FULL PRESCRIBING INFORMATION
WARNING: EMBRYO-FETAL TOXICITY
- Exposure to WELIREG during pregnancy can cause embryo-fetal harm.
- Verify pregnancy status prior to the initiation of WELIREG.
- Advise patients of these risks and the need for effective non-hormonal contraception. WELIREG can render some hormonal contraceptives ineffective [see Warnings and Precautions (5.3), Drug Interactions (7.2), Use in Specific Populations (8.1, 8.3)].
1 INDICATIONS AND USAGE
1.1 von Hippel-Lindau (VHL) disease
WELIREG is indicated for treatment of adult patients with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery.
1.2 Advanced Renal Cell Carcinoma (RCC)
WELIREG is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) following a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a vascular endothelial growth factor tyrosine kinase inhibitor (VEGF-TKI).
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of WELIREG is 120 mg administered orally once daily until disease progression or unacceptable toxicity. WELIREG should be taken at the same time each day and may be taken with or without food.
Advise patients to swallow tablets whole. Do not chew, crush, or split WELIREG prior to swallowing.
If a dose of WELIREG is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for WELIREG the next day. Do not take extra tablets to make up for the missed dose.
If vomiting occurs any time after taking WELIREG, do not retake the dose. Take the next dose on the next day.
2.2 Dosage Modifications for Adverse Reactions
Dosage modifications for WELIREG for adverse reactions are summarized in Table 1.
The recommended dose reductions are:
- First dose reduction: WELIREG 80 mg orally once daily
- Second dose reduction: WELIREG 40 mg orally once daily
- Third dose reduction: Permanently discontinue
| Adverse Reaction | Severity | Dosage Modification |
|---|---|---|
|
Anemia [see Warnings and Precautions (5.1)] | Hemoglobin <8 g/dL or transfusion indicated |
|
| Life-threatening or urgent intervention indicated |
|
|
|
Hypoxia [see Warnings and Precautions (5.2)] | Decreased oxygen saturation with exercise (e.g., pulse oximeter <88%) |
|
| Decreased oxygen saturation at rest (e.g., pulse oximeter <88% or PaO2 ≤55 mm Hg) or urgent intervention indicated |
|
|
| Life-threatening or recurrent symptomatic hypoxia |
|
|
|
Other Adverse Reactions [see Adverse Reactions (6)] | Grade 3 |
|
| Grade 4 |
|
3 DOSAGE FORMS AND STRENGTHS
Tablets: 40 mg, blue, oval shaped, film-coated, debossed with “177” on one side and plain on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Anemia
WELIREG can cause severe anemia that can require blood transfusion.
Monitor for anemia before initiation of, and periodically throughout, treatment with WELIREG. Transfuse patients as clinically indicated. For patients with hemoglobin <8g/dL, withhold WELIREG until ≥8g/dL, then resume at the same or reduced dose or permanently discontinue WELIREG, depending on the severity of anemia. For life threatening anemia or when urgent intervention is indicated, withhold WELIREG until hemoglobin ≥8g/dL, then resume at a reduced dose or permanently discontinue WELIREG [see Dosage and Administration (2.2)].
von Hippel-Lindau (VHL) disease
In LITESPARK-004, decreased hemoglobin occurred in 93% of patients and 7% had Grade 3 events [see Adverse Reactions (6.1)]. Median time to onset of anemia was 31 days (range: 1 day to 8.4 months).
The safety of erythropoiesis stimulating agents (ESAs) for treatment of anemia in patients with VHL disease treated with WELIREG has not been established. Randomized controlled trials in patients with cancer receiving myelosuppressive chemotherapy with ESAs have shown that ESAs increased the risks of death and serious cardiovascular reactions, and decreased progression-free survival and/or overall survival. See the prescribing information for ESAs for more information.
Advanced Renal Cell Carcinoma (RCC)
In LITESPARK-005, decreased hemoglobin occurred in 88% of patients and 29% had Grade 3 events [see Adverse Reactions (6.1)]. Median time to onset of anemia was 29 days (range: 1 day to 16.6 months). Of the patients with anemia, 22% received transfusions only, 20% of patients received ESAs only and 12% received both transfusion and ESAs.
5.2 Hypoxia
WELIREG can cause severe hypoxia that may require discontinuation, supplemental oxygen, or hospitalization [see Dosage and Administration (2.2)].
Monitor oxygen saturation before initiation of, and periodically throughout, treatment with WELIREG. For decreased oxygen saturation with exercise (e.g., pulse oximeter <88% or PaO2 ≤55 mm Hg), consider withholding WELIREG until pulse oximetry with exercise is greater than 88%, then resume at the same dose or at a reduced dose. For decreased oxygen saturation at rest (e.g., pulse oximeter <88% or PaO2 ≤55 mm Hg) or urgent intervention indicated, withhold WELIREG until resolved and resume at a reduced dose or discontinue. For life-threatening hypoxia or for recurrent symptomatic hypoxia, permanently discontinue WELIREG [see Dosage and Administration (2.2)].
