BOSENTAN- bosentan tablet, film coated
Alvogen Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use BOSENTAN TABLETS safely and effectively. See full prescribing information for BOSENTAN TABLETS.
BOSENTAN tablets, for oral use Initial U.S. Approval: 2001
WARNING: RISKS OF HEPATOTOXICITY and EMBRYO-FETAL TOXICITY
Bosentan Tablets are available only through a restricted distribution program called the Bosentan REMS Program because of these risks (5.3):
|
FULL PRESCRIBING INFORMATION
WARNING: RISKS OF HEPATOTOXICITY and EMBRYO-FETAL TOXICITY
Because of the risks of hepatotoxicity and birth defects, Bosentan Tablets are available only through a restricted program called the Bosentan REMS Program. The Bosentan REMS Program is a component of the Bosentan Tablets Risk Evaluation and Mitigation Strategy (REMS). Under the Bosentan REMS, prescribers, patients, and pharmacies must enroll in the program.[see Warnings and Precautions (5.3)].
Hepatotoxicity
In clinical studies, Bosentan Tablets caused at least 3-fold upper limit of normal (ULN) elevation of liver aminotransferases (ALT and AST) in about 11% of patients, accompanied by elevated bilirubin in a small number of cases. Because these changes are a marker for potential serious hepatotoxicity, serum aminotransferase levels must be measured prior to initiation of treatment and then monthly [see Dosage and Administration (2.4),Warnings and Precautions (5.1)]. In the postmarketing period, in the setting of close monitoring, rare cases of unexplained hepatic cirrhosis were reported after prolonged (> 12 months) therapy with Bosentan Tablets in patients with multiple comorbidities and drug therapies. There have also been reports of liver failure. The contribution of Bosentan Tablets in these cases could not be excluded.
In at least one case, the initial presentation (after > 20 months of treatment) included pronounced elevations in aminotransferases and bilirubin levels accompanied by non-specific symptoms, all of which resolved slowly over time after discontinuation of Bosentan Tablets. This case reinforces the importance of strict adherence to the monthly monitoring schedule for the duration of treatment and the treatment algorithm, which includes stopping Bosentan Tablets with a rise of aminotransferases accompanied by signs or symptoms of liver dysfunction [see Dosage and Administration (2.4)].
Elevations in aminotransferases require close attention [see Dosage and Administration (2.4)]. Bosentan Tablets should generally be avoided in patients with elevated aminotransferases (> 3 x ULN) at baseline because monitoring for hepatotoxicity may be more difficult. If liver aminotransferase elevations are accompanied by clinical symptoms of hepatotoxicity (such as nausea, vomiting, fever, abdominal pain, jaundice, or unusual lethargy or fatigue) or increases in bilirubin ≥ 2 x ULN, treatment with Bosentan Tablets should be stopped. There is no experience with the reintroduction of Bosentan Tablets in these circumstances.
Embryo-Fetal Toxicity
Bosentan Tablets are likely to cause major birth defects if used by pregnant females based on animal data [see Warnings and Precautions (5.2),Use in Specific Populations (8.1)]. Therefore, pregnancy must be excluded before the start of treatment with Bosentan Tablets. Throughout treatment and for one month after stopping Bosentan Tablets, females of reproductive potential must use two reliable methods of contraception unless the patient has an intrauterine device (IUD) or tubal sterilization, in which case no other contraception is needed. Hormonal contraceptives, including oral, injectable, transdermal, and implantable contraceptives should not be used as the sole means of contraception because these may not be effective in patients receiving Bosentan Tablets [see Drug Interactions (7.2)]. Obtain monthly pregnancy tests.
1 INDICATIONS AND USAGE
Bosentan Tablets are indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1):
- in adults to improve exercise ability and to decrease clinical worsening. Studies establishing effectiveness included predominantly patients with WHO Functional Class II-IV symptoms and etiologies of idiopathic or heritable PAH (60%), PAH associated with connective tissue diseases (21%), and PAH associated with congenital heart disease with left-to-right shunts (18%) [see Clinical Studies (14.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Required Monitoring
Healthcare professionals who prescribe Bosentan Tablets must enroll in the Bosentan REMS Program and must comply with the required monitoring to minimize the risks associated with Bosentan Tablets [see Warnings and Precautions (5.3)].
Obtain a pregnancy test in females of reproductive potential prior to Bosentan Tablets treatment, monthly during treatment and one month after stopping Bosentan Tablets. Initiate treatment with Bosentan Tablets in females of reproductive potential only after a negative pregnancy test [see Boxed Warning, Contraindications (4.1), Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
Measure liver aminotransferase levels prior to initiation of treatment and then monthly [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
Administer Bosentan Tablets orally following the dosing recommendations in Table 1. Doses above 125 mg twice daily did not appear to confer additional benefit sufficient to offset the increased risk of hepatotoxicity.
Table 1: Dosing Recommendations
| Initial 4 weeks | Maintenance (after 4 weeks) | |
| Patients >12 years of age and >40 kg | 62.5 mg twice daily | 125 mg twice daily |
| Patients >12 years of age and <40 kg | 62.5 mg twice daily | 62.5 mg twice daily |
2.4 Dosage Adjustments for Aminotransferase Elevations
If aminotransferase levels increase, adjust monitoring and treatment plan according to Table 2.
Discontinue Bosentan Tablets if liver aminotransferase elevations are accompanied by clinical symptoms of hepatotoxicity (such as nausea, vomiting, fever, abdominal pain, jaundice, or unusual lethargy or fatigue) or bilirubin ≥ 2 x Upper Limit of Normal (ULN). There is no experience with the reintroduction of Bosentan Tablets in these circumstances.
| ALT/AST levels | Treatment and monitoring recommendations |
| > 3 and ≤ 5 x ULN | Confirm by another aminotransferase test; if confirmed,
-in patients >12 years and >40 kg, reduce the daily dose to 62.5 mg twice daily or interrupt treatment, and monitor aminotransferase levels at least every 2 weeks. If the aminotransferase levels return to pretreatment values, treatment may continue or be reintroduced at 62.5 mg twice daily, with reassessment of aminotransferase levels within 3 days. |
| > 5 and ≤ 8 x ULN | Confirm by another aminotransferase test; if confirmed, stop treatment and monitor aminotransferase levels at least every 2 weeks. Once the aminotransferase levels return to pretreatment values, -in patients >12 years, consider reintroduction of treatment at 62.5 mg twice daily, with reassessment of aminotransferase levels within 3 days. |
| > 8 x ULN | Stop treatment permanently. There is no experience with reintroduction of Bosentan in these circumstances. |
2.5 Use with Ritonavir
Co-administration of Bosentan Tablets in Patients on Ritonavir
In patients who have been receiving ritonavir for at least 10 days, start Bosentan Tablets at the recommended initial dose once daily or every other day based upon individual tolerability [see Cytochrome P450 Drug Interactions (7.1)].
Co-administration of Ritonavir in Patients on Bosentan Tablets
Discontinue use of Bosentan Tablets at least 36 hours prior to initiation of ritonavir. After at least 10 days following the initiation of ritonavir, resume Bosentan Tablets at the recommended initial dose once daily or every other day based upon individual tolerability [see Cytochrome P450 Drug Interactions (7.1)].
2.6 Use in Patients with Pre-existing Hepatic Impairment
Avoid initiation of Bosentan Tablets in patients with aminotransferases >3 x ULN. No dose adjustment is required in patients with mildly impaired liver function [see Warnings and Precautions (5.3), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
62.5 mg tablets: round, biconvex, yellow, coated tablet, "A" debossed on one side and "2" debossed on the other side.
125 mg tablets: capsule-shaped, biconvex, yellow, coated tablet, "ALV" debossed on one side and "271" debossed on the other side.
4 CONTRAINDICATIONS
4.1 Pregnancy
Use of Bosentan Tablets is contraindicated in females who are or may become pregnant. To prevent pregnancy, females of reproductive potential must use two reliable forms of contraception during treatment and for one month after stopping Bosentan Tablets. [see Boxed Warning, Warnings and Precautions (5.2), Drug Interactions (7.2), Use in Specific Populations (8.1)].
