than 10 infections.
URINARY TRACT
Uncomplicated Urinary Tract Infections (cystitis) caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, or Staphylococcus saprophyticus. (See DOSAGE AND ADMINISTRATION and CLINICAL STUDIES—UNCOMPLICATED CYSTITIS.)
Complicated Urinary Tract Infections caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, Citrobacter diversus,1 or Enterobacter cloacae.1
NOTE: In clinical trials with patients experiencing complicated urinary tract infections (UTIs) due to P aeruginosa, 12 of 16 patients had the microorganism eradicated from the urine after therapy with lomefloxacin. None of the patients had concomitant bacteremia. Serum levels of lomefloxacin do not reliably exceed the MIC of Pseudomonas isolates. THE SAFETY AND EFFICACY OF LOMEFLOXACIN IN TREATING PATIENTS WITH PSEUDOMONAS BACTEREMIA HAVE NOT BEEN ESTABLISHED.
Appropriate culture and susceptibility tests should be performed before antimicrobial treatment in order to isolate and identify microorganisms causing infection and to determine their susceptibility to lomefloxacin. In patients with UTIs, therapy with Maxaquin film-coated tablets may be initiated before results of these tests are known; once these results become available, appropriate therapy should be continued. In patients with an acute bacterial exacerbation of chronic bronchitis, therapy should not be started empirically with lomefloxacin when there is a probability the causative pathogen is S pneumoniae.
Beta-lactamase production should have no effect on lomefloxacin activity.
Prevention / prophylaxis
Maxaquin is indicated preoperatively for the prevention of infection in the following situations:
- Transrectal prostate biopsy: to reduce the incidence of urinary tract infection, in the early and late postoperative periods (3–5 days and 3–4 weeks postsurgery).
- Transurethral surgical procedures: to reduce the incidence of urinary tract infection in the early postoperative period (3–5 days postsurgery).
Efficacy in decreasing the incidence of infections other than urinary tract infection has not been established. Maxaquin, like all drugs for prophylaxis of transurethral surgical procedures, usually should not be used in minor urologic procedures for which prophylaxis is not indicated (eg, simple cystoscopy or retrograde pyelography). (See Dosage and Administration.)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Maxaquin and other antibacterial drugs, Maxaquin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
CONTRAINDICATIONS
Maxaquin (lomefloxacin HCl) is contraindicated in persons with a history of hypersensitivity to lomefloxacin or any member of the quinolone group of antimicrobial agents.
WARNINGS
MODERATE TO SEVERE PHOTOTOXIC REACTIONS HAVE OCCURRED IN PATIENTS EXPOSED TO DIRECT OR INDIRECT SUNLIGHT OR TO ARTIFICIAL ULTRAVIOLET LIGHT (eg, sunlamps) DURING OR FOLLOWING TREATMENT WITH LOMEFLOXACIN. THESE REACTIONS HAVE ALSO OCCURRED IN PATIENTS EXPOSED TO SHADED OR DIFFUSE LIGHT, INCLUDING EXPOSURE THROUGH GLASS. PATIENTS SHOULD BE ADVISED TO DISCONTINUE LOMEFLOXACIN THERAPY AT THE FIRST SIGNS OR SYMPTOMS OF A PHOTOTOXICITY REACTION SUCH AS A SENSATION OF SKIN BURNING, REDNESS, SWELLING, BLISTERS, RASH, ITCHING, OR DERMATITIS.
These phototoxic reactions have occurred with and without the use of sunscreens or sunblocks. Single doses of lomefloxacin have been associated with these types of reactions. In a few cases, recovery was prolonged for several weeks. As with some other types of phototoxicity, there is the potential for exacerbation of the reaction on re-exposure to sunlight or artificial ultraviolet light prior to complete recovery from the reaction. In rare cases, reactions have recurred up to several weeks after stopping lomefloxacin therapy.
EXPOSURE TO DIRECT OR INDIRECT SUNLIGHT (EVEN WHEN USING SUNSCREENS OR SUNBLOCKS) SHOULD BE AVOIDED WHILE TAKING LOMEFLOXACIN AND FOR SEVERAL DAYS FOLLOWING THERAPY. LOMEFLOXACIN THERAPY SHOULD BE DISCONTINUED IMMEDIATELY AT THE FIRST SIGNS OR SYMPTOMS OF PHOTOTOXICITY. RISK OF PHOTOTOXICITY MAY BE REDUCED BY TAKING LOMEFLOXACIN IN THE EVENING (See Dosage and Administration.)