Advise patients to report signs and symptoms of hypoxia immediately to a healthcare provider.
von Hippel-Lindau (VHL) disease
In LITESPARK- 004, hypoxia occurred in 1.6% of patients [see Adverse Reactions (6.1)].
Advanced Renal Cell Carcinoma (RCC)
In LITESPARK- 005, hypoxia occurred in 15% of patients and 10% had Grade 3 events [see Adverse Reactions (6.1)]. Of the patients with hypoxia, 69% were treated with oxygen therapy. Median time to onset of hypoxia was 30.5 days (range: 1 day to 21.1 months).
5.3 Embryo-Fetal Toxicity
Based on findings in animals, WELIREG can cause fetal harm when administered to a pregnant woman. In an animal reproduction study, oral administration of belzutifan to pregnant rats during the period of organogenesis caused embryo-fetal lethality, reduced fetal body weight, and fetal skeletal malformations at maternal exposures ≥0.2 times the human exposures (AUC) at the recommended dose of 120 mg daily.
Advise pregnant women and females of reproductive potential of the potential risk to the fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with WELIREG and for 1 week after the last dose, since WELIREG can render some hormonal contraceptives ineffective [see Drug Interactions (7.1)]. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with WELIREG and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Anemia [see Warnings and Precautions (5.1)]
- Hypoxia [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
von Hippel-Lindau (VHL) disease
LITESPARK-004
The safety of WELIREG was evaluated in an open-label clinical trial (LITESPARK-004) in 61 patients with VHL disease who had at least one measurable solid tumor localized to the kidney [see Clinical Studies (14.1)]. Patients received WELIREG 120 mg orally once daily. The median duration of exposure to WELIREG was 68 weeks (range: 8.4 to 104.7 weeks).
Serious adverse reactions occurred in 15% of patients who received WELIREG, including anemia, hypoxia, anaphylaxis reaction, retinal detachment, and central retinal vein occlusion (1 patient each).
Permanent discontinuation of WELIREG due to adverse reactions occurred in 3.3% of patients. Adverse reactions which resulted in permanent discontinuation of WELIREG were dizziness and opioid overdose (1.6% each).
Dosage interruptions of WELIREG due to an adverse reaction occurred in 39% of patients. Adverse reactions which required dosage interruption in >2% of patients were fatigue, decreased hemoglobin, anemia, nausea, abdominal pain, headache, and influenza-like illness.
Dose reductions of WELIREG due to an adverse reaction occurred in 13% of patients. The most frequently reported adverse reaction which required dose reduction was fatigue (7%).
The most common (≥25%) adverse reactions, including laboratory abnormalities, that occurred in patients who received WELIREG were decreased hemoglobin, fatigue, increased creatinine, headache, dizziness, increased glucose, and nausea.
Table 2 summarizes the adverse reactions reported in patients treated with WELIREG in LITESPARK-004.
| Adverse Reaction | WELIREG (n=61) |
|
|---|---|---|
| All Grades*
(%) | Grade 3-4 (%) |
|
| General | ||
| Fatigue† | 64 | 5 |
| Nervous system | ||
| Headache† | 39 | 0 |
| Dizziness† | 38 | 0 |
| Gastrointestinal | ||
| Nausea | 31 | 0 |
| Constipation | 13 | 0 |
| Abdominal pain† | 13 | 0 |
| Eye Disorders | ||
| Visual impairment‡ | 21 | 3.3 |
| Infections | ||
| Upper respiratory tract infection † | 21 | 0 |
| Respiratory, Thoracic and Mediastinal | ||
| Dyspnea | 20 | 1.6 |
| Musculoskeletal and Connective Tissue | ||
| Arthralgia | 18 | 0 |
| Myalgia | 16 | 0 |
| Vascular | ||
| Hypertension | 13 | 3.3 |
| Metabolism and Nutrition | ||
| Weight increased | 12 | 1.6 |
Table 3 summarizes the laboratory abnormalities in LITESPARK-004.
| Laboratory Abnormality* | WELIREG (n=61) |
|
|---|---|---|
| Grades 1-4 % | Grades 3-4 % |
|
|
||
| Hematology | ||
| Decreased hemoglobin | 93 | 7 |
| Decreased leukocytes | 11 | 0 |
| Chemistry | ||
| Increased creatinine | 64 | 0 |
| Increased glucose | 34 | 4.9 |
| Increased ALT | 20 | 0 |
| Increased AST | 16 | 0 |
| Decreased calcium (corrected) | 10 | 0 |
| Decreased phosphate | 10 | 1.6 |
Advanced Renal Cell Carcinoma (RCC)
LITESPARK-005
The safety of WELIREG was evaluated in a randomized, active-controlled study (LITESPARK- 005) in 732 patients with advanced RCC that has progressed after prior PD-1 or PD-L1 checkpoint inhibitor and VEGF receptor targeted therapies [see Clinical Studies (14.2)]. Patients received 120 mg WELIREG (n=372) or 10 mg everolimus (n=360) by oral administration once daily. The median duration of exposure to WELIREG was 7.6 months (range 0.1 to 28.5 months).