4.2 Use with Cyclosporine A
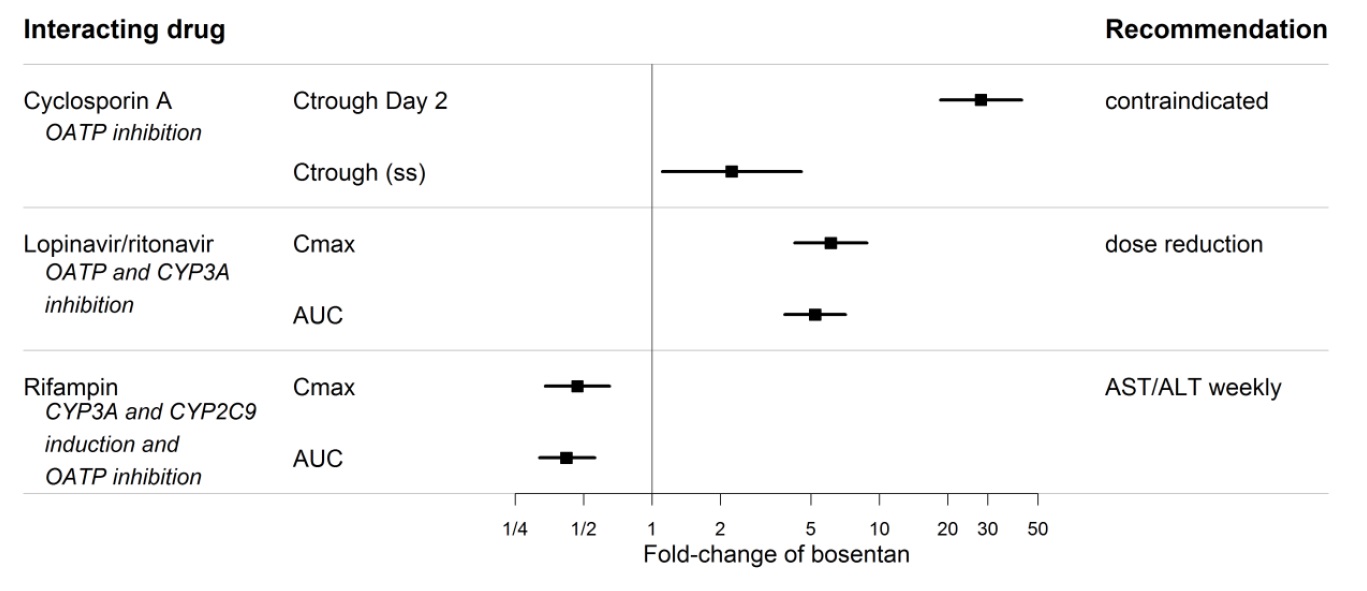
Co-administration of cyclosporine A and bosentan resulted in markedly increased plasma concentrations of bosentan. Therefore, concomitant use of Bosentan Tablets and cyclosporine A is contraindicated [see Cytochrome P450 Drug Interactions (7.1)].
4.3 Use with Glyburide
An increased risk of liver enzyme elevations was observed in patients receiving glyburide concomitantly with bosentan. Therefore co-administration of glyburide and Bosentan Tablets is contraindicated [see Cytochrome P450 Drug Interactions (7.1)].
4.4 Hypersensitivity
Bosentan Tablets are contraindicated in patients who are hypersensitive to bosentan or any component of the product. Observed reactions include Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), anaphylaxis, rash and angioedema [see Adverse Reactions (6.2), Description (11)].
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
ALT or AST > 3 x ULN were observed in 11% of Bosentan Tablets-treated patients (n = 658) compared to 2% of placebo-treated patients (n = 280). Three-fold increases were seen in 12% of 95 pulmonary arterial hypertension (PAH) patients on 125 mg twice daily and 14% of 70 PAH patients on 250 mg twice daily. Eight-fold increases were seen in 2% of PAH patients on 125 mg twice daily and 7% of PAH patients on 250 mg twice daily. Bilirubin increases to ≥ 3 x ULN were associated with aminotransferase increases in 2 of 658 (0.3%) of patients treated with Bosentan Tablets. The combination of hepatocellular injury (increases in aminotransferases of > 3 x ULN) and increases in total bilirubin (≥ 2 x ULN) is a marker for potential serious hepatotoxicity.
Elevations of AST or ALT associated with Bosentan Tablets are dose-dependent, occur both early and late in treatment, usually progress slowly, are typically asymptomatic, and usually have been reversible after treatment interruption or cessation. Aminotransferase elevations also may reverse spontaneously while continuing treatment with Bosentan Tablets.
Liver aminotransferase levels must be measured prior to initiation of treatment and then monthly and therapy adjusted accordingly [see Dosage and Administration (2.1, 2.4)]. Discontinue Bosentan Tablets if liver aminotransferase elevations are accompanied by clinical symptoms of hepatotoxicity (such as nausea, vomiting, fever, abdominal pain, jaundice, or unusual lethargy or fatigue) or increases in bilirubin ≥ 2 x ULN.
Avoid initiation of Bosentan Tablets in patients with elevated aminotransferases (> 3 x ULN) prior to drug initiation because monitoring hepatotoxicity in these patients may be more difficult [see Boxed Warning,Dosage and Administration (2.6), Use in Specific Populations (8.6)].
In WHO Functional Class II patients, consider whether the benefits of Bosentan Tablets are sufficient to offset the risk of hepatotoxicity, which may preclude future use as their disease progresses.
5.2 Embryo-fetal Toxicity
Based on data from animal reproduction studies, Bosentan Tablets may cause fetal harm when administered to a pregnant female and is contraindicated in females who are pregnant. Advise females of reproductive potential of the potential risk to a fetus. Obtain a pregnancy test prior to Bosentan Tablets treatment, monthly during treatment and for one month after stopping treatment. Advise females of reproductive potential to use two reliable forms of contraception during treatment with Bosentan Tablets and for at least one month after the last dose [see Dosage and Administration (2), Use in Specific Populations (8.1, 8.3)].
Bosentan Tablets are only available for females through a restricted program under REMS [see Warnings and Precautions (5.3)].
5.3 Prescribing and Distribution Program for Bosentan Tablets
Because of the risks of hepatotoxicity and birth defects, Bosentan Tablets are available only through a restricted program called the Bosentan REMS Program. As a component of the Bosentan Tablets REMS, prescribers, patients, and pharmacies must enroll in the program. [see Boxed Warning, Warnings and Precautions (5.1, 5.2), Contraindications (4.1)].
Required components of the Bosentan Tablets REMS are:
- Healthcare professionals who prescribe Bosentan Tablets must review the prescriber educational materials, enroll in the Bosentan REMS Program and comply with its requirements.
- Healthcare professionals must (1) review serum aminotransferases (ALT/AST) and bilirubin, and agree to order and monitor these tests monthly; and (2) for females of reproductive potential, confirm that the patient is not pregnant, and agree to order and monitor pregnancy tests monthly.
- To receive Bosentan Tablets, all patients must understand the risks and benefits, complete a patient enrollment form.
- Pharmacies that dispense Bosentan Tablets must enroll in the program and agree to comply with the Bosentan REMS Program requirements.
Further information about Bosentan Tablets and the Bosentan REMS Program is available at www.BosentanREMSProgram.com or 1-866-359-2612.
5.4 Fluid Retention
Peripheral edema is a known clinical consequence of PAH and worsening PAH and is also a known effect of Bosentan Tablets and other endothelin receptor antagonists. In PAH clinical trials with Bosentan Tablets, combined adverse events of fluid retention or edema were reported in 1.7% (placebo-corrected) of patients.
In addition, there have been numerous postmarketing reports of fluid retention in patients with pulmonary hypertension occurring within weeks after starting Bosentan Tablets. Patients required intervention with a diuretic, fluid management, or hospitalization for decompensating heart failure.
If clinically significant fluid retention develops, with or without associated weight gain, further evaluation should be undertaken to determine the cause, such as Bosentan Tablets or underlying heart failure, and the possible need for treatment or discontinuation of Bosentan Tablets [see Adverse Reactions (6.1), Clinical Studies (14.2)].
5.5 Pulmonary Veno-Occlusive Disease
If signs of pulmonary edema occur, consider the possibility of associated pulmonary veno-occlusive disease and consider whether Bosentan Tablets should be discontinued.
5.6 Decreased Sperm Counts
Decreased sperm counts have been observed in patients receiving Bosentan Tablets. Preclinical data also suggest that Bosentan Tablets, similar to other endothelin receptor antagonists, may have an adverse effect on spermatogenesis [see Adverse Reactions (6.1), Nonclinical Toxicology (13.1)].
5.7 Decreases in Hemoglobin and Hematocrit
Treatment with Bosentan Tablets can cause a dose-related decrease in hemoglobin and hematocrit. There have been postmarketing reports of decreases in hemoglobin concentration and hematocrit that have resulted in anemia requiring transfusion. It is recommended that hemoglobin concentrations be checked after 1 and 3 months, and every 3 months thereafter. If a marked decrease in hemoglobin concentration occurs, further evaluation should be undertaken to determine the cause and need for specific treatment [see Adverse Reactions (6.1)].
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [see Boxed Warning, Warnings and Precautions (5.1)]
- Embryo-fetal Toxicity [see Boxed Warning, Warnings and Precautions (5.2)]
- Fluid Retention [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Safety data on Bosentan Tablets was obtained from 13 clinical studies (9 placebo-controlled and 4 open-label) in 870 adult patients with PAH and other diseases. Doses up to 8 times the currently recommended clinical dose (125 mg twice daily) were administered for a variety of durations. The exposure to Bosentan Tablets in these trials ranged from 1 day to 4.1 years (n=94 for 1 year; n=61 for 1.5 years; and n=39 for more than 2 years). Exposure of PAH patients (n=328) to Bosentan Tablets ranged from 1 day to 1.7 years (n=174 more than 6 months and n=28 more than 12 months).
