FULL PRESCRIBING INFORMATION
WARNING: NEPHROTOXICITY, OTOTOXICITY, NEUROMUSCULAR BLOCKADE and FETAL HARM
- Nephrotoxicity has been reported with ZEMDRI. The risk of nephrotoxicity is greater in patients with impaired renal function, the elderly, and in those receiving concomitant nephrotoxic medications. Assess creatinine clearance in all patients prior to initiating therapy and daily during therapy [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)]. Therapeutic Drug Monitoring (TDM) is recommended for complicated urinary tract infection (cUTI) patients with CLcr less than 90 mL/min to avoid potentially toxic levels [see Dosage and Administration (2.3, 2.4)].
- Ototoxicity, manifested as hearing loss, tinnitus, and/or vertigo, has been reported with ZEMDRI. Symptoms of aminoglycoside-associated ototoxicity may be irreversible and may not become evident until after completion of therapy. Aminoglycoside-associated ototoxicity has been observed primarily in patients with a family history of hearing loss, patients with renal impairment, and in patients receiving higher doses and/or longer durations of therapy than recommended [see Warnings and Precautions (5.2)].
- Aminoglycosides have been associated with neuromuscular blockade. During therapy with ZEMDRI, monitor for adverse reactions associated with neuromuscular blockade, particularly in high-risk patients, such as patients with underlying neuromuscular disorders (including myasthenia gravis) or in patients concomitantly receiving neuromuscular blocking agents [see Warning and Precautions (5.3)].
- Aminoglycosides, including ZEMDRI, can cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
1. INDICATIONS AND USAGE
1.1 Complicated Urinary Tract Infections (cUTI), including Pyelonephritis
ZEMDRI is indicated in patients 18 years of age or older for the treatment of complicated urinary tract infections (cUTI), including pyelonephritis caused by the following susceptible microorganism(s): Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, and Enterobacter cloacae.
As only limited clinical safety and efficacy data for ZEMDRI are currently available, reserve ZEMDRI for use in cUTI patients who have limited or no alternative treatment options [see Clinical Studies (14.1)].
1.2 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZEMDRI and other antibacterial drugs, ZEMDRI should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage regimen of ZEMDRI is 15 mg/kg administered every 24 hours by intravenous (IV) infusion over 30 minutes in patients 18 years of age or older and with creatinine clearance (CLcr) greater than or equal to 90 mL/min (Table 1). The duration of therapy should be guided by the severity of infection and the patient's clinical status for up to 7 days. During treatment, dosage adjustments may be required based on change in renal function [see Dosage and Administration (2.3, 2.4)].
| cUTI Infection | Dosage Regimen † | Duration of Treatment |
|---|---|---|
|
||
| Complicated Urinary Tract Infections, including Pyelonephritis | 15 mg/kg every 24 hours | 4 to 7 days ‡ |
2.2 Monitoring of Renal Function
Assess creatinine clearance in all patients prior to initiating therapy and daily during therapy with ZEMDRI [see Dosage and Administration (2.3),Warnings and Precautions (5.1) and Use in Specific Populations (8.6)].
2.3 Dosage in Adult Patients With Renal Impairment
The recommended initial dosage regimen of ZEMDRI in adult patients with CLcr greater than or equal to 15 and less than 90 mL/min, estimated by the Cockcroft-Gault formula, is described in Table 2.
Patients with CLcr greater than or equal to 15 and less than 90 mL/min receiving ZEMDRI may require subsequent dosage adjustments based on change in renal function and/or Therapeutic Drug Monitoring (TDM) as appropriate [see Dosage and Administration (2.4)].
| Estimated CLcr * (mL/min) | Dosage † | Dosing Interval |
|---|---|---|
|
||
| Greater than or equal to 60 to less than 90 | 15 mg/kg | Every 24 hours |
| Greater than or equal to 30 to less than 60 | 10 mg/kg | Every 24 hours |
| Greater than or equal to 15 to less than 30 | 10 mg/kg | Every 48 hours |
There is insufficient information to recommend a dosage regimen in patients with CLcr less than 15 mL/min or on renal replacement therapy, including hemodialysis or continuous renal replacement therapy.
2.4 TDM in cUTI Patients With Renal Impairment
For cUTI patients with CLcr greater than or equal to 15 mL/min and less than 90 mL/min, TDM is recommended to maintain plasma trough concentrations below 3 mcg/mL. Measure plazomicin plasma trough concentration within approximately 30 minutes before administration of the second dose of ZEMDRI. Adjustment of the ZEMDRI dosage regimen based on TDM involves extending ZEMDRI dosing interval by 1.5 fold (i.e., from every 24 hours to every 36 hours or from every 48 hours to every 72 hours) for patients with plasma trough concentrations greater than or equal to 3 mcg/mL [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
2.5 Preparation of Diluted Solutions of ZEMDRI
ZEMDRI is supplied as a single-dose fliptop 10-mL vial that contains plazomicin sulfate equivalent to 500 mg plazomicin freebase in 10 mL Water for Injection (concentration of 50 mg/mL). The appropriate volume of ZEMDRI solution (50 mg/mL) for the required dose should be diluted in 0.9% Sodium Chloride Injection, USP or Lactated Ringer's Injection, USP to achieve a final volume of 50 mL for intravenous infusion. The stability of ZEMDRI solution in the compatible diluents is described below [see Dosage and Administration (2.7)].