Serious adverse reactions occurred in 38% of patients who received WELIREG. Serious adverse reactions in ≥2% of patients treated with WELIREG were hypoxia (7%), anemia (5%), pneumonia (3.5%), hemorrhage (3%), and pleural effusion (2.2%). Fatal adverse reactions occurred in 3.2% of patients who received WELIREG, including sepsis (0.5%) and hemorrhage (0.5%).
Permanent discontinuation of WELIREG due to adverse reactions occurred in 6% of patients. Adverse reactions which resulted in permanent discontinuation (≥0.5%) of WELIREG were hypoxia (1.1%), anemia (0.5%), and hemorrhage (0.5%).
Dosage interruptions of WELIREG due to an adverse reaction occurred in 39% of patients. Adverse reactions which required dosage interruption in ≥2% of patients were anemia (8%), hypoxia (5%), COVID-19 (4.3%), fatigue (3.2%), and hemorrhage (2.2%).
Dose reductions of WELIREG due to an adverse reaction occurred in 13% of patients. Adverse reactions which required dose reduction in ≥1% of patients were hypoxia (5%) and anemia (3.2%).
The most common (≥25%) adverse reactions, including laboratory abnormalities, that occurred in patients who received WELIREG were decreased hemoglobin, fatigue, musculoskeletal pain, increased creatinine, decreased lymphocytes, increased alanine aminotransferase, decreased sodium, increased potassium, and increased aspartate aminotransferase.
Table 4 summarizes the adverse reactions in LITESPARK-005.
| Adverse Reaction | WELIREG (n=372) | Everolimus (n=360) |
||
|---|---|---|---|---|
| All Grades*
(%) | Grade 3-4 (%) | All Grades*
(%) | Grade 3-4 (%) |
|
| Fatigue† | 43 | 3.2 | 41 | 6 |
| Edema† | 20 | 0.5 | 23 | 0.6 |
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal Pain† | 34 | 1.1 | 27 | 2.2 |
| Gastrointestinal | ||||
| Nausea | 17 | 0.5 | 11 | 0.3 |
| Constipation | 15 | 0 | 8 | 0 |
| Vomiting | 11 | 0.8 | 8 | 0.8 |
| Diarrhea† | 11 | 1.3 | 19 | 1.4 |
| Abdominal Pain† | 10 | 0.8 | 8 | 0.3 |
| Respiratory, Thoracic, and Mediastinal | ||||
| Dyspnea† | 16 | 1.6 | 16 | 2.5 |
| Hypoxia | 15 | 10 | 1.4 | 1.4 |
| Metabolism and Nutrition | ||||
| Decreased Appetite | 13 | 1.1 | 16 | 0 |
| Nervous Systems | ||||
| Headache† | 12 | 0.5 | 8 | 0.3 |
| Dizziness† | 11 | 0 | 1.9 | 0 |
Clinically relevant adverse reactions in <10% of patients who received WELIREG in LITESPARK-005 included hemorrhage (9%) [including intracranial/cerebral hemorrhage (0.8%)], rash (8%), hypertension (6%), visual impairment [including vision blurred (4%), visual acuity decreased (1.1%), visual impairment (0.5%), and retinal detachment (0.3%)] (6%) and increased weight (5%).
Table 5 summarizes the laboratory abnormalities in LITESPARK-005.
| Laboratory Test* | WELIREG | Everolimus | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Hematology | ||||
| Decreased hemoglobin | 88 | 29 | 76 | 17 |
| Decreased lymphocytes | 34 | 8 | 53 | 20 |
| Chemistry | ||||
| Increased creatinine | 34 | 4.7 | 43 | 5.1 |
| Increased alanine aminotransferase | 32 | 2.2 | 40 | 1.1 |
| Decreased sodium | 31 | 1.6 | 36 | 0.8 |
| Increased potassium | 29 | 2.5 | 20 | 2.8 |
| Increased aspartate aminotransferase | 27 | 2.2 | 38 | 2 |
| Decreased glucose | 22 | 1.1 | 19 | 1.1 |
| Decreased calcium | 21 | 1.1 | 45 | 3.1 |
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on WELIREG
UGT2B17 or CYP2C19 Inhibitors
Coadministration of WELIREG with inhibitors of UGT2B17 or CYP2C19 increases plasma exposure of belzutifan [see Clinical Pharmacology (12.3, 12.5)], which may increase the incidence and severity of adverse reactions of WELIREG. Monitor for anemia and hypoxia and reduce the dosage of WELIREG as recommended [see Dosage and Administration (2.2), Warnings and Precautions (5.1, 5.2), Adverse Reactions (6)].
