FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Maintenance Treatment of BRCA-mutated Recurrent Ovarian Cancer
Rubraca is indicated for the maintenance treatment of adult patients with a deleterious BRCAmutation (germline and/or somatic)- associated recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy.
1.2 BRCA-mutated Metastatic Castration-Resistant Prostate Cancer
Rubraca is indicated for the treatment of adult patients with a deleterious BRCAmutation (germline and/or somatic)-associated metastatic castration-resistant prostate cancer (mCRPC) who have been treated with androgen receptor-directed therapy and a taxane-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Rubraca [see Dosage and Administration ( 2.1)].
This indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies ( 14.2)] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Maintenance Treatment ofBRCA-mutated Recurrent Ovarian Cancer
Select patients for the maintenance treatment of recurrent ovarian cancer with Rubraca based on the presence of a deleterious BRCAmutation (germline and/or somatic) [ see Clinical Studies ( 14.1) ].
An FDA-approved test for the detection of deleterious germline and/or somatic BRCAmutations is not currently available.
Treatment ofBRCA-mutated mCRPC after Androgen Receptor-directed Therapy and Chemotherapy
Select patients for the treatment of mCRPC with Rubraca based on the presence of a deleterious BRCAmutation (germline and/or somatic) in plasma specimens [see Clinical Studies ( 14.2)]. A negative result from a plasma specimen does not mean that the patient's tumor is negative for BRCAmutations. Should the plasma specimen have a negative result, consider performing further genomic testing using tumor specimens as clinically indicated.
Information on the FDA-approved tests for the detection of a BRCAmutation in patients with ovarian cancer or with prostate cancer is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dose
The recommended dose of Rubraca is 600 mg (two 300 mg tablets) taken orally twice daily with or without food, for a total daily dose of 1,200 mg.
Continue treatment until disease progression or unacceptable toxicity.
If a patient misses a dose of Rubraca, instruct the patient to take the next dose at its scheduled time. Vomited doses should not be replaced.
Patients receiving Rubraca for mCRPC should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
2.3 Dose Modifications for Adverse Reactions
To manage adverse reactions, consider interruption of treatment or dose reduction. Recommended Rubraca dose modifications for adverse reactions are indicated in Table 1.
| Dose Reduction | Dose |
| Starting Dose | 600 mg twice daily (two 300 mg tablets) |
| First Dose Reduction | 500 mg twice daily (two 250 mg tablets) |
| Second Dose Reduction | 400 mg twice daily (two 200 mg tablets) |
| Third Dose Reduction | 300 mg twice daily (one 300 mg tablet) |
3 DOSAGE FORMS AND STRENGTHS
- Tablets (200 mg): blue, round, immediate-release, film-coated, debossed with “C2”.
- Tablets (250 mg): white, diamond, immediate-release, film-coated, debossed with “C25”.
- Tablets (300 mg): yellow, oval, immediate-release, film-coated, debossed with “C3”.
5 WARNINGS AND PRECAUTIONS
5.1 Myelodysplastic Syndrome/Acute Myeloid Leukemia
Myelodysplastic Syndrome (MDS)/Acute Myeloid Leukemia (AML) occur in patients treated with Rubraca, and are potentially fatal adverse reactions. In 1594 treated patients with ovarian cancer [see Adverse Reactions ( 6.1)] , MDS/AML occurred in 32 patients (2%), including those in long term follow-up. Of these, 14 occurred during treatment or during the 28-day safety follow-up (0.9%). The duration of Rubraca treatment prior to the diagnosis of MDS/AML ranged from < 2 months to approximately 72 months. The cases were typical of secondary MDS/cancer therapy-related AML; in all cases, patients had received previous platinum-containing chemotherapy regimens and/or other DNA damaging agents.
In ARIEL3, of patients with a germline and/or somatic BRCAmutation treated with Rubraca, MDS/AML occurred in 9 out of 129 (7%) patients treated with Rubraca and 4 out of 66 (6%) patients treated with placebo. The duration of therapy with Rubraca in patients who developed secondary MDS/cancer therapy-related AML varied from 1.2 to 4.7 years.
