FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.5 Paget's Disease of Bone
Zoledronic Acid Injection is indicated for treatment of Paget's disease of bone in men and women. Treatment is indicated in patients with Paget's disease of bone with elevations in serum alkaline phosphatase of two times or higher than the upper limit of the age-specific normal reference range, or those who are symptomatic, or those at risk for complications from their disease [see Clinical Studies (14.5)].
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Zoledronic Acid Injection must be administered as an intravenous infusion over no less than 15 minutes.
- Patients must be appropriately hydrated prior to administration of Zoledronic Acid Injection [see Warnings and Precautions (5.3)].
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Intravenous infusion should be followed by a 10 mL normal saline flush of the intravenous line.
- Administration of acetaminophen following Zoledronic Acid Injection administration may reduce the incidence of acute-phase reaction symptoms.
Caution: After removing the overwrap check for minute leaks by squeezing the inner bag firmly. If leaks are found, discard container as sterility may be compromised. Use only if solution is clear and the container is undamaged.
Preparation for Administration:
- Close flow control clamp of administration set.
- Remove cover from port at bottom of container.
- Insert piercing pin of administration set into port with a twisting motion until the pin is firmly seated. NOTE: See full directions on administration set carton.
- Suspend container from hanger.
- Squeeze and release drip chamber to establish proper fluid level in chamber during infusion of Zoledronic Acid Injection.
- Open flow control clamp to expel air from set. Close clamp.
- Regulate rate of administration with flow control clamp.
2.6 Treatment of Paget's Disease of Bone
The recommended dose is a 5 mg infusion. The infusion time must not be less than 15 minutes given over a constant infusion rate.
Re-treatment of Paget's Disease
After a single treatment with Zoledronic Acid Injection in Paget's disease an extended remission period is observed. Specific re-treatment data are not available. However, re-treatment with Zoledronic Acid Injection may be considered in patients who have relapsed, based on increases in serum alkaline phosphatase, or in those patients who failed to achieve normalization of their serum alkaline phosphatase, or in those patients with symptoms, as dictated by medical practice.
2.7 Laboratory Testing and Oral Examination Prior to Administration
- Prior to administration of each dose of Zoledronic Acid Injection, obtain a serum creatinine and creatinine clearance should be calculated based on actual body weight using Cockcroft-Gault formula before each Zoledronic Acid Injection dose. Zoledronic Acid Injection is contraindicated in patients with creatinine clearance less than 35 mL/min and in those with evidence of acute renal impairment. A 5 mg dose of Zoledronic Acid Injection administered intravenously is recommended for patients with creatinine clearance greater than or equal to 35 mL/min. There are no safety or efficacy data to support the adjustment of the Zoledronic Acid Injection dose based on baseline renal function. Therefore, no dose adjustment is required in patients with CrCl greater than or equal to 35 mL/min [see Contraindications (4), Warnings and Precautions (5.3)].
- A routine oral examination should be performed by the prescriber prior to initiation of Zoledronic Acid Injection treatment [see Warnings and Precautions (5.4)].
2.8 Calcium and Vitamin D Supplementation
- Instruct patients being treated for Paget's disease of bone on the importance of calcium and vitamin D supplementation in maintaining serum calcium levels, and on the symptoms of hypocalcemia. All patients should take 1500 mg elemental calcium daily in divided doses (750 mg two times a day, or 500 mg three times a day) and 800 international units vitamin D daily, particularly in the 2 weeks following Zoledronic Acid Injection administration [see Warnings and Precautions (5.2)].
2.9 Method of Administration
The Zoledronic Acid Injection infusion time must not be less than 15 minutes given over a constant infusion rate.
The intravenous infusion should be followed by a 10 mL normal saline flush of the intravenous line.
Zoledronic Acid Injection solution for infusion must not be allowed to come in contact with any calcium or other divalent cation-containing solutions, and should be administered as a single intravenous solution through a separate infusion line.
If refrigerated, allow the refrigerated solution to reach room temperature before administration. After opening, the solution is stable for 24 hours at 2°C to 8°C (36°F to 46°F) [see How Supplied/Storage and Handling (16)].
4 CONTRAINDICATIONS
Zoledronic Acid Injection is contraindicated in patients with the following conditions:
- Hypocalcemia [see Warnings and Precautions (5.2)].
- Creatinine clearance less than 35 mL/min and in those with evidence of acute renal impairment due to an increased risk of renal failure [see Warnings and Precautions (5.3)].
- Known hypersensitivity to zoledronic acid or any components of Zoledronic Acid Injection. Hypersensitivity reactions including urticaria, angioedema, and anaphylactic reaction/shock have been reported [see Adverse Reactions (6.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Drug Products with Same Active Ingredient
Zoledronic Acid Injection contains the same active ingredient found in Zometa, used for oncology indications, and a patient being treated with Zometa should not be treated with Zoledronic Acid Injection.