Treatment discontinuations due to adverse events other than those related to pulmonary hypertension during the clinical trials in adult patients with PAH were more frequent on Bosentan Tablets (6%; 15/258 patients) than on placebo (3%; 5/172 patients). In this database the only cause of discontinuations > 1% and occurring more often on Bosentan Tablets was abnormal liver function.
The adverse drug events that occurred in ≥3% of the Bosentan Tablets-treated patients and were more common on Bosentan Tablets in placebo-controlled trials in PAH at doses of 125 or 250 mg twice daily are shown in Table 3:
| Adverse Event | Bosentan Tablets n = 258 | Placebo n = 172 | ||
| No. | % | No. | % | |
| Respiratory Tract Infection** | 56 | 22% | 30 | 17% |
| Headache | 39 | 15% | 25 | 14% |
| Edema | 28 | 11% | 16 | 9% |
| Chest Pain | 13 | 5% | 8 | 5% |
| Syncope | 12 | 5% | 7 | 4% |
| Flushing | 10 | 4% | 5 | 3% |
| Hypotension | 10 | 4% | 3 | 2% |
| Sinusitis | 9 | 4% | 4 | 2% |
| Arthralgia | 9 | 4% | 3 | 2% |
| Serum Aminotransferases, abnormal | 9 | 4% | 3 | 2% |
| Palpitations | 9 | 4% | 3 | 2% |
| Anemia | 8 | 3% | - | - |
*Note: only AEs with onset from start of treatment to 1 calendar day after end of treatment are included. All reported events (at least 3%) are included except those too general to be informative, and those not reasonably associated with the use of the drug because they were associated with the condition being treated or are very common in the treated population.
** Respiratory Tract Infection combines the terms "Nasopharyngitis", "Upper Respiratory Tract Infection" and "Respiratory Tract Infection". Combined data from Study 351, BREATHE -1 and EARLY
Decreased Sperm Counts
An open-label, single arm, multicenter, safety study evaluated the effect on testicular function of Bosentan Tablets 62.5 mg twice daily for 4 weeks, followed by 125 mg twice daily for 5 months. Twenty-five male patients with WHO functional class III and IV PAH and normal baseline sperm count were enrolled. Twenty-three completed the study and 2 discontinued due to adverse events not related to testicular function. There was a decline in sperm count of at least 50% in 25% of the patients after 3 or 6 months of treatment with Bosentan Tablets. Sperm count remained within the normal range in all 22 patients with data after 6 months and no changes in sperm morphology, sperm motility, or hormone levels were observed. One patient developed marked oligospermia at 3 months and the sperm count remained low with 2 follow-up measurements over the subsequent 6 weeks. Bosentan Tablets was discontinued and after 2 months the sperm count had returned to baseline levels. Based on these findings and preclinical data from endothelin receptor antagonists, it cannot be excluded that endothelin receptor antagonists such as Bosentan Tablets have an adverse effect on spermatogenesis.
Decreases in Hemoglobin and Hematocrit
Treatment with Bosentan Tablets can cause a dose-related decrease in hemoglobin and hematocrit. It is recommended that hemoglobin concentrations be checked after 1 and 3 months, and every 3 months thereafter. If a marked decrease in hemoglobin concentration occurs, further evaluation should be undertaken to determine the cause and need for specific treatment.
The overall mean decrease in hemoglobin concentration for adult Bosentan Tablets-treated patients was 0.9 g/dL (change to end of treatment). Most of this decrease of hemoglobin concentration was detected during the first few weeks of Bosentan Tablets treatment and hemoglobin levels stabilized by 4 to 12 weeks of Bosentan Tablets treatment. In placebo-controlled studies of all uses of Bosentan Tablets, marked decreases in hemoglobin (> 15% decrease from baseline resulting in values < 11 g/dL) were observed in 6% of Bosentan Tablets-treated patients and 3% of placebo-treated patients. In patients with PAH treated with doses of 125 and 250 mg twice daily, marked decreases in hemoglobin occurred in 3% compared to 1% in placebo-treated patients.
A decrease in hemoglobin concentration by at least 1 g/dL was observed in 57% of Bosentan Tablets-treated patients as compared to 29% of placebo-treated patients. In 80% of those patients whose hemoglobin decreased by at least 1 g/dL, the decrease occurred during the first 6 weeks of Bosentan Tablets treatment.
During the course of treatment the hemoglobin concentration remained within normal limits in 68% of Bosentan Tablets-treated patients compared to 76% of placebo patients. The explanation for the change in hemoglobin is not known, but it does not appear to be hemorrhage or hemolysis.
6.2 Postmarketing Experience
There have been several postmarketing reports of angioedema associated with the use of Bosentan Tablets. The onset of the reported cases occurred within a range of 8 hours to 21 days after starting therapy. Some patients were treated with an antihistamine and their signs of angioedema resolved without discontinuing Bosentan Tablets.
The following additional adverse reactions have been reported during the postapproval use of Bosentan Tablets. Because these adverse reactions are reported from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Bosentan Tablets exposure:
Unexplained hepatic cirrhosis [see Boxed Warning]
Liver failure [see Boxed Warning]
Hypersensitivity, DRESS, and anaphylaxis [see Contraindications (4.4)]
Thrombocytopenia
Rash
Jaundice
Anemia requiring transfusion
Neutropenia and leukopenia
Nasal congestion
7 DRUG INTERACTIONS
7.1 Cytochrome P450 Drug Interactions
Bosentan is metabolized by CYP2C9 and CYP3A. Inhibition of these enzymes may increase the plasma concentration of bosentan [see Pharmacokinetics (12.3)]. Concomitant administration of both a CYP2C9 inhibitor (such as fluconazole or amiodarone) and a strong CYP3A inhibitor (e.g., ketoconazole, itraconazole) or a moderate CYP3A inhibitor (e.g., amprenavir, erythromycin, fluconazole, diltiazem) with Bosentan Tablets will likely lead to large increases in plasma concentrations of bosentan. Co-administration of such combinations of a CYP2C9 inhibitor plus a strong or moderate CYP3A inhibitor with Bosentan Tablets is not recommended.
Bosentan is an inducer of CYP3A and CYP2C9. Consequently plasma concentrations of drugs metabolized by these two isozymes will be decreased when Bosentan Tablets is co-administered. Bosentan had no relevant inhibitory effect on any CYP isozyme in vitro (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A). Consequently, Bosentan Tablets is not expected to increase the plasma concentrations of drugs metabolized by these enzymes.
Figure 1. CYP3A induction-mediated effect of bosentan on other drugs
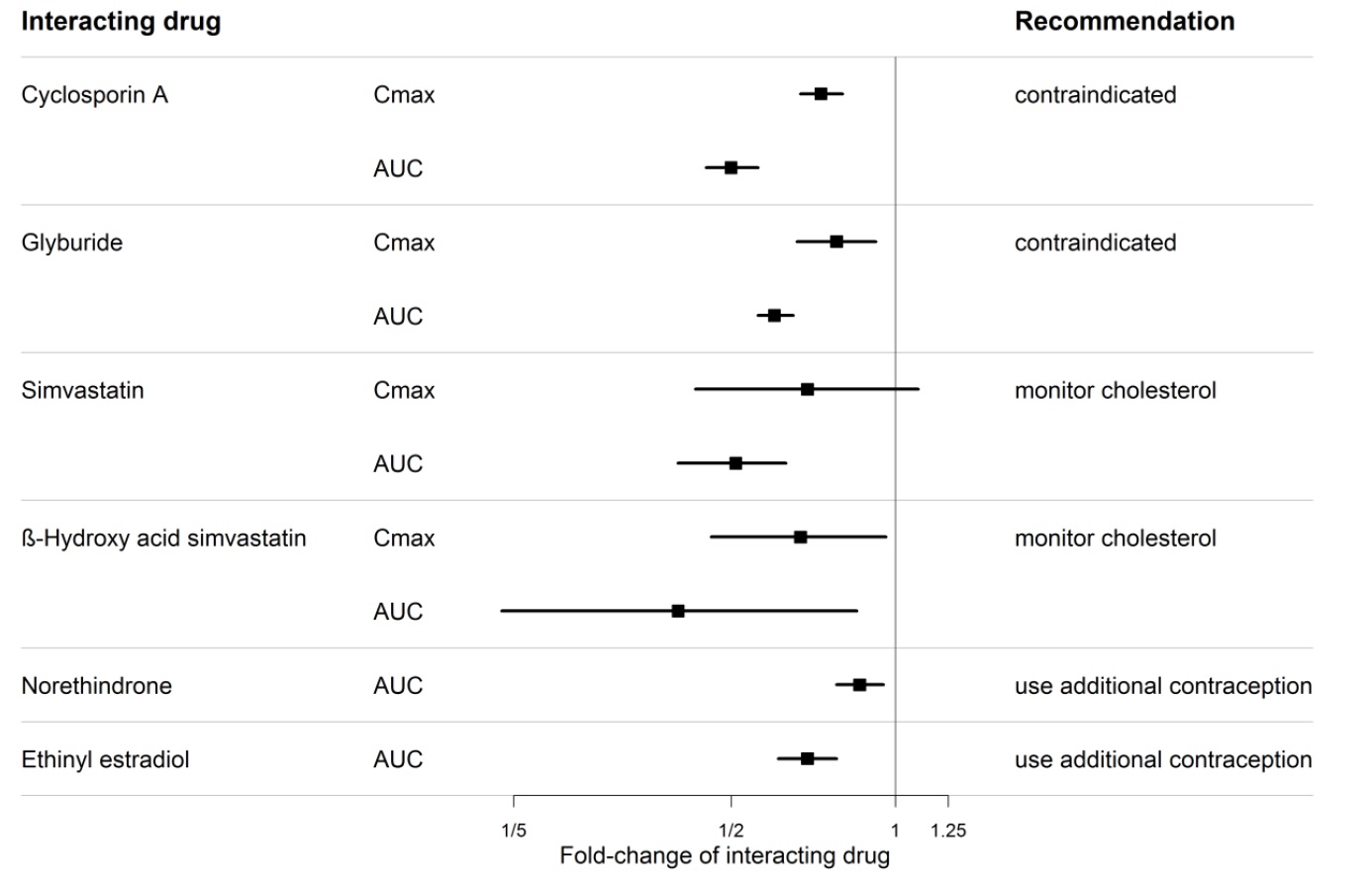
Figure 2. Effect of other drugs on bosentan
7.2 Hormonal Contraceptives
Hormonal contraceptives, including oral, injectable, transdermal, and implantable forms, may not be reliable when Bosentan Tablets are co-administered. Females should practice additional methods of contraception and not rely on hormonal contraception alone when taking Bosentan Tablets [see Use in Specific Populations (8.3)].