7.2 Effect of WELIREG on Other Drugs
Sensitive CYP3A4 Substrates
Coadministration of WELIREG with CYP3A4 substrates decreases concentrations of CYP3A substrates [see Clinical Pharmacology (12.3)], which may reduce the efficacy of these substrates. The magnitude of this decrease may be more pronounced in patients who are dual UGT2B17 and CYP2C19 poor metabolizers [see Clinical Pharmacology (12.3)]. Avoid coadministration of WELIREG with sensitive CYP3A4 substrates, for which minimal decrease in concentration may lead to therapeutic failures of the substrate. If coadministration cannot be avoided, increase the sensitive CYP3A4 substrate dosage in accordance with its Prescribing Information.
Hormonal Contraceptives
Coadministration of WELIREG with hormonal contraceptives may lead to contraceptive failure or an increase in breakthrough bleeding [see Clinical Pharmacology (12.3), Use in Specific Populations (8.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies, WELIREG can cause fetal harm when administered to a pregnant woman. There are no available data on the use of WELIREG in pregnant women to inform the drug-associated risk. In an animal reproduction study, oral administration of belzutifan to pregnant rats during the period of organogenesis caused embryo-fetal lethality, reduced fetal body weight, and fetal skeletal malformations at maternal exposures ≥0.2 times the human exposure (AUC) at the recommended dose of 120 mg daily (see Data). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Animal Data
In a pilot embryo-fetal development study, pregnant rats received oral doses of 6, 60, or 200 mg/kg/day of belzutifan during the period of organogenesis. Belzutifan caused embryo-fetal lethality at doses ≥60 mg/kg/day (approximately 1 time the human exposure at the recommended dose based on AUC). Reduced fetal body weights, fetal rib malformations, and reduced skeletal ossification occurred at doses of 6 and 60 mg/kg/day (approximately ≥0.2 times the human exposure at the recommended dose based on AUC).
8.2 Lactation
Risk Summary
There are no data on the presence of belzutifan or its metabolites in human milk or their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with WELIREG and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
WELIREG can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with WELIREG.
Contraception
Females
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with WELIREG and for 1 week after the last dose. WELIREG can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with WELIREG and for 1 week after the last dose.
Infertility
Based on findings in animals, WELIREG may impair fertility in males and females of reproductive potential [see Nonclinical Toxicology (13.1)]. The reversibility of the effect on fertility is unknown.
8.4 Pediatric Use
Safety and effectiveness of WELIREG have not been established in pediatric patients.
8.5 Geriatric Use
Of the patients who received WELIREG in LITESPARK-004, 3.3% were ≥65 years old [see Clinical Studies (14.1)] Clinical trials of WELIREG in patients with VHL did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently from younger patients.
Of the 372 patients who received WELIREG for advanced RCC in LITESPARK-005, 62% of patients younger than 65 years, 28% of patients were 65 to 74 years, and 10% were 75 years and over. No overall difference in efficacy was reported between patients who were ≥65 years of age and younger patients. Dose interruptions occurred in 48% of patients ≥65 years of age and in 34% of younger patients. Dose reductions occurred in 18% of patients ≥65 years of age and in 10% of younger patients.
8.6 Renal Impairment
No dosage modification of WELIREG is recommended in patients with mild (eGFR 60-89 mL/min/1.73 m2 estimated by MDRD) and moderate (eGFR 30-59 mL/min/1.73 m2) renal impairment [see Clinical Pharmacology (12.3)]. WELIREG has not been studied in patients with severe (eGFR 15-29 mL/min/1.73 m2) renal impairment.
8.7 Hepatic Impairment
No dosage modification of WELIREG is recommended in patients with mild [total bilirubin ≤ upper limit of normal (ULN) and aspartate aminotransferase (AST) > ULN or total bilirubin >1 to 1.5 x ULN and any AST] hepatic impairment. WELIREG has not been studied in patients with moderate or severe hepatic impairment (total bilirubin >1.5 x ULN and any AST) [see Clinical Pharmacology (12.3)].
8.8 Dual UGT2B17 and CYP2C19 Poor Metabolizers
Patients who are dual UGT2B17 and CYP2C19 poor metabolizers have higher belzutifan exposures, which may increase the incidence and severity of adverse reactions of WELIREG. Closely monitor for adverse reactions in patients who are dual UGT2B17 and CYP2C19 poor metabolizers [see Warnings and Precautions (5), Adverse Reactions (6), Clinical Pharmacology (12.5)].