THE SAFETY AND EFFICACY OF LOMEFLOXACIN IN PEDIATRIC PATIENTS AND ADOLESCENTS (UNDER THE AGE OF 18 YEARS), PREGNANT WOMEN, AND LACTATING WOMEN HAVE NOT BEEN ESTABLISHED. (See PRECAUTIONS—Pediatric Use, Pregnancy and Nursing Mothers subsections.) The oral administration of multiple doses of lomefloxacin to juvenile dogs at 0.3 times and to rats at 5.4 times the recommended adult human dose based on mg/m2 (0.6 and 34 times the recommended adult human dose based on mg/kg, respectively) caused arthropathy and lameness. Histopathologic examination of the weight-bearing joints of these animals revealed permanent lesions of the cartilage. Other quinolones also produce erosions of cartilage of weight-bearing joints and other signs of arthropathy in juvenile animals of various species. (See Animal Pharmacology.)
Convulsions have been reported in patients receiving lomefloxacin. Whether the convulsions were directly related to lomefloxacin administration has not yet been established. However, convulsions, increased intracranial pressure, and toxic psychoses have been reported in patients receiving other quinolones. Nevertheless, lomefloxacin has been associated with a possible increased risk of seizures compared to other quinolones. Some of these may occur with a relative absence of predisposing factors. Quinolones may also cause central nervous system (CNS) stimulation, which may lead to tremors, restlessness, lightheadedness, confusion, and hallucinations. If any of these reactions occurs in patients receiving lomefloxacin, the drug should be discontinued and appropriate measures instituted. However, until more information becomes available, lomefloxacin, like all other quinolones, should be used with caution in patients with known or suspected CNS disorders, such as severe cerebral arteriosclerosis, epilepsy, or other factors that predispose to seizures. (See Adverse Reactions.) Psychiatric disturbances, agitation, anxiety, and sleep disorders may be more common with lomefloxacin than other products in the quinolone class.
The safety and efficacy of lomefloxacin in the treatment of acute bacterial exacerbation of chronic bronchitis due to S pneumoniae have not been demonstrated. This product should not be used empirically in the treatment of acute bacterial exacerbation of chronic bronchitis when it is probable that S pneumoniae is a causative pathogen.
In clinical trials of complicated UTIs due to P aeruginosa, 12 of 16 patients had the microorganism eradicated from the urine after therapy with lomefloxacin. No patients had concomitant bacteremia. Serum levels of lomefloxacin do not reliably exceed the MIC of Pseudomonas isolates. THE SAFETY AND EFFICACY OF LOMEFLOXACIN IN TREATING PATIENTS WITH PSEUDOMONAS BACTEREMIA HAVE NOT BEEN ESTABLISHED.
Serious and occasionally fatal hypersensitivity (anaphylactoid or anaphylactic) reactions, some following the first dose, have been reported in patients receiving quinolone therapy. Some reactions were accompanied by cardiovascular collapse, loss of consciousness, tingling, pharyngeal or facial edema, dyspnea, urticaria, or itching. Only a few of these patients had a history of previous hypersensitivity reactions. Serious hypersensitivity reactions have also been reported following treatment with lomefloxacin. If an allergic reaction to lomefloxacin occurs, discontinue the drug. Serious acute hypersensitivity reactions may require immediate emergency treatment with epinephrine. Oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines, and airway management, including intubation, should be administered as indicated.
Pseudomembranous colitis has been reported with nearly all antibacterial agents, including lomefloxacin, and may range from mild to life-threatening in severity. Therefore, it is important to consider this diagnosis in patients who present with diarrhea subsequent to the administration of antibacterial agents. Treatment with antimicrobial agents alters the normal flora of the colon and may permit overgrowth of clostridia. Studies indicate that a toxin produced by Clostridium difficile is a primary cause of "antibiotic-associated colitis." After the diagnosis of pseudomembranous colitis has been established, therapeutic measures should be initiated. Mild cases of pseudomembranous colitis usually respond to discontinuation of drug alone. In moderate to severe cases, consideration should be given to management with fluids and electrolytes, protein supplementation, and treatment with an antibacterial drug clinically effective against C difficile colitis.