An interaction study demonstrated that co-administration of bosentan and a combination oral hormonal contraceptive produced average decreases of norethindrone and ethinyl estradiol levels of 14% and 31%, respectively. However, decreases in exposure were as much as 56% and 66%, respectively, in individual subjects.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on data from animal reproduction studies, Bosentan Tablets may cause fetal harm, including birth defects and fetal death, when administered to a pregnant female and is contraindicated during pregnancy [see Contraindications (4.1)]. There are limited data on Bosentan Tablets use in pregnant women. In animal reproduction studies, oral administration of bosentan to pregnant rats at 2 times the maximum recommended human dose (MRHD) on a mg/m2 basis caused teratogenic effects in rats, including malformations of the head, mouth, face, and large blood vessels [see Animal Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Bosentan was teratogenic in rats given oral doses two times the MRHD (on a mg/m2 basis). In an embryo-fetal toxicity study in rats, bosentan showed dose-dependent teratogenic effects, including malformations of the head, mouth, face and large blood vessels. Bosentan increased stillbirths and pup mortality at oral doses 2 and 10 times the MRHD (on a mg/m2 basis). Although birth defects were not observed in rabbits given oral doses of up to the equivalent of 10.5 g/day in a 70 kg person, plasma concentrations of bosentan in rabbits were lower than those reached in the rat. The similarity of malformations induced by bosentan and those observed in endothelin-1 knockout mice and in animals treated with other endothelin receptor antagonists indicates that embryo-fetal toxicity is a class effect of these drugs.
8.2 Lactation
Risk Summary
There are no data on the presence of bosentan in human milk, the effects on the breastfed infant, or the effect on milk production. Because of the potential for serious adverse reactions, such as fluid retention and hepatotoxicity, in breastfed infants from Bosentan Tablets, advise women not to breastfeed during treatment with Bosentan Tablets.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating Bosentan Tablets, monthly during treatment and one month after stopping treatment with Bosentan Tablets. The patient should contact her physician immediately for pregnancy testing if onset of menses is delayed or pregnancy is suspected. If the pregnancy test is positive, the physician and patient must discuss the risks to her, the pregnancy, and the fetus.
Contraception
Drug interaction studies show that bosentan reduces serum levels of the estrogen and progestin in oral contraceptives. Based on these findings, hormonal contraceptives (including oral, injectable, transdermal, and implantable contraceptives) may be less effective for preventing pregnancy in patients using Bosentan Tablets and should not be used as a patient’s only contraceptive method [see Drug Interactions (7.2)]. Females of reproductive potential using Bosentan Tablets must use two acceptable methods of contraception during treatment and for 1 month after treatment with Bosentan Tablets. Patients may choose one highly effective form of contraception (intrauterine devices (IUD) or tubal sterilization) or a combination of methods (hormone method with a barrier method or two barrier methods). If a partner’s vasectomy is the chosen method of contraception, a hormone or barrier method must be used along with this method. Counsel patients on pregnancy planning and prevention, including emergency contraception, or designate counseling by another healthcare provider trained in contraceptive counseling [see Boxed Warning].
Infertility
Males
Decreased sperm counts have been observed in patients receiving Bosentan Tablets. Based on these findings and findings in animals, Bosentan Tablets may impair fertility in males of reproductive potential.
It is not known whether effects on fertility would be reversible [see Warnings and Precautions (5.6), Adverse Reactions (6.1), Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Juvenile Animal Toxicity Data
In a juvenile rat toxicity study, rats were treated from Day 4 postpartum to adulthood (Day 69 postpartum). Decreased body weights, absolute weights of testes and epididymides, and reduced number of sperm in epididymides were observed after weaning. No effect on testis histology or sperm morphology and function was seen. The NOAEL was 4 times (at Day 4 postpartum) and 2 times (Day 69 postpartum) the human therapeutic exposure, respectively.
No effects on general development, sensory, cognitive function and reproductive performance were detected at the highest dose tested in juvenile rats, 7 times the therapeutic exposure in children with PAH.
8.5 Geriatric Use
Clinical studies of Bosentan Tablets did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
Because there is in vitro and in vivo evidence that the main route of excretion of bosentan is biliary, liver impairment could be expected to increase exposure (Cmax and AUC) of bosentan. The pharmacokinetics of Bosentan Tablets have not been evaluated in patients with severe liver impairment (Child-Pugh Class C). In patients with moderate hepatic impairment (Child-Pugh Class B), the systemic exposures to bosentan and its active metabolite increased significantly. Bosentan Tablets should generally be avoided in patients with moderate or severe liver impairment. Pharmacokinetics of bosentan were not altered in patients with mild impairment of hepatic function (Child-Pugh Class A) [see Dosage and Administration (2.6), Warnings and Precautions (5.1), Pharmacokinetics (12.3)].
8.7 Renal Impairment
The effect of renal impairment on the pharmacokinetics of bosentan is small and does not require dosing adjustment [see Pharmacokinetics (12.3)].
10 OVERDOSAGE
Bosentan has been given as a single dose of up to 2,400 mg in normal volunteers, or up to 2,000 mg/day for 2 months in patients, without any major clinical consequences. The most common side effect was headache of mild to moderate intensity. In the cyclosporine A interaction study, in which doses of 500 and 1,000 mg twice daily of bosentan were given concomitantly with cyclosporine A, trough plasma concentrations of bosentan increased 30-fold, resulting in severe headache, nausea, and vomiting, but no serious adverse events. Mild decreases in blood pressure and increases in heart rate were observed.
In the postmarketing period, there was one reported overdose of 10,000 mg of Bosentan Tablets taken by an adolescent male patient. He had symptoms of nausea, vomiting, hypotension, dizziness, sweating, and blurred vision. He recovered within 24 hours with blood pressure support.
Bosentan is unlikely to be effectively removed by dialysis due to the high molecular weight and extensive plasma protein binding.
11 DESCRIPTION
Bosentan is an endothelin receptor antagonist that belongs to a class of highly substituted pyrimidine derivatives, with no chiral centers. It is designated chemically as 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-[2,2´]-bipyrimidin-4-yl]- benzenesulfonamide monohydrate and has the following structural formula:
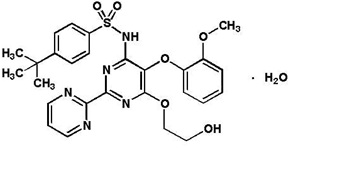
Bosentan has a molecular weight of 569.64 and a molecular formula of C27H29N5O6S•H2O. Bosentan is a white to yellowish powder. It is poorly soluble in water (1.0 mg/100 mL) and in aqueous solutions at low pH (0.1 mg/100 mL at pH 1.1 and 4.0; 0.2 mg/100 mL at pH 5.0). Solubility increases at higher pH values (43 mg/100 mL at pH 7.5). In the solid state, bosentan is very stable, is not hygroscopic and is not light sensitive.
Bosentan Tablets are available as 62.5 mg and 125 mg film-coated tablets for oral administration, and contain the following excipients: povidone, pregelatinized starch, sodium starch glycolate, and magnesium stearate. Additionally, the film coating contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, yellow iron oxide, and red iron oxide. Each 62.5 mg Bosentan Tablet contains 64.55 mg of bosentan, equivalent to 62.5 mg of anhydrous bosentan. Each 125 mg Bosentan Tablet contains 129.10 mg of bosentan, equivalent to 125 mg of anhydrous bosentan.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bosentan is a specific and competitive antagonist at endothelin receptor types ETA and ETB. Bosentan has a slightly higher affinity for ETA receptors than for ETB receptors. The clinical impact of dual endothelin blockage is unknown.