5.2 Hypocalcemia and Mineral Metabolism
Pre-existing hypocalcemia and disturbances of mineral metabolism (e.g., hypoparathyroidism, thyroid surgery, parathyroid surgery; malabsorption syndromes, excision of small intestine) must be effectively treated before initiating therapy with Zoledronic Acid Injection. Clinical monitoring of calcium and mineral levels (phosphorus and magnesium) is highly recommended for these patients [see Contraindications (4)].
Hypocalcemia following Zoledronic Acid Injection administration is a significant risk in Paget's disease. All patients should be instructed about the symptoms of hypocalcemia and the importance of calcium and vitamin D supplementation in maintaining serum calcium levels [see Dosage and Administration (2.8), Adverse Reactions (6.1), Patient Counseling Information (17)].
5.3 Renal Impairment
A single dose of Zoledronic Acid Injection should not exceed 5 mg and the duration of infusion should be no less than 15 minutes [see Dosage and Administration (2)].
Zoledronic Acid Injection is contraindicated in patients with creatinine clearance less than 35 mL/min and in those with evidence of acute renal impairment [see Contraindications (4)]. If history or physical signs suggest dehydration, Zoledronic Acid Injection therapy should be withheld until normovolemic status has been achieved [see Adverse Reactions (6.2)].
Zoledronic Acid Injection should be used with caution in patients with chronic renal impairment. Acute renal impairment, including renal failure, has been observed following the administration of zoledronic acid, especially in patients with pre-existing renal compromise, advanced age, concomitant nephrotoxic medications, concomitant diuretic therapy, or severe dehydration occurring before or after Zoledronic Acid Injection administration. Acute renal failure (ARF) has been observed in patients after a single administration. Rare reports of hospitalization and/or dialysis or fatal outcome occurred in patients with underlying moderate to severe renal impairment or with any of the risk factors described in this section [see Adverse Reactions (6.2)]. Renal impairment may lead to increased exposure of concomitant medications and/or their metabolites that are primarily renally excreted [see Drug Interactions (7.4)].
Creatinine clearance should be calculated based on actual body weight using Cockcroft-Gault formula before each Zoledronic Acid Injection dose. Transient increase in serum creatinine may be greater in patients with impaired renal function; interim monitoring of creatinine clearance should be performed in at-risk patients. Elderly patients and those receiving diuretic therapy are at increased risk of acute renal failure. These patients should have their fluid status assessed and be appropriately hydrated prior to administration of Zoledronic Acid Injection. Zoledronic Acid Injection should be used with caution with other nephrotoxic drugs [see Drug Interactions (7.3)]. Consider monitoring creatinine clearance in patients at-risk for ARF who are taking concomitant medications that are primarily excreted by the kidney [see Drug Interactions (7.4)].
5.4 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ) has been reported in patients treated with bisphosphonates, including zoledronic acid. Most cases have been in cancer patients treated with intravenous bisphosphonates undergoing dental procedures. A routine oral examination should be performed by the prescriber prior to initiation of bisphosphonate treatment. A dental examination with appropriate preventive dentistry should be considered prior to treatment with bisphosphonates in patients with a history of concomitant risk factors (e.g., cancer, chemotherapy, angiogenesis inhibitors, radiotherapy, corticosteroids, poor oral hygiene, pre-existing dental disease or infection, anemia, coagulopathy). The risk of ONJ may increase with duration of exposure to bisphosphonates. Concomitant administration of drugs associated with ONJ may increase the risk of developing ONJ.
While on treatment, patients with concomitant risk factors should avoid invasive dental procedures if possible. For patients who develop ONJ while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. The clinical judgment of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment [see Adverse Reactions (6.1)].
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical, low-energy, or low trauma fractures of the femoral shaft have been reported in bisphosphonate-treated patients. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution.
Atypical femur fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patients presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of bisphosphonate therapy should be considered, pending a risk/benefit assessment, on an individual basis.
5.6 Musculoskeletal Pain
In post-marketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain have been infrequently reported in patients taking bisphosphonates, including Zoledronic Acid Injection. The time to onset of symptoms varied from one day to several months after starting the drug. Consider withholding future Zoledronic Acid Injection treatment if severe symptoms develop. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate [see Adverse Reactions (6.2)].
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Paget's Disease of Bone
In the Paget's disease trials, two 6-month, double-blind, comparative, multinational studies of 349 men and women aged greater than 30 years with moderate to severe disease and with confirmed Paget's disease of bone, 177 patients were exposed to Zoledronic Acid Injection and 172 patients exposed to risedronate. Zoledronic Acid Injection was administered once as a single 5 mg dose in 100 mL solution infused over at least 15 minutes. Risedronate was given as an oral daily dose of 30 mg for 2 months.
The incidence of serious adverse events was 5.1% in the Zoledronic Acid Injection group and 6.4% in the risedronate group. The percentage of patients who withdrew from the study due to adverse events was 1.7% and 1.2% for the Zoledronic Acid Injection and risedronate groups, respectively.