QT interval prolongation/torsades de pointes
Rare cases of torsades de pointes have been spontaneously reported during post-marketing surveillance in patients receiving quinolones, including lomefloxacin. These rare cases were associated with one or more of the following factors: age over 60, female gender, underlying cardiac disease, and/or use of multiple medications. Lomefloxacin should be avoided in patients with known prolongation of the QT interval, patients with uncorrected hypokalemia, and patients receiving class IA (quinidine, procainamide), or class III (amiodarone, sotalol) antiarrhythmic agents.
Peripheral neuropathy
Rare cases of sensory or sensorimotor axonal polyneuropathy affecting small and/or large axons resulting in paresthesias, hypoesthesias, dysesthesias and weakness have been reported in patients receiving quinolones, including lomefloxacin. Lomefloxacin should be discontinued if the patient experiences symptoms of neuropathy including pain, burning, tingling, numbness, and/or weakness, or is found to have deficits in light touch, pain, temperature, position sense, vibratory sensation, and/or motor strength in order to prevent the development of an irreversible condition.
Tendon effects
Ruptures of the shoulder, hand, Achilles tendon or other tendons that required surgical repair or resulted in prolonged disability have been reported in patients receiving quinolones, including lomefloxacin. Postmarketing surveillance reports indicate that this risk may be increased in patients receiving concomitant corticosteroids, especially the elderly. Lomefloxacin should be discontinued if the patient experiences pain, inflammation, or rupture of a tendon. Patients should rest and refrain from exercise until the diagnosis of tendonitis or tendon rupture has been excluded. Tendon rupture can occur during or after therapy with quinolones, including lomefloxacin.
PRECAUTIONS
General
Alteration of the dosage regimen is recommended for patients with impairment of renal function (ClCr< 40 mL/min/1.73 m2). (See Dosage and Administration.)
Prescribing Maxaquin in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
Information for patients
Patients should be advised
- to avoid to the maximum extent possible direct or indirect sunlight (including exposure through glass and exposure through sunscreens and sunblocks) and artificial ultraviolet light (eg sunlamps) during treatment with lomefloxacin and for several days after therapy;
- that they may reduce the risk of developing phototoxicity from sunlight by taking the daily dose of lomefloxacin at least 12 hours before exposure to the sun (eg in the evening);
- to discontinue lomefloxacin therapy at the first signs or symptoms of phototoxicity reaction such as a sensation of skin burning, redness, swelling, blisters, rash, itching, or dermatitis;
- that a patient who has experienced a phototoxic reaction should avoid re-exposure to sunlight and artificial ultraviolet light until he has completely recovered from the reaction. In rare cases, reactions have recurred up to several weeks after stopping lomefloxacin therapy.
- to drink fluids liberally;
- that lomefloxacin can be taken without regard to meals;
- that mineral supplements or vitamins with iron or minerals should not be taken within the 2-hour period before or after taking lomefloxacin (see Drug Interactions);
- that sucralfate and antacids containing magnesium or aluminum, or Videx® (didanosine), chewable/buffered tablets or the pediatric powder for oral solution should not be taken within 4 hours before or 2 hours after taking lomefloxacin. (See PRECAUTIONS — Drug Interactions.)
- that lomefloxacin can cause dizziness and lightheadedness and, therefore, patients should know how they react to lomefloxacin before they operate an automobile or machinery or engage in activities requiring mental alertness and coordination;
- to discontinue treatment and inform their physician if they experience pain, inflammation, or rupture of a tendon, and to rest and refrain from exercise until the diagnosis of tendinitis or tendon rupture has been confidently excluded;
- that lomefloxacin may be associated with hypersensitivity reactions, even following the first dose, and to discontinue the drug at the first sign of a skin rash or other allergic reaction;
- that convulsions have been reported in patients taking quinolones, including lomefloxacin, and to notify their physician before taking this drug if there is a history of this condition.