Endothelin-1 (ET-1) is a neurohormone, the effects of which are mediated by binding to ETA and ETB receptors in the endothelium and vascular smooth muscle. ET-1 concentrations are elevated in plasma and lung tissue of patients with PAH, suggesting a pathogenic role for ET-1 in this disease.
12.3 Pharmacokinetics
General
After oral administration, maximum plasma concentrations of bosentan are attained within 3 to 5 hours and the terminal elimination half-life is about 5 hours in healthy adult subjects. The exposure to bosentan after intravenous and oral administration is about twice as high in adult patients with PAH as it is in healthy adult subjects.
Absorption
The absolute bioavailability of bosentan in normal volunteers is about 50% and is unaffected by food.
Bosentan is highly bound (> 98%) to plasma proteins, mainly albumin. Bosentan does not penetrate into erythrocytes. The volume of distribution is about 18 L.
Elimination
Metabolism
Bosentan has three metabolites, one of which is pharmacologically active and may contribute 10% to 20% of the effect of bosentan. Bosentan is an inducer of CYP2C9 and CYP3A and possibly also of CYP2C19. Upon multiple oral dosing, plasma concentrations in healthy adults decrease gradually to 50% to 65% of those seen after single dose administration, probably the effect of auto-induction of the metabolizing liver enzymes. Steady-state is reached within 3 to 5 days.
Excretion
Bosentan is eliminated by biliary excretion following metabolism in the liver. Less than 3% of an administered oral dose is recovered in urine. Total clearance after a single intravenous dose is about 4 L/h in patients with PAH.
Specific Populations
Hepatic Impairment
In vitro and in vivo evidence showing extensive hepatic metabolism of bosentan suggests that liver impairment could significantly increase exposure to bosentan. In a study comparing 8 patients with mild liver impairment (Child-Pugh Class A) to 8 controls, the single-and multiple-dose pharmacokinetics of bosentan were not altered in patients with mild hepatic impairment.
In another small (N=8) pharmacokinetic study, the steady-state AUC of bosentan was on average 4.7 times higher and the active metabolite Ro 48-5033 was 12.4 times higher in 5 patients with moderately impaired liver function (Child-Pugh Class B) and PAH associated with portal hypertension than in 3 patients with normal liver function and PAH of other etiologies.
The pharmacokinetics of Bosentan Tablets have not been evaluated in patients with severe liver impairment (Child-Pugh Class C) [see Dosage and Administration (2.2), Warnings and Precautions (5.1), Use in Specific Populations (8.6)].
Renal Impairment
In patients with severe renal impairment (creatinine clearance 15 mL/min to 30 mL/min), plasma concentrations of bosentan were essentially unchanged and plasma concentrations of the three metabolites were increased about 2-fold compared to subjects with normal renal function. These differences do not appear to be clinically important.
Drug Interactions
Ketoconazole
Co-administration of bosentan 125 mg twice daily and ketoconazole, a potent CYP3A inhibitor, increased the plasma concentrations of bosentan by approximately 100% in normal volunteers. No dose adjustment of Bosentan Tablets is necessary, but increased effects of Bosentan Tablets should be considered.
Warfarin
Co-administration of bosentan 500 mg twice daily for 6 days in normal volunteers decreased the plasma concentrations of both S-warfarin (a CYP2C9 substrate) and R-warfarin (a CYP3A substrate) by 29% and 38%, respectively. Clinical experience with concomitant administration of Bosentan Tablets and warfarin in patients with PAH did not show clinically relevant changes in INR or warfarin dose (baseline vs. end of the clinical studies), and the need to change the warfarin dose during the trials due to changes in INR or due to adverse events was similar among Bosentan Tablets-and placebo-treated patients.
Digoxin, Nimodipine, and Losartan
Bosentan has no significant pharmacokinetic interactions with digoxin and nimodipine, and losartan has no significant effect on plasma levels of bosentan.
Sildenafil
In normal volunteers, co-administration of multiple doses of 125 mg twice daily bosentan and 80 mg three times daily sildenafil resulted in a reduction of sildenafil plasma concentrations by 63% and increased bosentan plasma concentrations by 50%. The changes in plasma concentrations were not considered clinically relevant and dose adjustments are not necessary. This recommendation holds true when sildenafil is used for the treatment of PAH or erectile dysfunction.
Tadalafil
Bosentan (125 mg twice daily) reduced tadalafil (40 mg once per day) systemic exposure (AUC) by 42% and Cmax by 27% following multiple dose co-administration. Tadalafil did not affect the exposure (AUC and Cmax) of bosentan or its metabolites.
Figure 3. CYP Induction-mediated effect of bosentan on other drugs

Figure 4. Effects of other drugs on bosentan
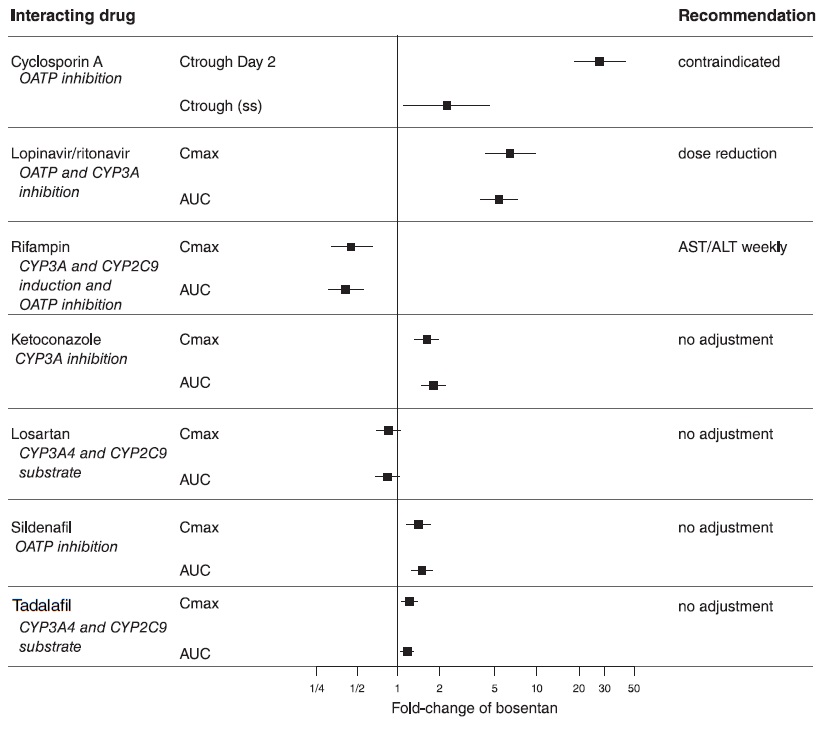
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Two years of dietary administration of bosentan to mice produced an increased incidence of hepatocellular adenomas and carcinomas in males at doses as low as 450 mg/kg/day (about 8 times the maximum recommended human dose [MRHD] of 125 mg twice daily, on a mg/m2 basis). In the same study, doses greater than 2,000 mg/kg/day (about 32 times the MRHD) were associated with an increased incidence of colon adenomas in both males and females. In rats, dietary administration of bosentan for two years was associated with an increased incidence of brain astrocytomas in males at doses as low as 500 mg/kg/day (about 16 times the MRHD). In a comprehensive battery of in vitro tests (the microbial mutagenesis assay, the unscheduled DNA synthesis assay, the V-79 mammalian cell mutagenesis assay, and human lymphocyte assay) and an in vivo mouse micronucleus assay, there was no evidence for any mutagenic or clastogenic activity of bosentan.
Impairment of Fertility/Testicular Function
The development of testicular tubular atrophy and impaired fertility has been linked with the chronic administration of certain endothelin receptor antagonists in rodents.
Treatment with bosentan at oral doses of up to 1,500 mg/kg/day (50 times the MRHD on a mg/m2 basis) or intravenous doses up to 40 mg/kg/day had no effects on sperm count, sperm motility, mating performance or fertility in male and female rats. An increased incidence of testicular tubular atrophy was observed in rats given bosentan orally at doses as low as 125 mg/kg/ day (about 4 times the MRHD and the lowest doses tested) for two years but not at doses as high as 1,500 mg/kg/day (about 50 times the MRHD) for 6 months. Effects on sperm count and motility were evaluated only in the much shorter duration fertility studies in which males had been exposed to the drug for 4 to 6 weeks. An increased incidence of tubular atrophy was not observed in mice treated for 2 years at doses up to 4,500 mg/kg/day (about 75 times the MRHD) or in dogs treated up to 12 months at doses up to 500 mg/kg/day (about 50 times the MRHD).
14 CLINICAL STUDIES
14.1 Pulmonary Arterial Hypertension
WHO Functional Class III-IV
Two randomized, double-blind, multi-center, placebo-controlled trials were conducted in 32 and 213 patients. The larger study (BREATHE-1) compared 2 doses (125 mg twice daily and 250 mg twice daily) of Bosentan Tablets with placebo. The smaller study (Study 351) compared 125 mg twice daily with placebo. Patients had severe (WHO functional Class III–IV) PAH: idiopathic or heritable PAH (72%) or PAH associated with scleroderma or other connective tissue diseases (21%), or to autoimmune diseases (7%). There were no patients with PAH associated with other conditions such as HIV disease or recurrent pulmonary emboli.