In TRITON2, MDS/AML was not observed in patients with mCRPC (n=209) regardless of homologous recombination deficiency (HRD) mutation [see Adverse Reactions ( 6.1)].
Do not start Rubraca until patients have recovered from hematological toxicity caused by previous chemotherapy (≤ Grade 1). Monitor complete blood counts for cytopenia at baseline and monthly thereafter for clinically significant changes during treatment. For prolonged hematological toxicities (> 4 weeks), interrupt Rubraca or reduce dose according to Table 1[see Dosage and Administration ( 2.3)] and monitor blood counts weekly until recovery. If the levels have not recovered to Grade 1 or less after 4 weeks or if MDS/AML is suspected, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Rubraca.
5.2 Embryo-Fetal Toxicity
Rubraca can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings from animal studies. In an animal reproduction study, administration of rucaparib to pregnant rats during the period of organogenesis resulted in embryo-fetal death at exposures that were 0.04 times the AUC 0-24hin patients receiving the recommended human dose of 600 mg twice daily. Apprise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of Rubraca [see Use in Specific Populations ( 8.1, 8.3) and Clinical Pharmacology ( 12.1)].
Based on findings from genetic toxicity and animal reproduction studies, advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 3 months following the last dose of Rubraca [see Use in Specific Populations ( 8.1, 8.3)].
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Myelodysplastic Syndrome/Acute Myeloid Leukemia [see Warnings and Precautions ( 5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population for patients with ovarian cancer in the WARNINGS AND PRECAUTIONS section reflect exposure to Rubraca at 600 mg orally twice daily in 1594 patients treated on clinical trials including ARIEL3. In this group, 57% of patients were exposed for 6 months or longer and 33% were exposed for greater than one year.
In the pooled safety population for patients with ovarian cancer, the most common adverse reactions in ≥ 10% of patients were nausea (68%), asthenia/fatigue (65%), anemia/hemoglobin decreased (45%), AST/ALT increased (39%), vomiting (36%), diarrhea (29%), decreased appetite (27%), thrombocytopenia/platelet count decreased (25%), dysgeusia (24%), neutropenia/neutrophil count decreased (21%), blood creatinine increased (17%), dyspnea (16%), dizziness (14%), dyspepsia(11%), photosensitivity reaction (10%), and leukopenia/white blood cell count decreased (10%).
Maintenance Treatment ofBRCA-mutated Recurrent Ovarian Cancer
The safety of Rubraca for the maintenance treatment of patients with BRCA-mutated recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer was investigated in ARIEL3, a randomized (2:1), double-blind, placebo-controlled study in which 195 patients with a deleterious BRCAmutation received either Rubraca 600 mg orally twice daily (n=129) or placebo (n=66) until disease progression or unacceptable toxicity. The median duration of study treatment was 13.6 months (range: < 1 month to 39 months) for patients who received Rubraca and 5.5 months for patients who received placebo.
Dose interruptions due to an adverse reaction of any grade occurred in 67% of patients receiving Rubraca and 14% of those receiving placebo; dose reductions due to an adverse reaction occurred in 57% of Rubraca patients and 6% of placebo patients. The most frequent adverse reactions leading to dose interruption or dose reduction of Rubraca were thrombocytopenia (25%), anemia (19%), AST/ALT increased (16%), fatigue/asthenia (14%), and nausea (10%). Discontinuation due to adverse reactions occurred in 15% of Rubraca patients and 5% of placebo patients. Specific adverse reactions that most frequently led to discontinuation in patients treated with Rubraca were thrombocytopenia (4%), nausea (3%), and anemia (2%). Table 2describes the adverse reactions occurring in ≥20% of patients; while Table 3describes the laboratory abnormalities occurring in ≥25% of patients occurring in ARIEL3.