ZEMDRI does not contain preservatives. Aseptic technique must be followed in preparing the infusion solution. Discard unused portion of the ZEMDRI vial.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
2.6 Stability of ZEMDRI Solution in Intravenous Fluids
After dilution, ZEMDRI solution for administration is stable for 24 hours at room temperature at concentrations of 2.5 mg/mL to 45 mg/mL in the following solutions:
- 0.9% Sodium Chloride Injection, USP
- Lactated Ringer's Injection, USP
2.7 Drug Compatibility
Compatibility of ZEMDRI for administration with other drugs has not been established. ZEMDRI should not be mixed with other drugs or physically added to solutions containing other drugs. Other medications should not be infused simultaneously with ZEMDRI through the same IV line.
3. DOSAGE FORMS AND STRENGTHS
ZEMDRI injection 500 mg/10 mL (50 mg/mL) is a sterile, clear, colorless to yellow solution supplied in a single-dose vial. Each single-dose vial contains plazomicin sulfate equivalent to 500 mg plazomicin freebase.
4. CONTRAINDICATIONS
ZEMDRI is contraindicated in patients with known hypersensitivity to any aminoglycoside [see Warnings and Precautions (5.5)].
5. WARNINGS AND PRECAUTIONS
5.1 Nephrotoxicity
Nephrotoxicity has been reported with the use of ZEMDRI [see Adverse Reactions (6.1)]. Most serum creatinine increases were ≤ 1 mg/dL above baseline and reversible.
In Trial 1, the incidence of adverse reactions associated with renal function (acute kidney injury, serum creatinine increased, chronic kidney disease, creatinine clearance decreased, renal failure, renal impairment) was 3.6% (11/303) in ZEMDRI-treated patients compared with 1.3% (4/301) in meropenem-treated patients [see Adverse Reactions (6.1)].
Serum creatinine increases of 0.5 mg/dL or greater above baseline occurred in 7% (21/300) of ZEMDRI-treated patients compared with 4% (12/297) of meropenem-treated patients. These increases mainly occurred in patients with CLcr ≤ 90 mL/min and were associated with a plazomicin trough level (Cmin) greater than or equal to 3 mcg/mL [see Adverse Reactions (6.1) and Clinical Pharmacology (12.2)].
Assess CLcr in all patients prior to initiating therapy and daily during therapy with ZEMDRI, particularly in those at increased risk of nephrotoxicity, such as those with renal impairment, the elderly, and those receiving concomitant potentially nephrotoxic medications. In the setting of worsening renal function, the benefit of continuing ZEMDRI should be assessed [see Dosage and Administration (2.2, 2.4), Adverse Reactions (6.1) and Use in Specific Populations (8.5, 8.6)].
Adjust the initial dosage regimen in cUTI patients with CLcr ≥ 15 mL/min and < 60 mL/min [see Dosage and Administration (2.3)]. For subsequent doses, TDM is recommended for patients with CLcr ≥15 mL/min and < 90 mL/min [see Dosage and Administration (2.4)].
5.2 Ototoxicity
Ototoxicity, manifested as hearing loss, tinnitus, and/or vertigo, has been reported with ZEMDRI. Symptoms of aminoglycoside-associated ototoxicity may be irreversible and may not become evident until after completion of therapy.
Regarding the incidence of adverse reactions associated with cochlear or vestibular function, in Trial 1, there was one case of reversible hypoacusis (1/303;0.3%) in ZEMDRI-treated patients and one case of tinnitus (1/301;0.3%) in meropenem-treated patients [see Adverse Reactions (6.1)]. In Trial 2, one case each of irreversible tinnitus and reversible vertigo was reported in ZEMDRI-treated patients, and one case of an abnormal audiogram occurred in a levofloxacin-treated patient [see Adverse Reactions (6.1)].
Aminoglycoside-associated ototoxicity has been observed primarily in patients with a family history of hearing loss (excluding age-related hearing loss), patients with renal impairment, and in patients receiving higher doses and/or for longer periods than recommended. In Trial 1 and Trial 2, patients with a history of hearing loss, with the exception of age-related hearing loss, were excluded. The benefit-risk of ZEMDRI therapy should be considered in these patients.
5.3 Neuromuscular Blockade
Aminoglycosides have been associated with exacerbation of muscle weakness in patients with underlying neuromuscular disorders, or delay in recovery of neuromuscular function in patients receiving concomitant neuromuscular blocking agents.
During therapy with ZEMDRI, monitor for adverse reactions associated with neuromuscular blockade, particularly in high-risk patients, such as patients with underlying neuromuscular disorders (including myasthenia gravis) or those patients concomitantly receiving neuromuscular blocking agents.