In both studies, Bosentan Tablets or placebo was added to patients' current therapy, which could have included a combination of digoxin, anticoagulants, diuretics, and vasodilators (e.g., calcium channel blockers, ACE inhibitors), but not epoprostenol. Bosentan Tablets were given at a dose of 62.5 mg twice daily for 4 weeks and then at 125 mg twice daily or 250 mg twice daily for either 12 (BREATHE-1) or 8 (Study 351) additional weeks. The primary study endpoint was 6-minute walk distance. In addition, symptoms and functional status were assessed. Hemodynamic measurements were made at 12 weeks in Study 351.
The mean age was about 49 years. About 80% of patients were female, and about 80% were Caucasian. Patients had been diagnosed with pulmonary hypertension for a mean of 2.4 years.
Submaximal Exercise Ability
Results of the 6-minute walk distance at 3 months (Study 351) or 4 months (BREATHE-1) are shown in Table 4.
Table 4. Effects of Bosentan Tablets on 6-minute walk distance
| Distance in meters: mean ± standard deviation. Changes are to week 16 for BREATHE-1 and to week 12 for Study 351. (a) p=0.01; by Wilcoxon; (b) p=0.0001; by Wilcoxon; (c) p=0.02; by Student's t-test. |
|||||
| BREATHE-1 | Study 351 | ||||
| Bosentan Tablets 125 mg twice daily (n = 74) | Bosentan Tablets 250 mg twice daily (n = 70) | Placebo (n = 69) | Bosentan Tablets 125 mg twice daily (n = 21) | Placebo (n = 11) |
|
| Baseline | 326 ± 73 | 333 ± 75 | 344 ± 76 | 360 ± 86 | 355 ± 82 |
| End point | 353 ± 115 | 379 ± 101 | 336 ± 129 | 431 ± 66 | 350 ± 147 |
| Change from baseline | 27 ± 75 | 46 ± 62 | -8 ± 96 | 70 ± 56 | -6 ± 121 |
| Placebo – subtracted | 35 (a) | 54 (b) | 76 (c) | ||
In both trials, treatment with Bosentan Tablets resulted in a significant increase in exercise ability. The improvement in walk distance was apparent after 1 month of treatment (with 62.5 mg twice daily) and fully developed by about 2 months of treatment (Figure 5). It was maintained for up to 7 months of double-blind treatment. Walking distance was somewhat greater with 250 mg twice daily, but the potential for increased hepatotoxicity causes this dose not to be recommended [see Dosage and Administration (2.1)]. There were no apparent differences in treatment effects on walk distance among subgroups analyzed by demographic factors, baseline disease severity, or disease etiology, but the studies had little power to detect such differences.
Figure 5. Mean Change in 6-min Walk Distance (BREATHE-1)
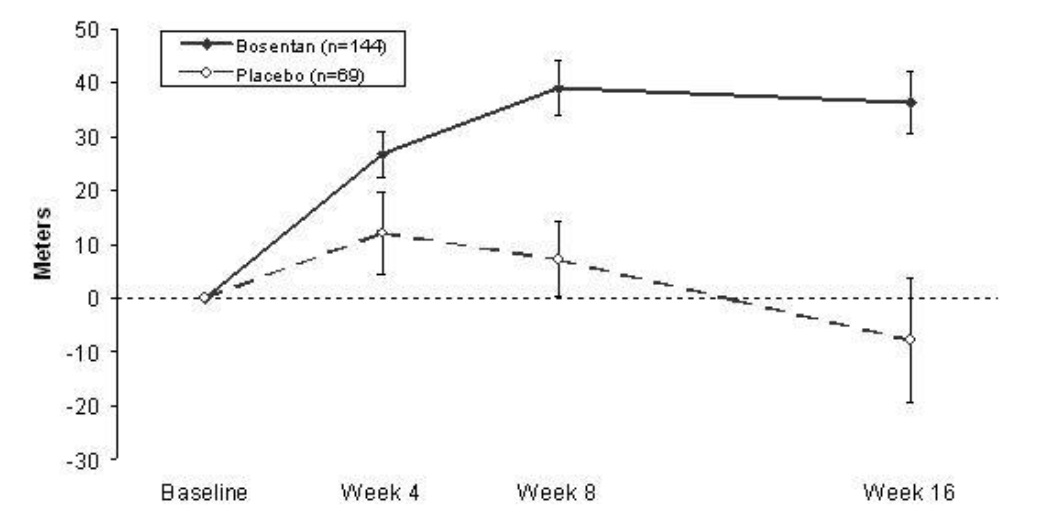
Change from baseline in 6-minute walking distance from start of therapy to week 16 in the placebo and combined Bosentan Tablets (125 mg twice daily and 250 mg twice daily) groups. Values are expressed as mean ± standard error of the mean.
Hemodynamic Changes
Invasive hemodynamic parameters were assessed in Study 351. Treatment with Bosentan Tablets led to a significant increase in cardiac index (CI) associated with a significant reduction in pulmonary artery pressure (PAP), pulmonary vascular resistance (PVR), and mean right atrial pressure (RAP) (Table 5).
The relationship between hemodynamic effects and improvements in 6-minute walk distance is unknown.
Table 5: Change from Baseline to Week 12: Hemodynamic Parameters
| Bosentan Tablets 125 mg twice daily | Placebo | ||
| CI (L/min/m2) | n=20 | n=10 | |
| Baseline | 2.35±0.73 | 2.48±1.03 | |
| Absolute Change | 0.50±0.46 | -0.52±0.48 | |
| Treatment Effect | 1.02 (a) | ||
| Mean PAP (mmHg) | n=20 | n=10 | |
| Baseline | 53.7±13.4 | 55.7±10.5 | |
| Absolute Change | -1.6±5.1 | 5.1±8.8 | |
| Treatment Effect | -6.7(b) | ||
| PVR (dyn·sec·cm-5) | n=19 | n=10 | |
| Baseline | 896±425 | 942±430 | |
| Absolute Change | -223±245 | 191±235 | |
| Treatment Effect | -415(a) | ||
| Mean RAP (mmHg) | n=19 | n=10 | |
| Baseline | 9.7±5.6 | 9.9±4.1 | |
| Absolute Change | -1.3±4.1 | 4.9±4.6 | |
| Treatment Effect | -6.2(a) | ||
| Values shown are means ± SD (a) p≤0.001; (b) p<0.02 |
|||
Symptoms and Functional Status
Symptoms of PAH were assessed by Borg dyspnea score, WHO functional class, and rate of "clinical worsening." Clinical worsening was assessed as the sum of death, hospitalizations for PAH, discontinuation of therapy because of PAH, and need for epoprostenol. There was a significant reduction in dyspnea during walk tests (Borg dyspnea score), and significant improvement in WHO functional class in Bosentan Tablets-treated patients. There was a significant reduction in the rate of clinical worsening (Table 6 and Figure 6). Figure 6 shows the log-rank test reflecting clinical worsening over 28 weeks.