10 OVERDOSAGE
There is no specific treatment for WELIREG overdose. In cases of suspected overdose, withhold WELIREG and institute supportive care. Grade 3 hypoxia occurred at dosages of 120 mg twice a day and Grade 4 thrombocytopenia occurred at dosages of 240 mg once daily (approximately 2 times the recommended dosage).
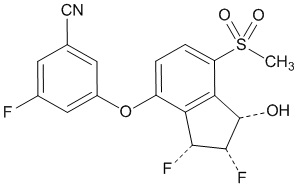
11 DESCRIPTION
Belzutifan is an inhibitor of hypoxia-inducible factor-2α (HIF-2α). The chemical name of belzutifan is 3-[[(1S,2S,3R)-2,3-Difluoro-2,3-dihydro-1-hydroxy-7-(methylsulfonyl)-1H-inden-4-yl]oxy]-5-fluorobenzonitrile. The molecular formula is C17H12F3NO4S and the molecular weight is 383.34 Daltons. The chemical structure is:
 |
Belzutifan is a white to light brown powder that is soluble in acetonitrile, dimethoxyethane, and acetone, sparingly soluble in ethyl acetate, very slightly soluble in isopropanol and toluene, and insoluble in water.
WELIREG is supplied as blue, film-coated tablets for oral use containing 40 mg of belzutifan together with croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, mannitol, microcrystalline cellulose, and silicon dioxide, as inactive ingredients. In addition, the film-coating contains FD&C Blue #2 aluminum lake, polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Belzutifan is an inhibitor of hypoxia-inducible factor 2 alpha (HIF-2α). HIF-2α is a transcription factor that plays a role in oxygen sensing by regulating genes that promote adaptation to hypoxia. Under normal oxygen levels, HIF-2α is targeted for ubiquitin-proteasomal degradation by VHL protein. Lack of functional VHL protein results in stabilization and accumulation of HIF-2α. Upon stabilization, HIF-2α translocates into the nucleus and interacts with hypoxia-inducible factor 1 beta (HIF-1β) to form a transcriptional complex that induces expression of downstream genes, including genes associated with cellular proliferation, angiogenesis, and tumor growth. Belzutifan binds to HIF-2α, and in conditions of hypoxia or impairment of VHL protein function, belzutifan blocks the HIF-2α-HIF-1β interaction, leading to reduced transcription and expression of HIF-2α target genes. In vivo, belzutifan demonstrated anti-tumor activity in mouse xenograft models of renal cell carcinoma.
12.2 Pharmacodynamics
Reductions in plasma levels of erythropoietin (EPO) were observed to be dose- and exposure-dependent at dosages up to 120 mg once daily. The maximum EPO suppression occurred following 2 weeks of consecutive dosing of WELIREG (mean percent decrease from baseline of approximately 60%). Mean EPO levels gradually returned to baseline values after 12 weeks of treatment.
The incidence of Grade 3 anemia increased with higher belzutifan exposure in patients with baseline hemoglobin levels <12 g/dL [see Warnings and Precautions (5.1)].
Cardiac Electrophysiology
At the recommended dosage, WELIREG does not cause large mean increases (i.e., >20 msec) in the QT interval.
12.3 Pharmacokinetics
The Cmax and AUC of belzutifan increase proportionally over a dose range of 20 mg to 120 mg WELIREG (0.17 to 1 times the approved recommended dose). The estimated geometric mean steady-state (CV%) Cmax is 1.5 μg/mL (45%) and AUC0-24h is 20.4 μg•hr/mL (62%) in patients treated with 120 mg WELIREG. Steady state is reached after approximately 3 days.
Absorption
The median Tmax occurs at 1 to 2 hours after WELIREG administration.
Effect of Food
A high-fat, high-calorie meal (total calories approximately 1000 kcal, 56 g fat, 55 g carbohydrate, and 31 g protein) delayed Tmax by approximately 2 hours with no clinically meaningful effect on Cmax, and AUC of belzutifan.
Distribution
The estimated mean (CV%) volume of distribution is 119 L (28%) Plasma protein binding of belzutifan is 45%. The blood-to-plasma concentration ratio of belzutifan is 0.88.
Elimination
The estimated mean (CV%) clearance is 6.0 L/hr (58%) and the mean elimination half-life is 14 hrs.
Metabolism
Belzutifan is primarily metabolized by UGT2B17 and CYP2C19 and to a lesser extent by CYP3A4 [see Clinical Pharmacology (12.5)].
Excretion
Following oral administration of radiolabeled belzutifan to healthy subjects, approximately 49.6% of the dose was excreted in urine and 51.7% in feces (primarily as inactive metabolites).
Specific Populations
Patients who are poor metabolizers of UGT2B17 and CYP2C19 had higher belzutifan AUC [see Clinical Pharmacology (12.5)].