5.4 Fetal Harm
Aminoglycosides, including ZEMDRI, can cause fetal harm when administered to a pregnant woman. Aminoglycosides cross the placenta, and streptomycin has been associated with several reports of total, irreversible, bilateral congenital deafness in pediatric patients exposed in utero. Patients who use ZEMDRI during pregnancy, or become pregnant while taking ZEMDRI should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
5.5 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions have been reported in patients receiving aminoglycoside antibacterial drugs. Before therapy with ZEMDRI is instituted, careful inquiry about previous hypersensitivity reactions to other aminoglycosides should be made. A history of hypersensitivity to other aminoglycosides is a contraindication to the use of ZEMDRI, because cross-sensitivity among aminoglycoside antibacterial drugs has been established. Discontinue ZEMDRI if an allergic reaction occurs.
5.6 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial drugs and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial drugs alters the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B that contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial drugs.
If CDAD is suspected or confirmed, antibacterial drugs not directed against C. difficile may need to be discontinued. Manage fluid and electrolyte levels as appropriate, supplement protein intake, monitor antibacterial treatment of C. difficile, and institute surgical evaluation as clinically indicated.
6. ADVERSE REACTIONS
The following important adverse reactions are discussed in greater detail in the Warnings and Precautions section:
- Nephrotoxicity [see Warnings and Precautions (5.1)]
- Ototoxicity [see Warnings and Precautions (5.2)]
- Neuromuscular Blockade [see Warnings and Precautions (5.3)]
- Fetal Harm [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.5)]
- Clostridium difficile-Associated Diarrhea [see Warnings and Precautions (5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be compared directly to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
ZEMDRI was evaluated in two comparator-controlled clinical trials (Trial 1, NCT02486627 and Trial 2, NCT01096849) in patients with cUTI, including pyelonephritis. In both trials, patients with CLcr greater than 60 mL/min received ZEMDRI 15 mg/kg IV once daily as a 30-minute infusion [see Clinical Studies (14.1)].
Trial 1 included 303 patients treated with ZEMDRI and 301 patients treated with meropenem. Patients were to receive 4 to 7 days of ZEMDRI (mean duration of 5.1 days). In some patients, parenteral therapy was followed by a switch to an oral antibacterial drug.
The median age of patients treated with ZEMDRI in Trial 1 was 62 years (range 18 to 90 years) and 45.2% of patients were 65 years of age or older. Patients treated with ZEMDRI were predominantly female (56.1%) and White (99.3%). A majority of patients (68.0%) had mild or moderate renal impairment (CLcr >30 to 90 mL/min) at baseline. Patients with CLcr of 30 mL/min or less were excluded.
Adverse Reactions Leading to Treatment Discontinuations in Trial 1
In Trial 1, treatment discontinuation from IV study drug due to an adverse reaction occurred in 2.0% of patients receiving ZEMDRI (6/303) and meropenem (6/301), respectively.
Common Adverse Reactions in Trial 1
Table 3 lists adverse reactions occurring in 1% or more of patients receiving ZEMDRI in Trial 1.
| Adverse Reactions | ZEMDRI (N=303) n (%) | Meropenem *
(N=301) n (%) |
|---|---|---|
| Decreased Renal Function † | 11 (3.6) | 4 (1.3) |
| Diarrhea | 7 (2.3) | 5 (1.7) |
| Hypertension | 7 (2.3) | 7 (2.3) |
| Headache | 4 (1.3) | 9 (3.0) |
| Nausea | 4 (1.3) | 4 (1.3) |
| Vomiting | 4 (1.3) | 3 (1.0) |
| Hypotension | 3 (1.0) | 2 (0.7) |
The adverse reactions profile for the cUTI patients in Trial 2 were similar to those observed in Trial 1.
Nephrotoxicity Reported in Trial 1
In Trial 1, serum creatinine increases of 0.5 mg/dL or greater above baseline occurred in 7.0% (21/300) of ZEMDRI-treated patients compared with 4.0% (12/297) of meropenem-treated patients. Of these, the incidence during IV therapy was 3.7% (11/300) vs 3.0% (9/297) in ZEMDRI- and meropenem-treated patients, respectively. By the last follow-up visit (between 8 to 43 days after completion of IV therapy), the majority of ZEMDRI-treated patients (9/11) and all meropenem treated patients (9/9) with serum creatinine increases while on therapy had fully recovered renal function. Serum creatinine increases of 0.5 mg/dL or greater above baseline were observed following completion of IV therapy. These increases were generally ≤ 1.0 mg/dL above baseline and recovered by the next measurement.
In cUTI patients with CLcr of greater than 30 and less than or equal to 90 mL/min, 9.7% (20/207) ZEMDRI-treated and 4.1% (9/217) meropenem-treated patients had serum creatinine increases of 0.5 mg/dL or greater above baseline. In cUTI patients with CLcr greater than 90 mL/min, 1.1% (1/93) ZEMDRI-treated and 3.8% (3/80) of meropenem-treated patients had serum creatinine increases of 0.5 mg/dL or greater above baseline [see Use in Specific Populations (8.6)].
Ototoxicity
Pure tone audiometry was evaluated in Phase 1 trials and in Trial 2. Treatment associated ototoxicity could not be definitively excluded according to the American Speech-Language-Hearing Association criteria1 in 2.2% (4/182) of ZEMDRI-exposed and 2.0% (1/49) of comparator- or placebo-exposed adults.