Adverse reactions occurring in at least 2% of the Paget's patients receiving Zoledronic Acid Injection (single 5 mg intravenous infusion) or risedronate (30 mg oral daily dose for 2 months) over a 6-month study period are listed by system organ class in Table 1.
| 5 mg IV Zoledronic Acid Injection | 30 mg/day x 2 Months risedronate | |
| % | % | |
| System Organ Class | (N = 177) | (N = 172) |
| Infections and Infestations | ||
| Influenza | 7 | 5 |
| Metabolism and Nutrition Disorders | ||
| Hypocalcemia | 3 | 1 |
| Anorexia | 2 | 2 |
| Nervous System Disorders | ||
| Headache | 11 | 10 |
| Dizziness | 9 | 4 |
| Lethargy | 5 | 1 |
| Paresthesia | 2 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Dyspnea | 5 | 1 |
| Gastrointestinal Disorders | ||
| Nausea | 9 | 6 |
| Diarrhea | 6 | 6 |
| Constipation | 6 | 5 |
| Dyspepsia | 5 | 4 |
| Abdominal Distension | 2 | 1 |
| Abdominal Pain | 2 | 2 |
| Vomiting | 2 | 2 |
| Abdominal Pain Upper | 1 | 2 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash | 3 | 2 |
| Musculoskeletal, Connective Tissue and Bone Disorders | ||
| Arthralgia | 9 | 11 |
| Bone Pain | 9 | 5 |
| Myalgia | 7 | 4 |
| Back Pain | 4 | 7 |
| Musculoskeletal Stiffness | 2 | 1 |
| General Disorders and Administrative Site Conditions | ||
| Influenza-like Illness | 11 | 6 |
| Pyrexia | 9 | 2 |
| Fatigue | 8 | 4 |
| Rigors | 8 | 1 |
| Pain | 5 | 4 |
| Peripheral Edema | 3 | 1 |
| Asthenia | 2 | 1 |
Laboratory Findings
In the Paget's disease trials, early, transient decreases in serum calcium and phosphate levels were observed. Approximately 21% of patients had serum calcium levels less than 8.4 mg/dL 9 to 11 days following Zoledronic Acid Injection administration.
Renal Impairment
In clinical trials in Paget's disease there were no cases of renal deterioration following a single 5 mg 15-minute infusion [see Warnings and Precautions (5.3)].
Acute Phase Reaction
The signs and symptoms of acute phase reaction (influenza-like illness, pyrexia, myalgia, arthralgia, and bone pain) were reported in 25% of patients in the Zoledronic Acid Injection-treated group compared to 8% in the risedronate-treated group. Symptoms usually occur within the first 3 days following Zoledronic Acid Injection administration. The majority of these symptoms resolved within 4 days of onset.
Osteonecrosis of the Jaw
Osteonecrosis of the jaw has been reported with zoledronic acid [see Warnings and Precautions (5.4)].
6.2 Post-Marketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been identified during post approval use of Zoledronic Acid Injection:
Acute Phase Reactions
Fever, headache, flu-like symptoms, nausea, vomiting, diarrhea, arthralgia, and myalgia. Symptoms may be significant and lead to dehydration.
Acute Renal Failure
Acute renal failure requiring hospitalization and/or dialysis or with a fatal outcome have been rarely reported. Increased serum creatinine was reported in patients with 1) underlying renal disease, 2) dehydration secondary to fever, sepsis, gastrointestinal losses, or diuretic therapy, or 3) other risk factors such as advanced age, or concomitant nephrotoxic drugs in the post-infusion period. Transient rise in serum creatinine can be correctable with intravenous fluids.
Allergic Reactions
Allergic reactions with intravenous zoledronic acid including anaphylactic reaction/shock, urticaria, angioedema, Stevens-Johnson syndrome, toxic epidermal necrolysis, and bronchoconstriction have been reported.
Osteonecrosis of other bones
Cases of osteonecrosis of other bones (including femur, hip, knee, ankle, wrist and humerus) have been reported; causality has not been determined in the population treated with zoledronic acid.
7 DRUG INTERACTIONS
No in vivo drug interaction studies have been performed for Zoledronic Acid Injection. In vitro and ex vivo studies showed low affinity of zoledronic acid for the cellular components of human blood. In vitro mean zoledronic acid protein binding in human plasma ranged from 28% at 200 ng/mL to 53% at 50 ng/mL. In vivo studies showed that zoledronic acid is not metabolized, and is excreted into the urine as the intact drug.
7.1 Aminoglycosides
Caution is advised when bisphosphonates, including zoledronic acid, are administered with aminoglycosides, since these agents may have an additive effect to lower serum calcium level for prolonged periods. This effect has not been reported in zoledronic acid clinical trials.
7.2 Loop Diuretics
Caution should also be exercised when Zoledronic Acid Injection is used in combination with loop diuretics due to an increased risk of hypocalcemia.
7.3 Nephrotoxic Drugs
Caution is indicated when Zoledronic Acid Injection is used with other potentially nephrotoxic drugs such as nonsteroidal anti-inflammatory drugs.