Table 6: Incidence of Clinical Worsening, Intent To Treat Population
| BREATHE-1 | Study 351 | |||
| Bosentan Tablets 125/250 mg twice daily (n = 144) | Placebo (n = 69) | Bosentan Tablets 125 mg twice daily (n = 21) | Placebo (n = 11) | |
| Patients with clinical worsening [n (%)] | 9 (6%)(a) | 14 (20%) | 0 (0%)(b) | 3 (27%) |
| Death | 1 (1%) | 2 (3%) | 0 (0%) | 0 (0%) |
| Hospitalization for PAH | 6 (4%) | 9 (13%) | 0 (0%) | 3 (27%) |
| Discontinuation due to worsening of PAH | 5 (3%) | 6 (9%) | 0 (0%) | 3 (27%) |
| Receipt of epoprostenol(c) | 4 (3%) | 3 (4%) | 0 (0%) | 3 (27%) |
| Note: Patients may have had more than one reason for clinical worsening. (a)p=0.0015 vs. placebo by log-rank test. There was no relevant difference between the 125 mg and 250 mg twice daily groups. (b)p=0.033 vs. placebo by Fisher's exact test. (c)Receipt of epoprostenol was always a consequence of clinical worsening. |
||||
Figure 6. Time to Clinical Worsening (BREATHE-1)
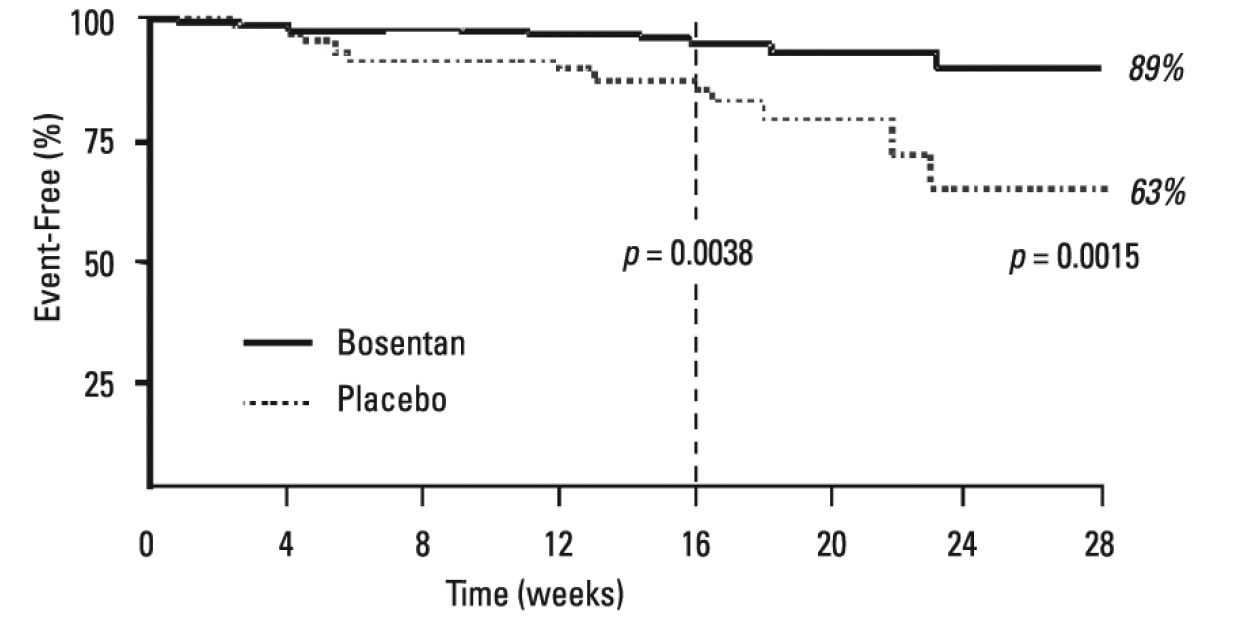
Time from randomization to clinical worsening with Kaplan-Meier estimate of the proportions of failures in BREATHE-1. All patients (n=144 in the Bosentan Tablets group and n=69 in the placebo group) participated in the first 16 weeks of the study. A subset of this population (n=35 in the Bosentan Tablets group and 13 in the placebo group) continued double-blind therapy for up to 28 weeks.
WHO Functional Class II
In a randomized, double-blind, multicenter, placebo-controlled trial, 185 mildly symptomatic PAH patients with WHO Functional Class II (mean baseline 6-minute walk distance of 443 meters) received Bosentan Tablets 62.5 mg twice daily for 4 weeks followed by 125 mg twice daily (n = 93), or placebo (n = 92) for 6 months. Enrolled patients were treatment-naïve (n = 156) or on a stable dose of sildenafil (n = 29). The coprimary endpoints were change from baseline to month 6 in PVR and 6-minute walk distance. Time to clinical worsening (assessed as the sum of death, hospitalization due to PAH complications, or symptomatic progression of PAH), Borg dyspnea index, change in WHO functional class and hemodynamics were assessed as secondary endpoints.
Compared with placebo, Bosentan Tablets treatment was associated with a reduced incidence of worsening of at least one functional class (3% Bosentan Tablets vs. 13% placebo, p = 0.03), and improvement in hemodynamic variables (PVR, mPAP, TPR, cardiac index, and SVO2; p < 0.05). The + 19 m mean (+14 m median) increase in 6-minute walk distance with Bosentan Tablets vs. placebo was not significant (p = 0.08). There was a significant delay in time to clinical worsening (first seen primarily as symptomatic progression of PAH) with Bosentan Tablets compared with placebo (hazard ratio 0.2, p = 0.01). Findings were consistent in strata with or without treatment with sildenafil at baseline.
Long-term Treatment of PAH
Long-term follow-up of patients with Class III and IV PAH who were treated with Bosentan Tablets in open-label extensions of trials (N=235) showed that 93% and 84% of patients were still alive at 1 and 2 years, respectively, after the start of treatment.
These uncontrolled observations do not allow comparison with a group not given Bosentan Tablets and cannot be used to determine the long-term effect of Bosentan Tablets on mortality.
Pulmonary Arterial Hypertension in Adults related to Congenital Heart Disease with Left-to-Right Shunts
A small study (N=54) and its open label extension (N=37) of up to 40 weeks in adult patients with Eisenmenger physiology demonstrated effects of Bosentan Tablets on exercise and safety that were similar to those seen in other trials in patients with PAH (WHO Group 1).
14.2 Lack of Benefit in Congestive Heart Failure
Bosentan Tablets are not effective in the treatment of congestive heart failure with left ventricular dysfunction. In a pair of studies, 1613 subjects with NYHA Class III-IV heart failure, left ventricular ejection fraction <35%, on diuretics, ACE inhibitor, and other therapies, were randomized to placebo or Bosentan Tablets (62.5 mg twice daily titrated as tolerated to 125 mg twice daily) and followed for up to 70 weeks. Use of Bosentan Tablets was associated with no benefit on patient global assessment (the primary end point) or mortality. However, hospitalizations for heart failure were more common during the first 4 to 8 weeks after Bosentan Tablets was initiated. In a placebo-controlled trial of patients with severe chronic heart failure, there was an increased incidence of hospitalization for CHF associated with weight gain and increased leg edema during the first 4-8 weeks of treatment with Bosentan Tablets. Patients required intervention with a diuretic, fluid management, or hospitalization for decompensating heart failure.
16 HOW SUPPLIED/STORAGE AND HANDLING
Bosentan Tablets are supplied as:
62.5 mg round, biconvex, yellow, coated tablet, "A" debossed on one side and "2" debossed on the other side, packaged in a white high-density polyethylene bottle with a child-resistant closure.
NDC 47781-270-60: Bottle containing 60 tablets
125 mg capsule-shaped, biconvex, yellow, coated tablet, "ALV" debossed on one side and "271" debossed on the other side, packaged in a white high-density polyethylene bottle with a child-resistant closure.
NDC 47781-271-60: Bottle containing 60 tablets
Store at 20°C – 25°C (68°F – 77°F). Excursions are permitted between 15°C and 30°C (59°F and 86°F). [See USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Restricted access
Advise the patient that Bosentan Tablets are only available through a restricted access program called the Bosentan REMS Program.
As a component of the Bosentan Tablets REMS, prescribers must review the contents of the Bosentan Tablets Medication Guide with the patient before initiating Bosentan Tablets.
Instruct patients that the risks associated with Bosentan Tablets include:
-
Hepatotoxicity
Discuss with the patient the requirement to measure serum aminotransferases monthly. -
Embryo-fetal toxicity
Educate and counsel female patients of reproductive potential about the need to use reliable methods of contraception during treatment with Bosentan Tablets and for one month after treatment discontinuation. Females of reproductive potential must have monthly pregnancy tests and must use two different forms of contraception while taking Bosentan Tablets and for one month after discontinuing Bosentan Tablets [see Use in Specific Populations (8.1)].Females who have intrauterine devices (IUD) or tubal sterilization can use these contraceptive methods alone. Patients should be instructed to immediately contact their physician if they suspect they may be pregnant. Patients should seek additional contraceptive advice from a gynecologist or similar expert as needed.
Educate and counsel females of reproductive potential on the use of emergency contraception in the event of unprotected sex or contraceptive failure.
Advise pre-pubertal females to report any changes in their reproductive status immediately to her prescriber.
Advise patients to contact their gynecologist or healthcare provider if they want to change the form of birth control which is used to ensure that another acceptable form of birth control is selected.
Advise the patient that Bosentan Tablets are available only from specialty pharmacies that are enrolled in the Bosentan REMS Program.
Patients must sign the Bosentan Tablets Patient Enrollment and Consent Form to confirm that they understand the risks of Bosentan Tablets.
-
Lactation
Advise women not to breastfeed during treatment with Bosentan Tablets [see Use in Specific Populations (8.2)]. -
Infertility
Advise males of reproductive potential that Bosentan Tablets may impair fertility [see Warnings and Precautions (5.6), Adverse Reactions (6.1), Use in Specific Populations (8.3) andNonclinical Toxicology (13.1)].
Other Risks Associated with Bosentan Tablets
Instruct patients that the risks associated with Bosentan Tablets also include the following:
Decreases in hemoglobin and hematocrit – advise patients of the importance of hemoglobin testing
Decreases in sperm count
Fluid retention
Medication Guide
Bosentan (boe-SEN-tan) Tablets
Read the Medication Guide that comes with Bosentan Tablets before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about Bosentan Tablets?
Bosentan Tablets are only available through the Bosentan REMS Program. Before you begin taking Bosentan Tablets, you must read and agree to all of the instructions in the Bosentan REMS Program.
Bosentan Tablets can cause serious side effects including:
Liver damage.
- Liver damage may not cause symptoms at first. Only a blood test can show if you have early liver damage. You must have your blood tested to check your liver function before you start Bosentan Tablets and each month after that. Your healthcare provider will order these tests. Regular blood tests are important because they will help your healthcare provider adjust or stop your treatment before there is permanent damage.