Other Adverse Reactions Reported with ZEMDRI
The following selected adverse reactions were reported in more than one ZEMDRI-treated patient in Trials 1 and 2 and are not described elsewhere in the labeling:
Gastrointestinal disorders: constipation, gastritis
Laboratory Investigations: alanine aminotransferase increased
Metabolism and nutrition disorders: hypokalemia
Nervous system disorders: dizziness
Renal and urinary disorders: hematuria
Respiratory, thoracic and mediastinal disorders: dyspnea
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Aminoglycosides, including ZEMDRI, can cause fetal harm when administered to a pregnant woman. There are no available data on the use of ZEMDRI in pregnant women to inform a drug associated risk of adverse developmental outcomes. Published literature reports of streptomycin, an aminoglycoside, state that it can cause total, irreversible, bilateral congenital deafness in children whose mothers received streptomycin during pregnancy. No drug-related visceral or skeletal malformations were observed in pregnant rats and rabbits administered subcutaneous plazomicin during organogenesis at maternal exposures approximately 0.8-fold (rats) and 2.5-fold (rabbits) of the human AUC at the clinical dose of 15 mg/kg/day. Auditory function of offspring was not measured in animal studies (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, plazomicin doses of 0, 8, 25, or 50 mg/kg/day administered subcutaneously during organogenesis did not cause drug-related visceral or skeletal malformations, or reduce survival of fetuses. The mid and high doses caused maternal toxicity (reductions in food consumption and body weight gain; increased kidney weight). The high dose resulted in maternal exposure (AUC) approximately 0.8-fold the human AUC at the clinical dose of 15 mg/kg once daily.
In an embryo-fetal development study in rabbits, plazomicin administered subcutaneously at doses of 0, 10, 30, or 50 mg/kg/day did not cause visceral or skeletal malformations or reduced fetal survival. At the high dose, significant maternal toxicity was observed (including renal injury and lethality) and exposure was approximately 2.5-fold the human AUC at the recommended clinical dose.
In a pre- and postnatal development study in rats, maternal animals received subcutaneous plazomicin at 0, 3, 8, or 30 mg/kg/day from the start of organogenesis through lactation. There were no adverse effects on maternal function or pre- and postnatal survival, development, behavior, or reproductive function of the offspring at up to 30 mg/kg/day (0.32-fold human AUC at the clinical daily dose of 15 mg/kg).
8.2 Lactation
Risk Summary
There are no data on the presence of ZEMDRI in human milk, the effects on the breastfed infant, or the effects on milk production. Plazomicin was detected in rat milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZEMDRI and any potential adverse effects on the breastfed infant from ZEMDRI or from the underlying maternal condition.
Data
In a pre- and postnatal development study in rats, low concentrations of plazomicin in maternal milk were detected, with mean concentrations representing 2% to 4% of maternal plasma concentrations. In nursing pups, the systemic exposure (AUC) to plazomicin through lactational exposure was approximately 0.04% of maternal systemic exposure.
8.4. Pediatric Use
The safety and effectiveness of ZEMDRI in patients less than 18 years of age have not been established.
8.5 Geriatric Use
Of the 425 patients treated with ZEMDRI in Trials 1 and 2, 40% (170/425) were 65 years of age and older, including 17.2% (73/425) patients 75 years of age and older. In Trial 1, for ZEMDRI- treated patients ≥ 65 years old, the incidence rate of adverse reactions was 27% (37/137) versus 18.9% (27/143) in the meropenem-treated patients ≥ 65 years old. For ZEMDRI- treated patients < 65 years old, the incidence rate of adverse reactions was 13.3% (22/166) versus 24.1% (38/158) in the meropenem-treated patients < 65 years old.
The rate of adverse reactions associated with renal function for the ZEMDRI-treated patients ≥ 65 years old was 6.6% (9/137) versus 2.8% (4/143) in the meropenem-treated patients. For ZEMDRI- treated patients < 65 years old, the incidence rate of adverse reactions associated with renal function was 1.2% (2/166), versus 0% (0/158) in the meropenem-treated patients [see Clinical Studies (14.1) and Adverse Reactions (6.1)].
ZEMDRI is substantially excreted by the kidneys, and the risk of adverse reactions to ZEMDRI may be greater in patients with renal impairment. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and renal function should be monitored. Dosage adjustment in elderly patients should take into account renal function and plazomicin concentrations as appropriate [see Dosage and Administration (2.2, 2.3, 2.4) and Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Plazomicin total body clearance was significantly decreased in patients with CLcr greater than or equal to 15 to less than 60 mL/min compared to patients with CLcr greater than or equal to 60 mL/min [see Clinical Pharmacology (12.3)]. Monitor CLcr daily and adjust ZEMDRI dosage accordingly [see Dosage and Administration (2.2)]. There is insufficient information to recommend a dosage regimen in patients with CLcr less than 15 mL/min or on renal replacement therapy, including hemodialysis or continuous renal replacement therapy.