7.4 Drugs Primarily Excreted by the Kidney
Renal impairment has been observed following the administration of zoledronic acid in patients with pre-existing renal compromise or other risk factors [see Warnings and Precautions (5.3)]. In patients with renal impairment, the exposure to concomitant medications that are primarily renally excreted (e.g., digoxin) may increase. Consider monitoring serum creatinine in patients at risk for renal impairment who are taking concomitant medications that are primarily excreted by the kidney.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data on the use of Zoledronic Acid Injection in pregnant women are insufficient to inform a drug-associated risk of adverse maternal or fetal outcomes. Discontinue Zoledronic Acid Injection when pregnancy is recognized.
In animal reproduction studies, daily subcutaneous administration of zoledronic acid to pregnant rats during organogenesis resulted in increases in fetal skeletal, visceral, and external malformations, decreases in postimplantation survival, and decreases in viable fetuses and fetal weight starting at doses equivalent to 2 times the recommended human 5 mg intravenous dose (based on AUC). Subcutaneous administration of zoledronic acid to rabbits during organogenesis did not cause adverse fetal effects at up to 0.4 times the human 5 mg intravenous dose (based on body surface area, mg/m2), but resulted in maternal mortality and abortion associated with hypocalcemia starting at doses equivalent to 0.04 times the human 5 mg intravenous dose. Subcutaneous dosing of female rats from before mating through gestation and lactation and allowed to deliver caused maternal dystocia and periparturient mortality, increases in stillbirths and neonatal deaths, and reduced pup body weight starting at doses equivalent to 0.1 times the human 5 mg intravenous dose (based on AUC). (see Data).Bisphosphonates are incorporated into the bone matrix, from which they are gradually released over a period of years. The amount of bisphosphonate incorporated into adult bone, and available for release into the systemic circulation is directly related to the dose and duration of bisphosphonate use. Consequently, based on the mechanism of action of bisphosphonates, there is a potential risk of fetal harm, predominantly skeletal, if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous versus oral) on the risk has not been studied.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
In pregnant rats given daily subcutaneous doses of zoledronic acid of 0.1, 0.2, or 0.4 mg/kg during organogenesis, fetal skeletal, visceral, and external malformations, increases in pre-and post-implantation loss, and decreases in viable fetuses and fetal weight were observed at 0.2 and 0.4 mg/kg/day (equivalent to 2 and 4 times the human 5 mg intravenous dose, based on AUC). Adverse fetal skeletal effects at 0.4 mg/kg/day (4 times the human 5 mg dose) included unossified or incompletely ossified bones, thickened, curved or shortened bones, wavy ribs, and shortened jaw. Other adverse fetal effects at this dose included reduced lens, rudimentary cerebellum, reduction or absence of liver lobes, reduction of lung lobes, vessel dilation, cleft palate, and edema. Skeletal variations were observed in all groups starting at 0.1 mg/kg/day (1.2 times the human 5 mg dose). Signs of maternal toxicity including reduced body weight and food consumption were observed at 0.4 mg/kg/day (4 times the human 5 mg dose).
In pregnant rabbits given daily subcutaneous doses of zoledronic acid of 0.01, 0.03, or 0.1 mg/kg during gestation no adverse fetal effects were observed up to 0.1 mg/kg/day (0.4 times the human 5 mg intravenous dose, based on body surface area, mg/m2). Maternal mortality and abortion were observed in all dose groups (starting at 0.04 times the human 5 mg dose). Adverse maternal effects were associated with drug-induced hypocalcemia.
In female rats given daily subcutaneous doses of 0.01, 0.03, or 0.1 mg/kg, beginning 15 days before mating and continuing through gestation, parturition and lactation, dystocia and periparturient mortality were observed in pregnant rats allowed to deliver starting at 0.01 mg/kg/day (0.1 times the human 5 mg intravenous dose, based on AUC). Also, there was an increase in stillbirths and a decrease in neonate survival starting at 0.03 mg/kg/day (0.3 times the human 5 mg dose), while the number of viable newborns and pup body weight on postnatal Day 7 were decreased at 0.1 mg/kg/day (equivalent to the human 5 mg dose). Maternal and neonatal deaths were considered related to drug-induced periparturient hypocalcemia.
8.2 Lactation
Risk Summary
There are no data on the presence of zoledronic acid in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breast-feeding should be considered along with the mother's clinical need for zoledronic acid and any potential adverse effects on the breast-fed child from zoledronic acid or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
There are no data available in humans. Female fertility may be impaired based on animal studies demonstrating adverse effects of zoledronic acid on fertility parameters [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Zoledronic Acid Injection is not indicated for use in children.