- Tell your healthcare provider if you have had liver problems, including liver problems while taking other medicines. Call your healthcare provider right away if you have any of these symptoms of liver problems while taking Bosentan Tablets:
- nausea
- vomiting
- fever
- unusual tiredness
- stomach area (abdominal) pain
- yellowing of the skin or the whites of your eyes (jaundice)
Serious birth defects.
-
Bosentan Tablets can cause serious birth defects if taken during pregnancy. You must not be pregnant when you start taking Bosentan Tablets or during Bosentan Tablets treatment. Serious birth defects from Bosentan Tablets can happen early in pregnancy. Females who are able to get pregnant must have a negative pregnancy test before starting treatment with Bosentan Tablets, each month during treatment with Bosentan Tablets and 1 month after stopping treatment with Bosentan Tablets.
- Talk to your healthcare provider about your menstrual cycle. Your healthcare provider will decide when to do a pregnancy test and will order a pregnancy test for you depending on your menstrual cycle.
- Females who are able to get pregnant are females who:
- have entered puberty, even if they have not started their menstrual period, and
- have a uterus, and
- have not gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed.
- Females who are not able to get pregnant are females who:
- have not yet entered puberty, or
- do not have a uterus, or
- have gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed or
- are infertile for other medical reasons and this infertility is permanent and cannot be reversed.
-
Females who are able to get pregnant must use two acceptable forms of birth control during treatment with Bosentan Tablets, and for one month after stopping Bosentan Tablets because the medicine may still be in the body.
- If you have had a tubal sterilization or have an IUD (intrauterine device), these methods can be used alone and no other form of birth control is needed.
- Talk with your healthcare provider or gynecologist (a doctor who specializes in female reproduction) to find out about options for acceptable birth control that you may use to prevent pregnancy during treatment with Bosentan Tablets.
- If you decide that you want to change the form of birth control that you use, talk with your healthcare provider or gynecologist to be sure that you choose another acceptable form of birth control.
See the chart below for Acceptable Birth Control Options during treatment with Bosentan Tablets.
- Do not have unprotected sex. Talk to your healthcare provider or pharmacist right away if you have unprotected sex or if you think your birth control has failed. Your healthcare provider may talk with you about using emergency birth control.
- Tell your healthcare provider right away if you miss a menstrual period or think you may be pregnant.
If you are the parent or caregiver of a female child who started taking Bosentan Tablets before reaching puberty, you should check your child regularly to see if she is developing signs of puberty. Tell your healthcare provider right away if you notice that she is developing breast buds or any pubic hair. Your healthcare provider should decide if your child has reached puberty. Your child may reach puberty before having her first menstrual period.
Acceptable birth control options
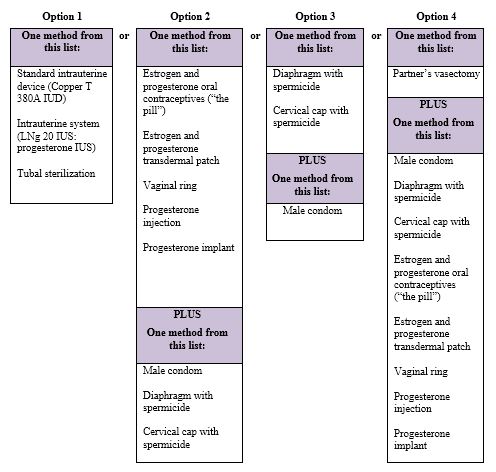
See “What are the possible side effects of Bosentan Tablets?” for more information about side effects.
What are Bosentan Tablets?
Bosentan Tablets are a prescription medicine used to treat people with certain types of pulmonary arterial hypertension (PAH), which is high blood pressure in the vessels of the lungs.
Bosentan Tablets can improve your ability to exercise and can slow the worsening of your physical condition and symptoms. Bosentan Tablets lowers high blood pressure in your lungs and lets your heart pump blood more efficiently.
Bosentan Tablets are only:
Prescribed by healthcare providers who are enrolled in the Bosentan REMS Program.
Available to people who understand and agree to enroll in the Bosentan REMS Program.
Who should not take Bosentan Tablets?
Do not take Bosentan Tablets if you:
-
are pregnant, plan to become pregnant, or become pregnant during Bosentan Tablets treatment. Bosentan Tablets can cause serious birth defects. All females should read the birth defects section of "What is the most important information I should know about Bosentan Tablets?"
- take any of these medicines:
- cyclosporine A used to treat psoriasis and rheumatoid arthritis, and to prevent rejection of heart, liver, and kidney transplants
- glyburide used to treat diabetes
- are allergic to bosentan or any of the ingredients in Bosentan Tablets. See the end of this Medication Guide for a complete list of the ingredients in Bosentan Tablets. If you have a rash, hives or your lips swell after taking Bosentan Tablets, it may be a sign of allergy. You should stop taking your Bosentan Tablets and talk to your healthcare provider.
What should I tell my healthcare provider before taking Bosentan Tablets?
Bosentan Tablets may not be right for you. Tell your healthcare provider about all your medical conditions, including if you:
- have liver problems.
- are breast-feeding or plan to breast feed. It is not known if Bosentan Tablets passes into your milk. You and your healthcare provider should decide if you will take Bosentan Tablets or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Bosentan Tablets and other medicines may affect how each other works and cause side effects. Especially tell your healthcare provider if you take:
- hormone-based birth control, such as pills, shots, patches, and implants. These birth control methods may not work as well when taken with Bosentan Tablets.
- simvastatin or other "-statin" medicines used to lower cholesterol
- rifampin used for tuberculosis
- ketoconazole, fluconazole, itraconazole, or voriconazole used for fungal infections
- warfarin sodium used to prevent blood clots
- ritonavir used to treat HIV
There may be more than one brand name medicine. Ask your healthcare provider if you are not sure if your medicine is one that is listed above.
Know the medicines you take. Keep a list of them and show it to your healthcare provider or pharmacist when you get a new medicine.
How should I take Bosentan Tablets?
Your healthcare provider will give you detailed information about the Bosentan REMS Program.
- Bosentan Tablets will be mailed to you by a specialty pharmacy. You will only receive a 30-day supply of Bosentan Tablets at one time.
- Take Bosentan Tablets exactly as prescribed.
- Your healthcare provider will tell you how much Bosentan Tablets to take and when to take it.
- In most cases, you will take 1 tablet in the morning and 1 in the evening.
- You can take Bosentan Tablets orally with or without food.
- If you take more than the prescribed dose of Bosentan Tablets, call your healthcare provider right away.
- If you miss a dose of Bosentan Tablets, take your tablet as soon as you remember. Do not take 2 doses at the same time. If it is almost time for your next dose, skip the missed dose. Just take the next dose at your regular time.
- Do not stop taking Bosentan Tablets unless your healthcare provider tells you to. Suddenly stopping your treatment may cause your symptoms to get worse. If you need to stop taking Bosentan Tablets, speak with your healthcare provider about the right way to stop.
What are the possible side effects of Bosentan Tablets?
Bosentan Tablets can cause serious side effects, including:
- See "What is the most important information I should know about Bosentan Tablets?"
- Fluid retention and swelling of your ankles and legs. Bosentan Tablets can cause your body to hold too much water, and you may get swelling of your ankles and legs. Tell your healthcare provider if you have swelling of your ankles and legs that happens either with or without weight gain, or if you have more trouble with your breathing than normal. Your healthcare provider will look for the cause of this.
- Lower Sperm Count. Some men who take Bosentan Tablets may have lower sperm counts. This may affect your ability to father a child. Tell your healthcare provider if fertility is a concern for you.
- Low red blood cell levels (anemia). Your healthcare provider will do blood tests to check your red blood cells during treatment with Bosentan Tablets.
The most common side effects of Bosentan Tablets include:
- respiratory tract infection
- headache
- fainting
- flushing
- low blood pressure
- inflamed nose passages (sinusitis)
- joint pain
- irregular heart beats
Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of Bosentan Tablets. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Bosentan Tablets?
- Store Bosentan Tablets at room temperature between 68°F to 77°F (20°C to 25°C).
Keep Bosentan Tablets and all medicines out of the reach of children.
General information about Bosentan Tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Bosentan Tablets for a condition for which it was not prescribed. Do not give Bosentan Tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about Bosentan Tablets. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about Bosentan Tablets that is written for health professionals. For more information, call Alvogen at 1-866-770-3024.
What are the ingredients in Bosentan Tablets?
Active ingredient: bosentan
Inactive ingredients: povidone, pregelatinized starch, sodium starch glycolate, and magnesium stearate.
Additionally, the film coating contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, yellow iron oxide, and red iron oxide.


| BOSENTAN
bosentan tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| BOSENTAN
bosentan tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Alvogen Inc. (008057330) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Norwich Pharmaceuticals, Inc. | 132218731 | analysis(47781-270, 47781-271) , manufacture(47781-270, 47781-271) , pack(47781-270, 47781-271) | |