For patients with CLcr greater than or equal to 15 mL/min and less than 90 mL/min, TDM is recommended. Monitor plazomicin trough concentrations and adjust ZEMDRI dosage accordingly [see Dosage and Administration (2.3, 2.4)].
10. OVERDOSAGE
In the event of overdosage, ZEMDRI should be discontinued and supportive care is advised. Maintenance of glomerular filtration and careful monitoring of renal function is recommended. Hemodialysis may aid in the removal of ZEMDRI from the blood, especially if renal function is, or becomes, compromised. No clinical information is available on the use of hemodialysis to treat ZEMDRI overdosage.
11. DESCRIPTION
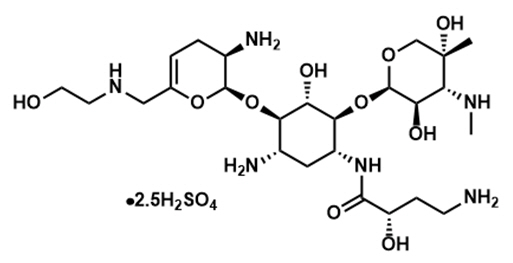
ZEMDRI contains plazomicin sulfate, a semi-synthetic aminoglycoside antibacterial derived from sisomicin. The chemical name of plazomicin sulfate is (2"R,3"R,4"R,5"R)-2"-[(1S,2S,3R,4S,6R)-4-amino-6-[(2'"S)-4'"-amino-2'"-hydroxybutanamido)amino]-3-[(2'S,3'R)-3'-amino-6'-((2-hydroxyethylamino)methyl)-3',4'-dihydro-2H-pyran-2'-yloxy]-2-hydroxycyclohexyloxy]-5''-methyl-4''-(methylamino)tetrahydro-2H-pyran-3'',5''-diol sulfate. Plazomicin sulfate contains a theoretical 2.5 molar equivalents of sulfate relative to the freebase, based on complete protonation. The molecular weight of plazomicin sulfate is calculated based on 1:2.5 stoichiometry. The corresponding empirical formula is C25H48N6O10∙2.5 H2SO4 (plazomicin sulfate) and the molecular weight of the plazomicin sulfate salt is 837.89 g/mol and the molecular weight of the freebase is 592.69 g/mol.
Figure 1: Chemical Structure of Plazomicin Sulfate
ZEMDRI injection 500 mg/10 mL is a sterile, clear, colorless-to-yellow liquid for intravenous administration supplied in 10-mL single-dose Type 1 glass vials. Each vial contains plazomicin sulfate equivalent to 500 mg plazomicin freebase at a concentration of 50 mg/mL adjusted to pH 6.5. Each vial also contains Water for Injection and sodium hydroxide for pH adjustment. This sterile, nonpyrogenic solution is formulated without preservatives.
12. CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
The ratio of area under the plasma concentration-time curve to the minimum inhibitory concentration (AUC:MIC) for plazomicin has been shown to best correlate with efficacy in animal and in vitro models of infection against Enterobacteriaceae.
Exposure- Response Relationship for Nephrotoxicity in cUTI Patients
Based on exposure-response analysis for nephrotoxicity, defined as serum creatinine increases greater than or equal to 0.5 mg/dL from baseline, using the data from two cUTI clinical trials (Trial 1 and Trial 2), development of nephrotoxicity was associated with estimated plazomicin exposure (i.e., the plasma trough concentration [Cmin]) in patients with CLcr greater than 30 mL/min and less than or equal to 90 mL/min (N=243). The incidence of nephrotoxicity was higher in patients with plazomicin Cmin greater than or equal to 3 mcg/mL (36%, 10/28) compared to patients with plazomicin Cmin less than 3 mcg/mL (5%, 11/215).
Cardiac Electrophysiology
The effect of ZEMDRI on the QTc interval was evaluated in a Phase 1 randomized, placebo and positive controlled, double-blind, single-dose, crossover thorough QTc study in 56 healthy adult subjects. At a single dose of 20 mg/kg (1.3 times the maximum recommended dose), ZEMDRI did not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
The pharmacokinetic (PK) parameters of plazomicin are similar for single- and multiple-dose administration of ZEMDRI in healthy subjects. No appreciable accumulation of plazomicin was observed following multiple IV infusions of 15 mg/kg administered every 24 hours in subjects with normal renal function. The AUC, maximum plasma concentration (Cmax), and Cmin increased in proportion to the dose over the dose range of 4 to 15 mg/kg. The plazomicin AUC, Cmax, and Cmin are summarized in Table 4.
| Healthy Subjects *
Geometric mean (±SD) N=54 | cUTI Patients †
Geometric mean (±SD) N=87 |
|
|---|---|---|
| AUC (mcg∙h/mL) | 257 (±67.0) | 226 (±113) |
| Cmax (mcg/mL) | 73.7 (±19.7) | 51.0 (±26.7) |
| Cmin (mcg/mL) | 0.3 (±0.2) | 0.5 (±1.2) |
Distribution
The mean (±SD) volume of distribution of plazomicin in healthy adults and cUTI patients is 17.9 (±4.8) and 30.8 (±12.1) L, respectively. The average binding of plazomicin to human plasma proteins is approximately 20%. The degree of protein binding was concentration-independent across the range tested in vitro (5 to 100 mcg/mL).