The safety and effectiveness of zoledronic acid was studied in a one-year active controlled trial of 152 pediatric subjects (74 receiving zoledronic acid). The enrolled population was subjects with severe osteogenesis imperfecta, aged 1 to 17 years, 55% male, 84% Caucasian, with a mean lumbar spine BMD of 0.431 gm/cm2, which is 2.7 standard deviations below the mean for age-matched controls (BMD Z-score of -2.7). At one year, increases in BMD were observed in the zoledronic acid treatment group. However, changes in BMD in individual patients with severe osteogenesis imperfecta did not necessarily correlate with the risk for fracture or the incidence or severity of chronic bone pain. The adverse events observed with zoledronic acid use in children did not raise any new safety findings beyond those previously seen in adults treated for Paget's disease of bone including osteonecrosis of the jaw (ONJ) and renal impairment. However, adverse reactions seen more commonly in pediatric patients included pyrexia (61%), arthralgia (26%), hypocalcemia (22%) and headache (22%). These reactions, excluding arthralgia, occurred most frequently within three days after the first infusion and became less common with repeat dosing. No cases of ONJ or renal impairment were observed in this study. Because of long-term retention in bone, Zoledronic Acid Injection should only be used in children if the potential benefit outweighs the potential risk.
Plasma zoledronic acid concentration data was obtained from 10 patients with severe osteogenesis imperfecta (4 in the age group of 3 to 8 years and 6 in the age group of 9 to 17 years) infused with 0.05 mg/kg dose over 30 minutes. Mean Cmax and AUC(0-last) was 167 ng/mL and 220 ng.h/mL respectively. The plasma concentration time profile of zoledronic acid in pediatric patients represent a multi-exponential decline, as observed in adult cancer patients at an approximately equivalent mg/kg dose.
8.5 Geriatric Use
Of the patients receiving Zoledronic Acid Injection in the Paget's disease studies, 132 patients were 65 years of age or over, while 68 patients were at least 75 years of age.
Because decreased renal function occurs more commonly in the elderly, special care should be taken to monitor renal function.
8.6 Renal Impairment
Zoledronic Acid Injection is contraindicated in patients with creatinine clearance less than 35 mL/min and in those with evidence of acute renal impairment. There are no safety or efficacy data to support the adjustment of the Zoledronic Acid Injection dose based on baseline renal function. Therefore, no dosage adjustment is required in patients with a creatinine clearance of greater than or equal to 35 mL/min [see Warnings and Precautions (5.3), Clinical Pharmacology (12.3)]. Risk of acute renal failure may increase with underlying renal disease and dehydration secondary to fever, sepsis, gastrointestinal losses, diuretic therapy, advanced age, etc. [see Adverse Reactions (6.2)].
10 OVERDOSAGE
Clinical experience with acute overdosage of zoledronic acid (Zoledronic Acid Injection) solution for intravenous infusion is limited. Patients who have received doses higher than those recommended should be carefully monitored. Overdosage may cause clinically significant renal impairment, hypocalcemia, hypophosphatemia, and hypomagnesemia. Clinically relevant reductions in serum levels of calcium, phosphorus, and magnesium should be corrected by intravenous administration of calcium gluconate, potassium or sodium phosphate, and magnesium sulfate, respectively.
Single doses of Zoledronic Acid Injection should not exceed 5 mg and the duration of the intravenous infusion should be no less than 15 minutes [see Dosage and Administration (2)].
11 DESCRIPTION
Zoledronic Acid Injection contains zoledronic acid, a bisphosphonic acid which is an inhibitor of osteoclastic bone resorption. Zoledronic acid is designated chemically as (1-Hydroxy-2-imidazol-1-yl-phosphonoethyl) phosphonic acid monohydrate and its structural formula is:
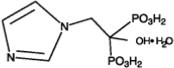
Zoledronic acid monohydrate is a white crystalline powder. Its molecular formula is C5H10N2O7P2 • H2O and a molar mass of 290.1 g/Mol. Zoledronic acid monohydrate is highly soluble in 0.1N sodium hydroxide solution, sparingly soluble in water and 0.1N hydrochloric acid, and practically insoluble in organic solvents. The pH of the Zoledronic Acid Injection solution for infusion is approximately 6.0 to 7.0.
Zoledronic Acid Injection is available as a sterile solution in bags for intravenous infusion. One bag with 100 mL solution contains 5.330 mg of zoledronic acid monohydrate, equivalent to 5 mg zoledronic acid on an anhydrous basis.
Inactive Ingredients: 4950 mg of mannitol, USP; and 30 mg of sodium citrate, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Zoledronic Acid Injection is a bisphosphonate and acts primarily on bone. It is an inhibitor of osteoclast-mediated bone resorption.
The selective action of bisphosphonates on bone is based on their high affinity for mineralized bone. Intravenously administered zoledronic acid rapidly partitions to bone and localizes preferentially at sites of high bone turnover. The main molecular target of zoledronic acid in the osteoclast is the enzyme farnesyl pyrophosphate synthase. The relatively long duration of action of zoledronic acid is attributable to its high binding affinity to bone mineral.
12.3 Pharmacokinetics
Pharmacokinetic data in patients with Paget's disease of bone are not available.