There were no clinically significant differences in the pharmacokinetics of belzutifan based on age (19 to 90 years), sex, ethnicity (non-Hispanic, Hispanic), race (White, Black, Asian, Native American, Pacific Islander), body weight (42 to 166 kg), mild to moderate renal impairment (eGFR 30-89 mL/min/1.73 m2 estimated by MDRD), or mild hepatic impairment (total bilirubin ≤ ULN with AST > ULN or total bilirubin > ULN to 1.5 x ULN with any AST). The effect of severe renal impairment (eGFR 15-29 mL/min/1.73 m2) and moderate to severe hepatic impairment (total bilirubin > 1.5 x ULN and any AST) have not been studied.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Belzutifan on CYP3A Substrates: Coadministration of WELIREG 120 mg once daily with midazolam (a sensitive CYP3A4 substrate) decreased the midazolam AUC by 40% and the Cmax by 34%. Midazolam AUC is predicted to decrease up to 70% in patients with higher belzutifan concentrations (e.g., dual poor metabolizers) [see Clinical Pharmacology (12.5)].
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Belzutifan does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4.
Belzutifan does not induce CYP1A2 or CYP2B6.
Transporter Systems: Belzutifan is a substrate of P-gp, OATP1B1, and OATP1B3, but is not a substrate of BCRP.
Belzutifan inhibits MATE2K. Belzutifan does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, or MATE1.
12.5 Pharmacogenomics
Patients who are UGT2B17, CYP2C19, or dual UGT2B17 and CYP2C19 poor metabolizers are estimated to have 2.5-, 1.3-, or 3.2-fold higher belzutifan steady state AUC0-24h, respectively compared to patients who are UGT2B17 normal (extensive) metabolizers and CYP2C19 non-poor (ultrarapid, rapid, normal, and intermediate) metabolizers [see Use in Specific Populations (8.7)].
UGT2B17 poor metabolizers who are homozygous for the UGT2B17*2 allele have no UGT2B17 enzyme activity. CYP2C19 poor metabolizers (such as *2/*2, *3/*3, *2/*3) have significantly reduced or absent CYP2C19 enzyme activity. Approximately 15% of White, 6% of Black or African American, and up to 77% of certain Asian populations are UGT2B17 poor metabolizers. Approximately 2% of White, 5% of Black or African American, and up to 19% of certain Asian populations are CYP2C19 poor metabolizers. Approximately 0.4% of White, 0.3% of Black or African American, and up to 15% of certain Asian populations are dual UGT2B17 and CYP2C19 poor metabolizers.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with belzutifan.
Belzutifan was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. Belzutifan was not clastogenic in either an in vitro micronucleus assay or an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with belzutifan. In repeat-dose toxicity studies up to 3-month duration, belzutifan-related findings included degeneration/atrophy of testes and hypospermia and cellular debris of the epididymis in rats administered ≥2 mg/kg/day (approximately 0.1 times the human exposure at the recommended dose of 120 mg daily). Findings in testes and epididymis were associated with decreased sperm count and motility and abnormal sperm morphology at ≥6 mg/kg/day (approximately 0.2 times the human exposure at the recommended dose of 120 mg daily) and did not reverse by the end of the recovery period. Belzutifan had no adverse effects on female reproductive organs in repeat-dose toxicity studies up to 3-month duration; however, belzutifan caused embryo-fetal lethality (post-implantation loss) in pregnant rats given oral doses ≥60 mg/kg/day (approximately 1 time the human exposure at the recommended dose based on AUC) during the period of organogenesis [see Use in Specific Population (8.1)].
14 CLINICAL STUDIES
14.1 von Hippel-Lindau (VHL) disease
The efficacy of WELIREG was evaluated in LITESPARK-004 (NCT03401788), an open-label clinical trial in 61 patients with VHL-associated RCC diagnosed based on a VHL germline alteration and with at least one measurable solid tumor localized to the kidney as defined by response evaluation criteria in solid tumors (RECIST) v1.1. Enrolled patients had other VHL-associated tumors including CNS hemangioblastomas and pNET. CNS hemangioblastomas and pNET in these patients were diagnosed based on the presence of at least one measurable solid tumor in brain/spine or pancreas, respectively, as defined by RECIST v1.1 and identified by IRC. The study excluded patients with metastatic disease. Patients received WELIREG 120 mg once daily until progression of disease or unacceptable toxicity.
The study population characteristics were: median age 41 years [range 19-66 years], 3.3% age 65 or older; 53% male; 90% were White, 3.3% were Black or African-American, 1.6% were Asian, and 1.6% were Native Hawaiian or other Pacific Islander; 82% had an ECOG PS of 0, 16% had an ECOG PS of 1, and 1.6% had an ECOG PS of 2; and 84% had VHL Type I Disease. The median diameter of RCC target lesions per central independent review committee (IRC) was 2.2 cm (range 1-6.1). Median time from initial radiographic diagnosis of VHL-associated RCC tumors that led to enrollment on LITESPARK-004 to the time of treatment with WELIREG was 17.9 months (range 2.8-96.7). Seventy-seven percent of patients had prior surgical procedures for RCC.