- that antibacterial drugs including Maxaquin should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Maxaquin is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by Maxaquin or other antibacterial drugs in the future.
Drug interactions
Theophylline
In three pharmacokinetic studies including 46 normal, healthy subjects, theophylline clearance and concentration were not significantly altered by the addition of lomefloxacin. In clinical studies where patients were on chronic theophylline therapy, lomefloxacin had no measurable effect on the mean distribution of theophylline concentrations or the mean estimates of theophylline clearance. Though individual theophylline levels fluctuated, there were no clinically significant symptoms of drug interaction.
Antacids and sucralfate
Sucralfate and antacids containing magnesium or aluminum, as well as formulations containing divalent and trivalent cations such as Videx® (didanosine), chewable/buffered tablets or the pediatric powder for oral solution can form chelation complexes with lomefloxacin and interfere with its bioavailability. Sucralfate administered 2 hours before lomefloxacin resulted in a slower absorption (mean Cmax decreased by 30% and mean Tmax increased by 1 hour) and a lesser extent of absorption (mean AUC decreased by approximately 25%). Magnesium- and aluminum-containing antacids, administered concomitantly with lomefloxacin, significantly decreased the bioavailability (48%) of lomefloxacin. Separating the doses of antacid and lomefloxacin minimizes this decrease in bioavailability; therefore, administration of these agents should precede lomefloxacin dosing by 4 hours or follow lomefloxacin dosing by at least 2 hours.
Caffeine
Two hundred mg of caffeine (equivalent to 1 to 3 cups of American coffee) was administered to 16 normal, healthy volunteers who had achieved steady-state blood concentrations of lomefloxacin after being dosed at 400 mg qd. This did not result in any statistically or clinically relevant changes in the pharmacokinetic parameters of either caffeine or its major metabolite, paraxanthine. No data are available on potential interactions in individuals who consume greater than 200 mg of caffeine per day or in those, such as the geriatric population, who are generally believed to be more susceptible to the development of drug-induced CNS-related adverse effects. Other quinolones have demonstrated moderate to marked interference with the metabolism of caffeine, resulting in a reduced clearance, a prolongation of plasma half-life, and an increase in symptoms that accompany high levels of caffeine.
Cimetidine
Cimetidine has been demonstrated to interfere with the elimination of other quinolones. This interference has resulted in significant increases in half-life and AUC. The interaction between lomefloxacin and cimetidine has not been studied.
Cyclosporine
Elevated serum levels of cyclosporine have been reported with concomitant use of cyclosporine with other members of the quinolone class. Interaction between lomefloxacin and cyclosporine has not been studied.
Omeprazole
No clinically significant changes in lomefloxacin pharmacokinetics (AUC, Cmax, or Tmax) were observed when a single dose of lomefloxacin 400 mg was given after multiple doses of omeprazole (20 mg qd) in 13 healthy volunteers. Changes in omeprazole pharmacokinetics were not studied.
Phenytoin
No significant differences were observed in mean phenytoin AUC, Cmax, Cmin or Tmax (although Cmax increased by 11%) when extended phenytoin sodium capsules (100 mg tid) were coadministered with lomefloxacin (400 mg qd) for five days in 15 healthy males. Lomefloxacin is unlikely to have a significant effect on phenytoin metabolism.
Probenecid
Probenecid slows the renal elimination of lomefloxacin. An increase of 63% in the mean AUC and increases of 50% and 4%, respectively, in the mean Tmax and mean Cmax were noted in 1 study of 6 individuals.
Terfenadine
No clinically significant changes occurred in heart rate or corrected QT intervals, or in terfenadine metabolite or lomefloxacin pharmacokinetics, during concurrent administration of lomefloxacin and terfenadine at steady-state in 28 healthy males.
Warfarin
Quinolones may enhance the effects of the oral anticoagulant, warfarin, or its derivatives. When these products are administered concomitantly, prothrombin or other suitable coagulation tests should be monitored closely. However, no clinically or statistically significant differences in prothrombin time ratio or warfarin enantiomer pharmacokinetics were observed in a small study of 7 healthy males who received both warfarin and lomefloxacin under steady-state conditions.