Distribution: Single or multiple (every 28 days) 5-minute or 15-minute infusions of 2, 4, 8 or 16 mg zoledronic acid were given to 64 patients with cancer and bone metastases. The post-infusion decline of zoledronic acid concentrations in plasma was consistent with a triphasic process showing a rapid decrease from peak concentrations at end-of-infusion to less than 1% of Cmax 24 hours post infusion with population half-lives of t1/2α 0.24 hour and t1/2β 1.87 hours for the early disposition phases of the drug. The terminal elimination phase of zoledronic acid was prolonged, with very low concentrations in plasma between Days 2 and 28 post infusion, and a terminal elimination half-life t1/2γ of 146 hours. The area under the plasma concentration versus time curve (AUC0-24h) of zoledronic acid was dose proportional from 2 to 16 mg. The accumulation of zoledronic acid measured over three cycles was low, with mean AUC0-24h ratios for cycles 2 and 3 versus 1 of 1.13 ± 0.30 and 1.16 ± 0.36, respectively.
In vitro and ex vivo studies showed low affinity of zoledronic acid for the cellular components of human blood. In vitro mean zoledronic acid protein binding in human plasma ranged from 28% at 200 ng/mL to 53% at 50 ng/mL.
Metabolism: Zoledronic acid does not inhibit human P450 enzymes in vitro. Zoledronic acid does not undergo biotransformation in vivo. In animal studies, less than 3% of the administered intravenous dose was found in the feces, with the balance either recovered in the urine or taken up by bone, indicating that the drug is eliminated intact via the kidney. Following an intravenous dose of 20 nCi 14C-zoledronic acid in a patient with cancer and bone metastases, only a single radioactive species with chromatographic properties identical to those of parent drug was recovered in urine, which suggests that zoledronic acid is not metabolized.
Excretion: In 64 patients with cancer and bone metastases on average (± SD) 39 ± 16% of the administered zoledronic acid dose was recovered in the urine within 24 hours, with only trace amounts of drug found in urine post Day 2. The cumulative percent of drug excreted in the urine over 0 to 24 hours was independent of dose. The balance of drug not recovered in urine over 0 to 24 hours, representing drug presumably bound to bone, is slowly released back into the systemic circulation, giving rise to the observed prolonged low plasma concentrations. The 0 to 24 hour renal clearance of zoledronic acid was 3.7 ± 2.0 L/h.
Zoledronic acid clearance was independent of dose but dependent upon the patient's creatinine clearance. In a study in patients with cancer and bone metastases, increasing the infusion time of a 4 mg dose of zoledronic acid from 5 minutes (n=5) to 15 minutes (n=7) resulted in a 34% decrease in the zoledronic acid concentration at the end of the infusion ([mean ± SD] 403 ± 118 ng/mL vs. 264 ± 86 ng/mL) and a 10% increase in the total AUC (378 ± 116 ng x h/mL vs. 420 ± 218 ng x h/mL). The difference between the AUC means was not statistically significant.
Pediatrics: Zoledronic Acid Injection is not indicated for use in children [see Pediatric Use (8.4)].
Geriatrics: The pharmacokinetics of zoledronic acid was not affected by age in patients with cancer and bone metastases whose age ranged from 38 years to 84 years.
Race: The pharmacokinetics of zoledronic acid was not affected by race in patients with cancer and bone metastases.
Hepatic Impairment: No clinical studies were conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of zoledronic acid.
Renal Impairment: The pharmacokinetic studies conducted in 64 cancer patients represented typical clinical populations with normal to moderately-impaired renal function. Compared to patients with creatinine clearance greater than 80 mL/min (N=37), patients with creatinine clearance = 50 to 80 mL/min (N=15) showed an average increase in plasma AUC of 15%, whereas patients with creatinine clearance = 30 to 50 mL/min (N=11) showed an average increase in plasma AUC of 43%. No dosage adjustment is required in patients with a creatinine clearance of greater than or equal to 35 mL/min. Zoledronic Acid Injection is contraindicated in patients with creatinine clearance less than 35 mL/min and in those with evidence of acute renal impairment due to an increased risk of renal failure [see Contraindications (4), Warnings and Precautions (5.3), Use in Specific Populations (8.6)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Lifetime carcinogenicity bioassays were conducted in mice and rats. Mice were given daily oral doses of zoledronic acid of 0.1, 0.5, or 2 mg/kg/day for 2 years. There was an increased incidence of Harderian gland adenomas in males and females in all treatment groups (starting at doses equivalent to 0.002 times the human 5 mg intravenous dose, based on body surface area, mg/m2). Rats were given daily oral doses of zoledronic acid of 0.1, 0.5, or 2 mg/kg/day for 2 years. No increased incidence of tumors was observed at any dose (up to to 0.1 times the human intravenous dose of 5 mg, based on body surface area, mg/m2).
Mutagenesis: Zoledronic acid was not genotoxic in the Ames bacterial mutagenicity assay, in the Chinese hamster ovary cell assay, or in the Chinese hamster gene mutation assay, with or without metabolic activation. Zoledronic acid was not genotoxic in the in vivo rat micronucleus assay.