The major efficacy endpoint for the treatment of VHL-associated RCC was objective response rate (ORR) measured by radiology assessment using Response Evaluation Criteria In Solid Tumors (RECIST) v1.1 as assessed by IRC. Additional efficacy endpoints included duration of response (DoR), and time to response (TTR).
Table 6 summarizes the efficacy results for VHL-associated RCC in LITESPARK-004.
| Efficacy Outcome Measure | WELIREG n=61 |
|---|---|
| + Denotes ongoing response. | |
|
|
| Objective Response Rate, % (n)
(95% CI) | 49% (30)*
(36, 62) |
| Complete response | 0% |
| Partial response | 49% |
| Duration of Response | |
| Median in months (range) | Not reached (2.8+, 22+) |
| % (n) with DoR ≥ 12 months | 56% (17/30) |
For VHL-associated RCC, median TTR was 8 months (range 2.7, 19).
Table 7 summarizes the efficacy results for VHL-associated pNET or CNS hemangioblastomas in LITESPARK-004.
| Endpoint | Patients with CNS Hemangioblastomas n=24* | Patients with pNET n=12* |
|---|---|---|
| + Denotes ongoing response. | ||
|
||
| Objective Response Rate, % (n)
(95% CI) | 63%, (15) (41, 81) | 83% (10) (52, 98) |
| Complete response | 4% (1) | 17% (2) |
| Partial response | 58% (14) | 67% (8) |
| Duration of Response | ||
| Median in months (range) | Not reached (3.7+, 22+) | Not reached (11+, 19+) |
| % (n) with DoR ≥12 months | 73% (11/15) | 50% (5/10) |
For VHL-associated CNS hemangioblastomas, TTR was 3.1 months (range 2.5, 11). For VHL-associated pNET, median TTR was 8.1 months (range 2.7, 11).
Decreases in size of CNS hemangioblastoma-associated peri-tumoral cysts and syringes were observed.
14.2 Advanced Renal Cell Carcinoma (RCC)
The efficacy of WELIREG was evaluated in LITESPARK-005 (NCT04195750), an open-label, randomized, active-controlled clinical trial in 746 patients with unresectable, locally advanced or metastatic clear cell RCC that progressed following PD-1 or PD-L1 checkpoint inhibitor and VEGF receptor targeted therapies either in sequence or in combination.
Patients could have received up to 3 prior treatment regimens and were required to have measurable disease per RECIST v1.1. Patients were randomized in a 1:1 ratio to receive 120 mg WELIREG or 10 mg everolimus by oral administration once daily. Randomization was stratified by IMDC risk categories (favorable versus intermediate versus poor) and number of prior VEGF receptor targeted therapies (1 versus 2-3). Patients were evaluated radiologically at Week 9 from the date of randomization, then every 8 weeks through Week 49, and every 12 weeks thereafter.
The study population characteristics were: median age 63 years [range 22 to 90 years], 42% age 65 or older; 78% male; 79% White; 12% Asian; 1% Black or African American; 11% Hispanic or Latino; 44% ECOG performance status 0 and 55% ECOG performance status 1. Prior therapies: 13% patients had 1 prior line of therapy, 43% had 2 prior lines of therapy and 43% had 3 prior lines of therapy; 49% received 2 to 3 prior VEGF receptor targeted therapies. Patient distribution by IMDC risk categories was 22% favorable, 66% intermediate, and 12% poor. Common sites of metastasis in patients were 65% lung, 59% lymph nodes, and 49% bone.
The major efficacy endpoints were Progression Free Survival (PFS) measured by BICR using RECIST v1.1 and Overall Survival (OS). Additional efficacy endpoint included objective response rate (ORR) by BICR using RECIST v1.1.
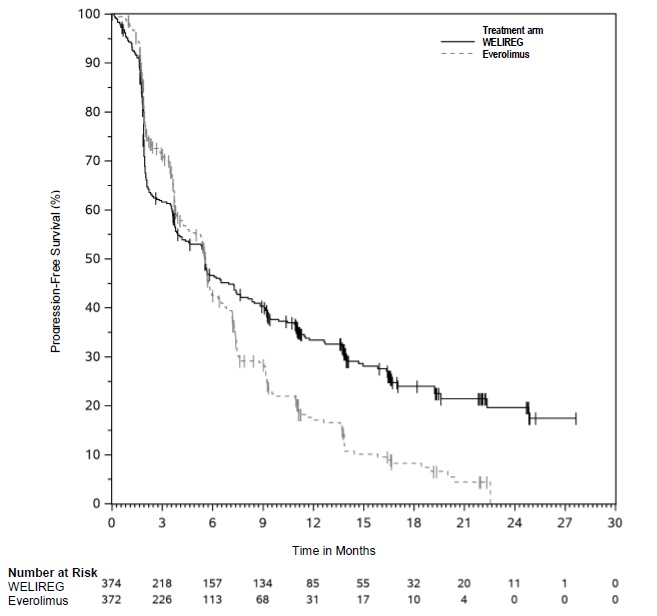
The trial demonstrated a statistically significant improvement in PFS for patients randomized to WELIREG compared with everolimus. Table 8 and Figure 1 summarize the efficacy results for LITESPARK-005.