Carcinogenesis, mutagenesis, impairment of fertility
Carcinogenesis
Hairless (Skh-1) mice were exposed to UVA light for 3.5 hours five times every two weeks for up to 52 weeks while concurrently being administered lomefloxacin. The lomefloxacin doses used in this study caused a phototoxic response. In mice treated with both UVA and lomefloxacin concomitantly, the time to development of skin tumors was 16 weeks. In mice treated concomitantly in this model with both UVA and other quinolones, the times to development of skin tumors ranged from 28 to 52 weeks.
Ninety-two percent (92%) of the mice treated concomitantly with both UVA and lomefloxacin developed well-differentiated squamous cell carcinomas of the skin. These squamous cell carcinomas were nonmetastatic and were endophytic in character. Two-thirds of these squamous cell carcinomas contained large central keratinous inclusion masses and were thought to arise from the vestigial hair follicles in these hairless animals.
In this model, mice treated with lomefloxacin alone did not develop skin or systemic tumors.
There are no data from similar models using pigmented mice and/or fully haired mice
The clinical significance of these findings to humans is unknown.
Mutagenesis
One in vitro mutagenicity test (CHO/HGPRT assay) was weakly positive at lomefloxacin concentrations ≥ 226 µg/mL and negative at concentrations < 226 µg/mL. Two other in vitro mutagenicity tests (chromosomal aberrations in Chinese hamster ovary cells, chromosomal aberrations in human lymphocytes) and two in vivo mouse micronucleus mutagenicity tests were all negative.
Pregnancy
Teratogenic effects. Pregnancy Category C
Reproductive function studies have been performed in rats at doses up to 8 times the recommended human dose based on mg/m2 (34 times the recommended human dose based on mg/kg), and no impaired fertility or harm to the fetus was reported due to lomefloxacin. Increased incidence of fetal loss in monkeys has been observed at approximately 3 to 6 times the recommended human dose based on mg/m2 (6 to 12 times the recommended human dose based on mg/kg). No teratogenicity has been observed in rats and monkeys at up to 16 times the recommended human dose exposure. In the rabbit, maternal toxicity and associated fetotoxicity, decreased placental weight, and variations of the coccygeal vertebrae occurred at doses 2 times the recommended human exposure based on mg/m2. There are, however, no adequate and well-controlled studies in pregnant women. Lomefloxacin should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing mothers
It is not known whether lomefloxacin is excreted in human milk. However, it is known that other drugs of this class are excreted in human milk and that lomefloxacin is excreted in the milk of lactating rats. Because of the potential for serious adverse reactions from lomefloxacin in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric use
The safety and effectiveness of lomefloxacin in pediatric patients and adolescents less than 18 years of age have not been established. Lomefloxacin causes arthropathy in juvenile animals of several species. (See Warnings and Animal Pharmacology.)
Geriatric use
Of the total number of subjects in clinical studies of lomefloxacin, 25% were ≥ 65 years and 9% were ≥ 75 years. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. (See Clinical Pharmacology — Pharmacokinetics in the geriatric population.)
ADVERSE REACTIONS
In clinical trials, most of the adverse events reported were mild to moderate in severity and transient in nature. During these clinical investigations, 5,623 patients received Maxaquin. In 2.2% of the patients, lomefloxacin was discontinued because of adverse events, primarily involving the gastrointestinal system (0.7%), skin (0.7%), or CNS (0.5%).
Adverse clinical events
The events with the highest incidence (≥ 1%) in patients, regardless of relationship to drug, were headache (3.6%), nausea (3.5%), photosensitivity (2.3%) [see Warnings], dizziness (2.1%), diarrhea (1.4%), and abdominal pain (1.2%).
Additional clinical events reported in < 1% of patients treated with Maxaquin, regardless of relationship to drug, are listed below:
Autonomic: increased sweating, dry mouth, flushing, syncope.
Body as a whole: fatigue, back pain, malaise, asthenia, chest pain, face edema, hot flashes, influenza-like symptoms, edema, chills, allergic reaction, anaphylactoid reaction, decreased heat tolerance.
Cardiovascular: tachycardia, hypertension, hypotension, myocardial infarction, angina pectoris, cardiac failure, bradycardia, arrhythmia, phlebitis, pulmonary embolism, extrasystoles, cerebrovascular disorder, cyanosis, cardiomyopathy.