Impairment of Fertility: Female rats were given daily subcutaneous doses of zoledronic acid of 0.01, 0.03, or 0.1 mg/kg beginning 15 days before mating and continuing through gestation. Inhibition of ovulation and a decrease in the number of pregnant rats were observed at 0.1 mg/kg/day (equivalent to the human 5 mg intravenous dose, based on AUC). An increase in preimplantation loss and a decrease in the number of implantations and live fetuses were observed at 0.03 and 1 mg/kg/day (0.3 to 1 times the human 5 mg human intravenous dose).
13.2 Animal Pharmacology
Bone Safety Studies: Zoledronic acid is a potent inhibitor of osteoclastic bone resorption. In the ovariectomized rat, single IV doses of zoledronic acid of 4 to 500 mcg/kg (0.1 to 3.5 times the human 5 mg intravenous dose, based on body surface area, mg/m2) suppressed bone turnover and protected against trabecular bone loss, cortical thinning and the reduction in vertebral and femoral bone strength in a dose-dependent manner. At a dose equivalent to human exposure at the 5 mg intravenous dose, the effect persisted for 8 months, which corresponds to approximately 8 remodeling cycles or 3 years in humans.
In ovariectomized rats and monkeys, weekly treatment with zoledronic acid dose-dependently suppressed bone turnover and prevented the decrease in cancellous and cortical BMD and bone strength, at yearly cumulative doses up to 3.5 times the human 5 mg intravenous dose, based on body surface area, mg/m2. Bone tissue was normal and there was no evidence of a mineralization defect, no accumulation of osteoid, and no woven bone.
14 CLINICAL STUDIES
14.5 Treatment of Paget's Disease of Bone
Zoledronic Acid Injection was studied in male and female patients with moderate to severe Paget's disease of bone, defined as serum alkaline phosphatase level at least twice the upper limit of the age-specific normal reference range at the time of study entry. Diagnosis was confirmed by radiographic evidence.
The efficacy of one infusion of 5 mg Zoledronic Acid Injection vs. oral daily doses of 30 mg risedronate for 2 months was demonstrated in two identically designed 6-month randomized, double-blind trials. The mean age of patients in the two trials was 70. Ninety-three percent (93%) of patients were Caucasian. Therapeutic response was defined as either normalization of serum alkaline phosphatase (SAP) or a reduction of at least 75% from baseline in total SAP excess at the end of 6 months. SAP excess was defined as the difference between the measured level and midpoint of normal range.
In both trials Zoledronic Acid Injection demonstrated a superior and more rapid therapeutic response compared with risedronate and returned more patients to normal levels of bone turnover, as evidenced by biochemical markers of formation (SAP, serum N-terminal propeptide of type I collagen [P1NP]) and resorption (serum CTx 1 [cross-linked C-telopeptides of type I collagen] and urine α-CTx).
The 6-month combined data from both trials showed that 96% (169/176) of Zoledronic Acid Injection-treated patients achieved a therapeutic response as compared with 74% (127/171) of patients treated with risedronate. Most Zoledronic Acid Injection patients achieved a therapeutic response by the Day 63 visit. In addition, at 6 months, 89% (156/176) of Zoledronic Acid Injection-treated patients achieved normalization of SAP levels, compared to 58% (99/171) of patients treated with risedronate (p<0.0001) (see Figure 1).
Figure 1. Therapeutic Response/Serum Alkaline Phosphatase (SAP) Normalization Over Time
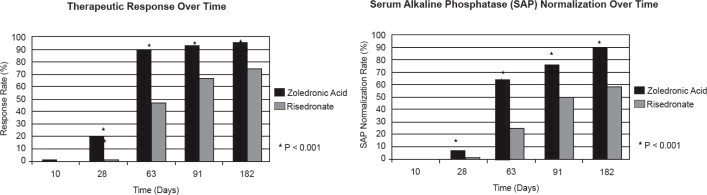
The therapeutic response to Zoledronic Acid Injection was similar across demographic and disease-severity groups defined by gender, age, previous bisphosphonate use, and disease severity. At 6 months, the percentage of Zoledronic Acid Injection-treated patients who achieved therapeutic response was 97% and 95%, respectively, in each of the baseline disease severity subgroups (baseline SAP less than 3xULN, greater than or equal to 3xULN) compared to 75% and 74%, respectively, for the same disease severity subgroups of risedronate-treated patients.
In patients who had previously received treatment with oral bisphosphonates, therapeutic response rates were 96% and 55% for Zoledronic Acid Injection and risedronate, respectively. The comparatively low risedronate response was due to the low response rate (7/23, 30%) in patients previously treated with risedronate. In patients naïve to previous treatment, a greater therapeutic response was also observed with Zoledronic Acid Injection (98%) relative to risedronate (86%). In patients with symptomatic pain at screening, therapeutic response rates were 94% and 70% for Zoledronic Acid Injection and risedronate respectively. For patients without pain at screening, therapeutic response rates were 100% and 82% for Zoledronic Acid Injection and risedronate respectively.