Elimination
The mean (±SD) total body clearance of plazomicin in healthy adults and cUTI patients is 4.5 (±0.9) and 5.1 (±2.01) L/h, respectively. The mean (±SD) half-life of plazomicin was 3.5 h (±0.5) in healthy adults with normal renal function (n=54).
Excretion
Plazomicin is primarily excreted by the kidneys. Following a single 15 mg/kg IV dose of radiolabeled plazomicin in healthy subjects, 56% of the total administered radioactivity was recovered in urine within 4 hours, 89.1% was recovered within 168 hours, with less than 0.2% in feces. In total, 97.5% of the dose was recovered in the urine as unchanged plazomicin. The mean renal clearance (±SD) of plazomicin (4.6 [±1.2] L/h) was similar to total body clearance, suggesting that plazomicin is eliminated by the kidneys.
Specific Populations
No clinically significant differences in the pharmacokinetics of plazomicin were observed based on age (18 to 90 years of age), sex, or race/ethnicity. The pharmacokinetics of plazomicin in patients with hepatic impairment is unknown.
Patients with Renal Impairment
Following a single 7.5 mg/kg IV dose (0.5 times the recommended dose) of ZEMDRI as a 30-minute infusion, the geometric mean AUC0-inf of plazomicin in subjects with mild (CLcr 60 to <90 mL/min, n=6), moderate (CLcr 30 to <60 mL/min, n=6), and severe (CLcr 15 to <30 mL/min, n=6) renal impairment was 1.01-fold, 1.98-fold, and 4.42-fold higher, respectively, compared to subjects with normal renal function (CLcr ≥90 mL/min, n=6) [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Based on the population PK model, the recommended dosage of ZEMDRI was associated with a mean (±SD) Cmin of 1.0 (±1.3) and 1.7 (±1.4) mcg/mL in cUTI patients with mild (CLcr 60 to <90 mL/min, n=104) and moderate (CLcr 30 to <60 mL/min, n=89) renal impairment, respectively. The mean (±SD) area under the curve from time zero to 24 hours (AUC0-24h) was 261 (±102) and 224 (±147) mcg∙h/mL in cUTI patients with mild (CLcr 60 to <90 mL/min, n=104) and moderate (CLcr 30 to <60 mL/min, n=89) renal impairment, respectively. There were insufficient data to calculate Cmin and AUC0-24h for patients with severe renal impairment (CLcr 15 to <30 mL/min).
Geriatric Patients
No clinically relevant trend in plazomicin exposure (Cmax and AUC0-24h) was observed with regard to age alone. Higher Cmin in elderly subjects (65 to 90 years of age) as compared to non-elderly adult subjects (18 to 64 years of age) was mainly attributable to age-related changes in renal function [see Dosage and Administration (2.2) and Use in Specific Populations (8.5)].
Drug Interaction Studies
Clinical Studies
Based on the results of a clinical drug-drug interaction (DDI) study that evaluated the effect of a single dose of plazomicin (15 mg/kg) on the single dose plasma PK of metformin, plazomicin did not affect the PK of metformin, which is a substrate of OCT and MATE transporters.
In Vitro Studies
Drug-Metabolizing Enzymes
Plazomicin does not inhibit the following cytochrome P450 isoforms: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5. Plazomicin does not induce CYP1A2, CYP2B6, and CYP3A4.
Membrane Transporters
Plazomicin is not a substrate of P-gp or BCRP transporters. Plazomicin does not inhibit the following hepatic and renal transporters in vitro at clinically relevant concentrations: P-gp, BCRP, BSEP, MRP2, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, and OCT2. Plazomicin selectively inhibited the MATE1 and MATE2-K renal transporter in vitro with an IC50 value of 1300 and 338 mcg/mL, respectively.
12.4 Microbiology
Mechanism of Action
Plazomicin is an aminoglycoside that acts by binding to bacterial 30S ribosomal subunit, thereby inhibiting protein synthesis. Plazomicin has concentration-dependent bactericidal activity as measured by time kill studies. In vitro studies demonstrated a plazomicin post-antibiotic effect ranging from 0.2 to 2.6 hours at 2× MIC against Enterobacteriaceae.
Resistance
Resistance to aminoglycosides includes production of aminoglycoside modifying enzymes (AMEs), alteration of the ribosomal target through production of 16S rRNA methyltransferases, up-regulation of efflux pumps and reduced permeability into bacterial cell due to loss of outer membrane porins.
Plazomicin is not inhibited by most AMEs known to affect gentamicin, amikacin and tobramycin, including acetyltransferases (AACs), phosphotransferases (APHs) and nucleotidyltransferases (ANTs). Plazomicin, like other aminoglycosides, is inactive against bacterial isolates that produce 16S rRNA methyltransferases. Plazomicin may have reduced activity against Enterobacteriaceae that overexpress certain efflux pumps (e.g., acrAB-tolC) or lower expression of porins (e.g., ompF or ompK36).