Central and peripheral nervous system: tremor, vertigo, paresthesias, twitching, hypertonia, convulsions, hyperkinesia, coma.
Gastrointestinal: dyspepsia, vomiting, flatulence, constipation, gastrointestinal bleeding, dysphagia, stomatitis, tongue discoloration, gastrointestinal inflammation.
Hearing: earache, tinnitus.
Hematologic: purpura, lymphadenopathy, thrombocythemia, anemia, thrombocytopenia, increased fibrinolysis.
Hepatic: abnormal liver function.
Metabolic: thirst, hyperglycemia, hypoglycemia, gout.
Musculoskeletal: arthralgia, myalgia, leg cramps.
Ophthalmologic: abnormal vision, conjunctivitis, photophobia, eye pain, abnormal lacrimation.
Psychiatric: insomnia, nervousness, somnolence, anorexia, depression, confusion, agitation, increased appetite, depersonalization, paranoid reaction, anxiety, paroniria, abnormal thinking, concentration impairment.
Reproductive system: Female: vaginal moniliasis, vaginitis, leukorrhea, menstrual disorder, perineal pain, intermenstrual bleeding. Male: epididymitis, orchitis.
Resistance mechanism: viral infection, moniliasis, fungal infection.
Respiratory: respiratory infection, rhinitis, pharyngitis, dyspnea, cough, epistaxis, bronchospasm, respiratory disorder, increased sputum, stridor, respiratory depression.
Skin/Allergic: pruritus, rash, urticaria, skin exfoliation, bullous eruption, eczema, skin disorder, acne, skin discoloration, skin ulceration, angioedema. (See also Body as a whole.)
Special senses: taste perversion.
Urinary: hematuria, micturition disorder, dysuria, strangury, anuria.
Adverse laboratory events
Changes in laboratory parameters, listed as adverse events, without regard to drug relationship include:
Hematologic: monocytosis (0.2%), eosinophilia (0.1%), leukopenia (0.1%), leukocytosis (0.1%).
Renal: elevated BUN (0.1%), decreased potassium (0.1%), increased creatinine (0.1%).
Hepatic: elevations of ALT (SGPT) (0.4%), AST (SGOT) (0.3%), bilirubin (0.1%), alkaline phosphatase (0.1%).
Additional laboratory changes occurring in < 0.1% in the clinical studies included: elevation of serum gamma glutamyl transferase, decrease in total protein or albumin, prolongation of prothrombin time, anemia, decrease in hemoglobin, thrombocythemia, thrombocytopenia, abnormalities of urine specific gravity or serum electrolytes, increased albumin, elevated ESR, albuminuria, macrocytosis.
Post-Marketing Adverse Events
Post-marketing adverse events
Adverse events reported from worldwide marketing experience with lomefloxacin are: anaphylaxis, cardiopulmonary arrest, laryngeal or pulmonary edema, ataxia, cerebral thrombosis, hallucinations, painful oral mucosa, pseudomembranous colitis, hemolytic anemia, hepatitis, tendinitis, diplopia, photophobia, phobia, exfoliative dermatitis, hyperpigmentation, Stevens-Johnson syndrome, toxic epidermal necrolysis, dysgeusia, interstitial nephritis, polyuria, renal failure, urinary retention, and vasculitis.
Quinolone-class adverse events
Additional quinolone-class adverse events include: peripheral neuropathy, torsades de pointes, erythema nodosum, hepatic necrosis, possible exacerbation of myasthenia gravis, dysphasia, nystagmus, intestinal perforation, manic reaction, renal calculi, acidosis and hiccough.
Laboratory adverse events include: agranulocytosis, elevation of serum triglycerides, elevation of serum cholesterol, elevation of blood glucose, elevation of serum potassium, albuminuria, candiduria, and crystalluria.
OVERDOSAGE
Information on overdosage in humans is limited. In the event of acute overdosage, the stomach should be emptied by inducing vomiting or by gastric lavage, and the patient should be carefully observed and given supportive treatment. Adequate hydration must be maintained. Hemodialysis or peritoneal dialysis is unlikely to aid in the removal of lomefloxacin as < 3% is removed by these modalities.