Bone histology was evaluated in 7 patients with Paget's disease 6 months after being treated with Zoledronic Acid Injection 5 mg. Bone biopsy results showed bone of normal quality with no evidence of impaired bone remodeling and no evidence of mineralization defect.
16 HOW SUPPLIED/STORAGE AND HANDLING
Zoledronic Acid Injection is supplied as follows:
| NDC | Zoledronic Acid Injection (0.05 mg per mL) | Package Factor |
| 25021-830-82 | 5 mg per 100 mL ready-to-infuse solution | 1 bag per carton |
| in a flexible plastic container (bag) |
Handling
After entering the IV administration port, the solution is stable for 24 hours at 2° to 8°C (36° to 46°F). If refrigerated, allow the refrigerated solution to reach room temperature before administration.
Storage Conditions
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° and 30°C (59° and 86°F). [See USP Controlled Room Temperature.]
Single-use only. Discard unused portion.
Sterile, Nonpyrogenic, Preservative-free, DEHP-free, PVC-free.
The container closure is not made with natural rubber latex.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Information for Patients
Patients should be made aware that Zoledronic Acid Injection contains the same active ingredient (zoledronic acid) found in Zometa®, and that patients being treated with Zometa should not be treated with Zoledronic Acid Injection.
Zoledronic Acid Injection is contraindicated in patients with creatinine clearance less than 35 mL/min [see Contraindications (4)].
Before being given Zoledronic Acid Injection, patients should tell their doctor if they have kidney problems and what medications they are taking.
Zoledronic Acid Injection should not be given if the patient is pregnant or plans to become pregnant, or if she is breast-feeding [see Warnings and Precautions (5.6)].
There have been reports of bronchoconstriction in aspirin-sensitive patients receiving bisphosphonates, including Zoledronic Acid Injection. Before being given Zoledronic Acid Injection, patients should tell their doctor if they are aspirin-sensitive.
If the patient had surgery to remove some or all of the parathyroid glands in their neck, or had sections of their intestine removed, or are unable to take calcium supplements they should tell their doctor.
Zoledronic Acid Injection is given as an infusion into a vein by a nurse or a doctor, and the infusion time must not be less than 15 minutes.
On the day of treatment the patient should eat and drink normally, which includes drinking at least 2 glasses of fluid such as water within a few hours prior to the infusion, as directed by their doctor, before receiving Zoledronic Acid Injection.
After getting Zoledronic Acid Injection it is strongly recommended patients with Paget's disease take calcium in divided doses (for example, 2 to 4 times a day) for a total of 1500 mg calcium a day to prevent low blood calcium levels. This is especially important for the two weeks after getting Zoledronic Acid Injection [see Warnings and Precautions (5.2)].
Patients should be aware of the most commonly associated side effects of therapy. Patients may experience one or more side effects that could include: fever, flu-like symptoms, myalgia, arthralgia, and headache. Most of these side effects occur within the first 3 days following the dose of Zoledronic Acid Injection. They usually resolve within 3 days of onset but may last for up to 7 to 14 days. Patients should consult their physician if they have questions or if these symptoms persist. The incidence of these symptoms decreased markedly with subsequent doses of Zoledronic Acid Injection.
Administration of acetaminophen following Zoledronic Acid Injection administration may reduce the incidence of these symptoms.
Physicians should inform their patients that there have been reports of persistent pain and/or a non-healing sore of the mouth or jaw, primarily in patients treated with bisphosphonates for other illnesses. During treatment with Zoledronic Acid Injection, patients should be instructed to maintain good oral hygiene and undergo routine dental check-ups. If they experience any oral symptoms, they should immediately report them to their physician or dentist.
Severe and occasionally incapacitating bone, joint, and/or muscle pain have been infrequently reported in patients taking bisphosphonates, including Zoledronic Acid Injection. Consider withholding future Zoledronic Acid Injection treatment if severe symptoms develop.
Atypical femur fractures in patients on bisphosphonate therapy have been reported; patients with thigh or groin pain should be evaluated to rule out a femoral fracture.
Brands listed are the trademarks of their respective owners.
SAGENT®
Mfd. for SAGENT Pharmaceuticals
Schaumburg, IL 60195 (USA)
Made in Switzerland
©2020 Sagent Pharmaceuticals, Inc.
Revised: August 2020
MEDICATION GUIDE
Zoledronic Acid (ZOE-le-DRON-ik AS-id) Injection
This Patient Information has been approved by the U.S. Food and Drug Administration.
PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – Bag Label
NDC 25021-830-82
100 mL
Zoledronic Acid Injection
5 mg per 100 mL
(0.05 mg per mL)
Rx only
For Intravenous Infusion
Do not mix with calcium-containing solutions
Infusion time must not be less than 15 minutes
Single Use Only – Discard Unused Portion