Plazomicin has no in vitro activity against streptococci (including Streptococcus pneumoniae), enterococci (including Enterococcus faecalis, E. faecium), anaerobes, Stenotrophomonas maltophilia and Acinetobacter spp and variable activity against Pseudomonas aeruginosa.
Activity of plazomicin was demonstrated in vitro against Enterobacteriaceae in the presence of certain beta-lactamases, including extended-spectrum beta-lactamases (TEM, SHV, CTX-M, AmpC), serine carbapenemases (KPC-2, KPC-3), and oxacillinase (OXA-48). Bacteria producing metallo-beta-lactamases often co-express 16S rRNA methyltransferase, conferring resistance to plazomicin.
Interaction With Other Antimicrobials
In vitro studies have demonstrated that against Enterobacteriaceae isolates, no antagonism was observed for plazomicin in combination with clindamycin, colistin, daptomycin, fosfomycin, levofloxacin, linezolid, rifampin, tigecycline and vancomycin; few isolates showed synergy with ceftazidime, meropenem and piperacillin-tazobactam. The clinical significance of these findings is unknown.
Animal Infection Models
Plazomicin demonstrated activity in animal models of infection (e.g., thigh infection, lung infection, and septicemia) caused by either amikacin-non-susceptible, gentamicin-non-susceptible, or beta-lactamase producing Enterobacteriaceae.
Antimicrobial Activity
ZEMDRI has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1)]
Aerobic Bacteria
Gram-negative Bacteria
- Escherichia coli
- Klebsiella pneumoniae
- Proteus mirabilis
- Enterobacter cloacae
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for plazomicin against isolates of similar genus or organism group. However, the efficacy of ZEMDRI in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
Long term carcinogenicity studies in animals have not been conducted with plazomicin.
Mutagenesis
Plazomicin was negative for mutagenicity in an Ames test and did not induce chromosome aberrations in cultured human peripheral blood lymphocytes. In vivo, a mouse bone marrow micronucleus assay showed no evidence of clastogenic potential.
Impairment of Fertility
In a fertility and early embryonic development study, male and female rats received subcutaneous plazomicin at 0, 8, 25, or 50 mg/kg/day from prior to pairing through the mating and postmating period. Parental toxicity (reduced food consumption and body weight gain, and gross kidney changes) was observed at the mid and high doses. Plazomicin had no adverse effects on fertility in male rats at up to 50 mg/kg/day, resulting in an exposure (AUC) approximately 0.8-fold the human AUC at the clinical dose of 15 mg/kg once daily. In female rats, there were no effects on estrous cyclicity or reproductive performance including mating indices, fertility and fecundity indices, and copulatory intervals. At 25 and 50 mg/kg/day, female rats had fewer corpora lutea, leading to fewer uterine implantation sites and viable embryos per dam. The no observed effect level (NOEL) for fertility and reproductive performance in female rats was 8 mg/kg/day (0.1-fold human AUC).
14. CLINICAL STUDIES
14.1 Complicated Urinary Tract Infections, Including Pyelonephritis
A total of 609 adults hospitalized with cUTI (including pyelonephritis) were randomized in a multinational, double-blind, noninferiority trial comparing ZEMDRI (15 mg/kg IV once daily as a 30-minute infusion) to meropenem (1 g intravenously every 8 hours as a 30-minute infusion) (Trial 1, NCT02486627). Switch to an oral antibacterial drug, such as levofloxacin, was allowed after a minimum of 4 and maximum of 7 days of IV therapy for a total of 7 to 10 days of treatment.
Efficacy was assessed in the microbiological modified intent-to-treat (mMITT) population, which included all patients who received study medication and had at least 1 baseline uropathogen. The mMITT population excluded patients with organisms resistant to study drugs. Patient demographic and baseline characteristics were balanced between treatment groups in the mMITT population. The mMITT population consisted of 388 patients with cUTI, including 162 (41.8%) with pyelonephritis. The median age was 64 years, 52.8% were female and 99.5% were White. The majority of the patients (99%) were from Eastern Europe; 3 patients were from the United States. Concomitant bacteremia was identified in 25 (13.1%) and 23 (11.7%) patients at baseline in the ZEMDRI and meropenem groups, respectively. The median treatment duration of IV study drug was 6 days in both groups.
ZEMDRI demonstrated efficacy for composite cure at Day 5 and the Test of Cure (TOC) visit (Table 5). Composite cure at Day 5 was defined as resolution or improvement of clinical cUTI symptoms and a microbiological outcome of eradication (all baseline uropathogens reduced to <104 colony-forming units [CFU]/mL). Composite cure at the TOC visit (Day 17 ± 2 from the first dose of study drug) was defined as resolution of clinical cUTI symptoms and a microbiological outcome of eradication.