Clinical signs of acute toxicity in rodents progressed from salivation to tremors, decreased activity, dyspnea, and clonic convulsions prior to death. These signs were noted in rats and mice as lomefloxacin doses were increased.
DOSAGE AND ADMINISTRATION
Maxaquin (lomefloxacin HCl) may be taken without regard to meals. Sucralfate and antacids containing magnesium or aluminum, or Videx® (didanosine), chewable/buffered tablets or the pediatric powder for oral solution should not be taken within 4 hours before or 2 hours after taking lomefloxacin. Risk of reaction to solar UVA light may be reduced by taking Maxaquin at least 12 hours before exposure to the sun (eg, in the evening). (See Clinical Pharmacology.)
See Indications and Usage for information on appropriate pathogens and patient populations.
Treatment
Patients with normal renal function
The recommended daily dose of Maxaquin is described in the following chart:
| Infection | Unit Dose | Frequency | Duration | Daily Dose |
|---|---|---|---|---|
| Acute bacterial exacerbation of chronic bronchitis | 400 mg | qd | 10 days | 400 mg |
| Uncomplicated cystitis in females caused by E coli | 400 mg | qd | 3 days | 400 mg |
| (see CLINICAL STUDIES—UNCOMPLICATED CYSTITIS.) | ||||
| Uncomplicated cystitis caused by K pneumoniae, P mirabilis, or S Saprophyticus | 400 mg | qd | 10 days | 400 mg |
| Complicated UTI | 400 mg | qd | 14 days | 400 mg |
Elderly patients
No dosage adjustment is needed for elderly patients with normal renal function (ClCr≥ 40 mL/min/1.73 m2).
Patients with impaired renal function
Lomefloxacin is primarily eliminated by renal excretion. (See Clinical Pharmacology.) Modification of dosage is recommended in patients with renal dysfunction. In patients with a creatinine clearance > 10 mL/min/1.73 m2 but < 40 mL/min/1.73 m2, the recommended dosage is an initial loading dose of 400 mg followed by daily maintenance doses of 200 mg (1/2 tablet) once daily for the duration of treatment. It is suggested that serial determinations of lomefloxacin levels be performed to determine any necessary alteration in the appropriate next dosing interval.
If only the serum creatinine is known, the following formula may be used to estimate creatinine clearance.
Men: (weight in kg) × (140 – age)
(72) × serum creatinine (mg/dL)
Women: (0.85) × (calculated value for men)
Dialysis patients
Hemodialysis removes only a negligible amount of lomefloxacin (3% in 4 hours). Hemodialysis patients should receive an initial loading dose of 400 mg followed by daily maintenance doses of 200 mg (1/2 tablet) once daily for the duration of treatment.
Patients with cirrhosis
Cirrhosis does not reduce the nonrenal clearance of lomefloxacin. The need for a dosage reduction in this population should be based on the degree of renal function of the patient and on the plasma concentrations. (See Clinical Pharmacology and Dosage and Administration—Patients with impaired renal function.)
Prevention / prophylaxis
The recommended dose of Maxaquin is described in the following chart:
| Procedure | Dose | Oral Administration |
|---|---|---|
|
||
| Transrectal prostate biopsy | 400 mg single dose | 1–6 hours prior to procedure |
| Transurethral surgical procedures* | 400 mg single dose | 2–6 hours prior to procedure |
HOW SUPPLIED
Maxaquin (lomefloxacin HCl) is supplied as a scored, film-coated tablet containing the equivalent of 400 mg of lomefloxacin base present as the hydrochloride. The tablet is oval, white, and film-coated with "MAXAQUIN 400" debossed on one side and scored on the other side and is supplied in:
| NDC Number | Size |
|---|---|
| 0025-5501-01 | bottle of 20 |
Store at 59° to 77°F (15° to 25°C).