| Efficacy Outcome Measure | WELIREG n=374 | Everolimus n=372 |
|---|---|---|
| Progression-Free Survival (PFS) | ||
| Number of events, n (%) | 257 (69%) | 262 (70%) |
| Progressive disease | 234 (63%) | 222 (60%) |
| Death | 23 (6%) | 40 (11%) |
| Median in months (95% CI) * | 5.6 (3.9, 7.0) | 5.6 (4.8, 5.8) |
| Hazard ratio † (95% CI) | 0.75 (0.63, 0.90) | |
| p-Value ‡ | 0.0008 | |
| Confirmed Objective Response Rate | ||
| Number of patients with measurable disease at baseline | 373 | 364 |
| ORR % (n) (95% CI) | 22% (82) (18, 27) | 4% (13) (2, 6) |
| Complete response | 3% (10) | 0% (0) |
| Partial response | 19% (72) | 4% (13) |
| p-Value§ | <0.0001 | |
Among the 82 patients treated with WELIREG who achieved a confirmed response based on BICR per RECIST 1.1, 25 (30%) patients had a duration of response ≥12 months. OS results were immature. At the time of the subsequent pre-specified analysis, 59% of the patients had died in the randomized population.
 |
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
WELIREG tablets are supplied as 40 mg blue, oval shaped, film-coated, debossed with “177” on one side and plain on the other side, available in:
- bottles of 90 tablets with child-resistant closure: NDC 0006-5331-01.
The bottle also contains two desiccant canisters. Do not eat.
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Anemia
Inform patients that WELIREG can cause severe anemia that may require blood transfusions and that red blood cell levels will be monitored routinely during treatment. Advise patients to contact their healthcare provider if the patient experiences any symptoms suggestive of anemia [see Warnings and Precautions (5.1)].
Hypoxia
Inform patients that WELIREG can cause severe hypoxia that may require discontinuation, supplemental oxygen, or hospitalization; and that oxygen levels will be monitored routinely during treatment. Advise patients to contact their healthcare provider if the patient experiences any symptoms suggestive of hypoxia [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3) and Use in Specific Population (8.1)].
- Advise females of reproductive potential to use effective non-hormonal contraception during treatment with WELIREG and for 1 week after the last dose [see Use in Specific Populations (8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with WELIREG and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with WELIREG and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise male and female patients that WELIREG may impair fertility [see Use in Specific Populations (8.3)].
Dosage and Administration
Instruct patients to take their dose of WELIREG at the same time each day (once daily). Advise patients WELIREG can be taken with or without food. Each tablet should be swallowed whole [see Dosage and Administration (2.1)].
Manufactured for: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
For patent information: www.msd.com/research/patent
Copyright © 2021-2023 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates.
All rights reserved.
uspi-mk6482-t-2312r002
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 01/2024 |
|
MEDICATION GUIDE |
|
| What is the most important information I should know about WELIREG?
WELIREG may cause serious side effects, including:
See “What are the possible side effects of WELIREG?” for more information about side effects. |
|
|
What is WELIREG? WELIREG is a prescription medicine used to treat adults with:
It is not known if WELIREG is safe and effective in children. |
|
|
Before taking WELIREG, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. WELIREG and certain other medicines can affect each other and cause serious side effects. Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine. |
|
|
How should I take WELIREG?
|
|
|
What are the possible side effects of WELIREG? WELIREG may cause serious side effects, including:
The most common side effects of WELIREG in adults with VHL disease include: |
|
|
|
|
The most common side effects of WELIREG in adults with advanced RCC include: |
|
|
|
|
WELIREG may cause fertility problems in males and females, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of WELIREG. |
|
|
How should I store WELIREG?
Keep WELIREG and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of WELIREG. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use WELIREG for a condition for which it was not prescribed. Do not give WELIREG to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about WELIREG that is written for health professionals. |
|
|
What are the ingredients in WELIREG? Active ingredient: belzutifan Inactive ingredients: croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, mannitol, microcrystalline cellulose, and silicon dioxide. The film-coating contains FD&C Blue #2 aluminum lake, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. |
|
|
Manufactured for: Merck Sharp & Dohme LLC For patent information: www.msd.com/research/patent Copyright © 2021-2024 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. usmg-mk6482-t-2401r003 |
|