| Analysis Visit | ZEMDRI n/N (%) | Meropenem n/N (%) | Treatment Difference *
(95% CI) |
|---|---|---|---|
| Abbreviations: CI=confidence interval; TOC=test-of-cure; CI=95% confidence interval based on Newcombe method with continuity correction. | |||
|
|||
| Day 5 | 168/191 (88.0) | 180/197 (91.4) | -3.4 (-10.0, 3.1) |
| Clinical cure or improvement | 171/191 (89.5) | 182/197 (92.4) | |
| Microbiological eradication | 188/191 (98.4) | 193/197 (98.0) | |
| TOC | 156/191 (81.7) | 138/197 (70.1) | 11.6 (2.7, 20.3) |
| Clinical Cure | 170/191 (89.0) | 178/197 (90.4) | |
| Microbiological eradication | 171/191 (89.5) | 147/197 (74.6) | |
Microbiological eradication rates at the TOC visit by baseline uropathogen in the mMITT population are presented in Table 6. Composite Cure at the TOC visit in individuals with concomitant bacteremia at baseline was achieved in 72.0% (18/25) of patients in the ZEMDRI group and 56.5% (13/23) of patients in the meropenem group.
| Pathogen | ZEMDRI n/N (%) | Meropenem n/N (%) |
|---|---|---|
| All Enterobacteriaceae | 177/198 (89.4) | 157/208 (75.5) |
| Escherichia coli | 120/128 (93.8) | 106/142 (74.6) |
| Klebsiella pneumoniae | 27/33 (81.8) | 32/43 (74.4) |
| Proteus mirabilis | 9/11 (81.8) | 4/7 (57.1) |
| Enterobacter cloacae | 13/16 (81.3) | 3/3 (100.0) |
There were 52 baseline Enterobacteriaceae isolates in 51/189 (27%) patients in the ZEMDRI group that were non-susceptible (defined as intermediate or resistant) to gentamicin, or tobramycin or both. All of these isolates were susceptible to plazomicin and all but one was susceptible to amikacin (one isolate was intermediate to amikacin). The microbiological eradication rate at the TOC visit in this subset was 78.9% (41/52) in the ZEMDRI group. Note that certain resistance mechanisms can confer resistance to all aminoglycosides, including plazomicin [see Microbiology (12.4)].
15. REFERENCES
- American Speech-Language-Hearing Association. (1994). Audiologic management of individuals receiving cochleotoxic drug therapy [Guidelines]. Available from www.asha.org/policy.
16. HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ZEMDRI injection 500 mg/10 mL (50 mg/mL) is supplied in single-dose, 10-mL vials fitted with flip-off seals with royal blue polypropylene buttons as a clear, colorless to yellow, sterile solution. Each vial contains plazomicin sulfate equivalent to 500 mg plazomicin freebase at a concentration of 50 mg/mL plazomicin in Water for Injection. Each vial contains sodium hydroxide for pH adjustment to 6.5. The solution may become yellow in color; this does not indicate a decrease in potency.
| NDC number | Package/Volume | Units per carton | Plazomicin content |
|---|---|---|---|
| 71045-010-02 | Single use, fliptop vial, 10-mL | 10 | 500 mg in 10 mL (50 mg/mL) |
17. PATIENT COUNSELING INFORMATION
Nephrotoxicity
Advise patients, their families, or caregivers that nephrotoxicity has been reported with ZEMDRI therapy. Counsel patients to follow their physician's directions regarding renal function laboratory tests, maintenance of adequate hydration, and avoidance of potentially nephrotoxic agents while receiving ZEMDRI therapy [see Warnings and Precautions (5.1)].
Ototoxicity
Advise patients, their families, or caregivers that hearing loss, vertigo, and tinnitus have been reported with ZEMDRI therapy. Counsel patients to inform their physician if they experience changes in hearing or balance, or if they experience new onset or changes in preexisting buzzing or roaring in their ear(s), even if it occurs after the completion of ZEMDRI therapy [see Warnings and Precautions (5.2)].
Aggravation of Neuromuscular Disorders
Advise patients, their families, or caregivers that aggravation of muscle weakness has been reported for other aminoglycosides, particularly in patients with underlying neuromuscular disease or receiving neuromuscular blocking agents. Counsel patients to inform their physician if they have an underlying neuromuscular disorder such as myasthenia gravis or are receiving neuromuscular blocking agents [see Warnings and Precautions (5.3)].
Fetal Harm
Aminoglycosides, including ZEMDRI, can cause fetal harm when administered to a pregnant woman. Counsel women of childbearing potential about the potential risk of fetal harm if ZEMDRI is used during pregnancy. Advise pregnant women that aminoglycosides can cause irreversible congenital deafness when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Tell women of childbearing potential to notify their prescribing physician/ healthcare provider if they become pregnant during ZEMDRI treatment [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Advise patients, their families, or caregivers that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. Ask them about any previous hypersensitivity reactions to ZEMDRI or other aminoglycosides [see Warnings and Precautions (5.5)].
Potentially Serious Diarrhea
Advise patients, their families, or caregivers that diarrhea is a common problem caused by antibacterial drugs, including ZEMDRI. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, tell patient to contact his or her healthcare provider [see Warnings and Precautions (5.6)].
Antibacterial Resistance
Counsel patients, their families, or caregivers that antibacterial drugs, including ZEMDRI, should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When ZEMDRI is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by ZEMDRI or other antibacterial drugs in the future [see Warnings and Precautions (5.7)].