CLINICAL STUDIES—UNCOMPLICATED CYSTITIS
In three controlled clinical studies of uncomplicated cystitis in females, two performed in the United States and one in Canada, lomefloxacin was compared to other oral antimicrobial agents. In these studies, using very strict evaluability criteria and microbiological criteria at 5–9 days posttherapy follow-up, the following bacterial eradication outcomes were obtained:
STUDIES 1, 2, AND 3
| U.S. AND CANADIAN STUDIES | ||||
|---|---|---|---|---|
| Lomefloxacin 3-Day Treatment | Norfloxacin 3-Day Treatment | Ofloxacin 3-Day Treatment | Trimethoprim/ sulfamethoxazole 10-Day Treatment |
|
| E coli | 133/135 (99%) | 36/39 (92%) | 65/67 (97%) | 33/34 (97%) |
| K pneumoniae | 7/7 (100%) | 2/2 (100%) | 4/4 (100%) | 2/2 (100%) |
| P mirabilis | 8/8 (100%) | 1/1 (100%) | 2/2 (100%) | 1/1 (100%) |
| S saprophyticus | 11/11 (100%) | 3/3 (100%) | 1/1 (100%) | 0/0 |
STUDY 4
In a controlled clinical study of uncomplicated cystitis performed in Sweden, lomefloxacin 3-day treatment was compared with lomefloxacin 7-day treatment and norfloxacin 7-day treatment. In this study, using very strict evaluability criteria and microbiological criteria at 5–9 days post-therapy follow-up, the following bacterial eradication outcomes were obtained:
| SWEDISH STUDY | ||||
|---|---|---|---|---|
| Lomefloxacin 3-Day Treatment | Lomefloxacin 7-Day Treatment | Norfloxacin 7-Day Treatment |
||
| E coli | 101/109 (93%) | 102/104 (98%) | 108/110 (98%) | |
| K pneumoniae | 2/2 (100%) | 5/5 (100%) | 1/1 (100%) | |
| P mirabilis | 0/0 | 6/6 (100%) | 4/4 (100%) | |
| S saprophyticus | 11/17 (65%) | 23/23 (100%) | 16/16 (100%) | |
ANIMAL PHARMACOLOGY
Lomefloxacin and other quinolones have been shown to cause arthropathy in juvenile animals. Arthropathy, involving multiple diarthrodial joints, was observed in juvenile dogs administered lomefloxacin at doses as low as 4.5 mg/kg for 7 to 8 days (0.3 times the recommended human dose based on mg/m2 or 0.6 times the recommended human dose based on mg/kg). In juvenile rats, no changes were observed in the joints with doses up to 91 mg/kg for 7 days (2 times the recommended human dose based on mg/m2 or 11 times the recommended human dose based on mg/kg). (See Warnings.)
In a 13-week oral rat study, gamma globulin decreased when lomefloxacin was administered at less than the recommended human exposure. Beta globulin decreased when lomefloxacin was administered at 0.6 to 2 times the recommended human dose based on mg/m2. The A/G ratio increased when lomefloxacin was administered at 6 to 20 times the human dose. Following a 4-week recovery period, beta globulins in the females and A/G ratios in the females returned to control values. Gamma globulin values in the females and beta and gamma globulins and A/G ratios in the males were still statistically significantly different from control values. No effects on globulins were seen in oral studies in dogs or monkeys in the limited number of specimens collected.
Twenty-seven NSAIDs, administered concomitantly with lomefloxacin, were tested for seizure induction in mice at approximately 2 times the recommended human dose based on mg/m2. At a dose of lomefloxacin equivalent to the recommended human exposure based on mg/m2 (10 times the human dose based on mg/kg), only fenbufen, when coadministered, produced an increase in seizures.
Crystalluria and ocular toxicity, seen with some related quinolones, were not observed in any lomefloxacin-treated animals, either in studies designed to look for these effects specifically or in subchronic and chronic toxicity studies in rats, dogs, and monkeys.
Long-term, high-dose systemic use of other quinolones in experimental animals has caused lenticular opacities; however, this finding was not observed with lomefloxacin.
REFERENCES
- National Committee for Clinical Laboratory Standards, Performance Standards for Antimicrobial Disk Susceptibility Tests—4th ed. Approved Standard NCCLS Document M2–A4, vol 10, No. 7, NCCLS, Villanova, Pa, 1990.
- National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically—2nd ed. Approved Standard NCCLS Document M7–A2, vol 10, No. 8, NCCLS, Villanova, Pa, 1990.