|
aNational Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE version 4.03) |
||||
|
bConsists of grouped related terms that reflect the medical concept of the adverse reaction |
||||
| Adverse reactions | Rubraca
N=129 | Placebo
N=66 |
||
| Grades 1-4a
% | Grades 3-4
% | Grades 1-4a
% | Grades 3-4
% |
|
| Gastrointestinal Disorders | ||||
| Nausea | 79 | 2 | 29 | 0 |
| Abdominal pain/distention b | 48 | 3 | 49 | 2 |
| Constipation | 39 | 4 | 36 | 2 |
| Vomiting | 37 | 4 | 14 | 0 |
| Diarrhea | 34 | 2 | 18 | 0 |
| Stomatitis b | 28 | 0.8 | 12 | 0 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue/asthenia | 74 | 9 | 52 | 5 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash b | 45 | 0 | 23 | 0 |
| Nervous System Disorders | ||||
| Dysgeusia | 33 | 0 | 6 | 0 |
| Headache | 22 | 0 | 15 | 2 |
| Investigations | ||||
| AST/ALT elevation | 33 | 16 | 3 | 0 |
| Blood and Lymphatic System Disorders | ||||
| Anemia | 41 | 26 | 6 | 0 |
| Thrombocytopenia | 35 | 6 | 3 | 0 |
| Neutropenia | 22 | 8 | 6 | 0 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||
| Nasopharyngitis/Upper respiratory tract infection b | 29 | 0 | 20 | 2 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 23 | 2 | 14 | 0 |
Adverse reactions occurring < 20% of patients treated with Rubraca include insomnia (19%), dyspnea (17%), dizziness (15%), pyrexia (15%), dyspepsia (12%), peripheral edema (12%), and depression (11%).
|
aPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1. |
||||
|
bNCI CTCAE version 4.03. |
||||
| Laboratory Parametera | Rubraca
N=129 | Placebo
N=66 |
||
| Grades 1-4b
% | Grades 3-4
% | Grades 1-4b
% | Grades 3-4
% |
|
| Chemistry | ||||
| Increase in creatinine | 96 | 0 | 89 | 0 |
| Increase in ALT | 67 | 11 | 6 | 0 |
| Increase in AST | 59 | 1 | 6 | 0 |
| Increase in cholesterol | 39 | 3 | 20 | 0 |
| Increase in Alkaline Phosphatase | 39 | 0 | 3 | 0 |
| Hematology | ||||
| Decrease in hemoglobin | 61 | 18 | 14 | 2 |
| Decrease in platelets | 47 | 2 | 8 | 0 |
| Decrease in leukocytes | 39 | 3 | 23 | 0 |
| Decrease in neutrophils | 38 | 6 | 18 | 2 |
| Decrease in lymphocytes | 33 | 7 | 14 | 2 |
Treatment ofBRCA-mutated mCRPC after Androgen Receptor-directed Therapy and Chemotherapy
The safety of Rubraca 600 mg twice daily was evaluated in a single arm trial (TRITON2) [see Clinical Studies ( 14.2)] . TRITON2 enrolled 209 patients with HRD-positive mCRPC, including 115 with BRCA-mutated mCRPC. Among the patients with BRCA-mutated mCRPC, the median duration of Rubraca treatment was 6.5 months (range 0.5 to 26.7).
There were 2 (1.7%) patients with adverse reactions leading to death, one each attributed to acute respiratory distress syndrome and pneumonia.
Dose interruptions due to an adverse reaction occurred in 57% of patients receiving Rubraca. Adverse reactions requiring dose interruption in >3% of patients included anemia, thrombocytopenia, asthenia/fatigue, nausea, vomiting, neutropenia, ALT/AST increased, creatinine increased, decreased appetite, acute kidney injury, and hypophosphatemia.
Dose reductions due to an adverse reaction occurred in 41% of patients receiving Rubraca. Adverse reactions requiring dose reduction in >3% of patients were anemia (14%), asthenia/fatigue (10%), thrombocytopenia (7%), nausea (6%), decreased appetite (4%), and rash (3%).
Discontinuation due to adverse reactions occurred in 8% of patients receiving Rubraca. None of the adverse reactions leading to discontinuation of Rubraca (ECG QT prolonged, acute respiratory distress syndrome, anemia, balance disorder, cardiac failure, decreased appetite/fatigue/weight decreased, leukopenia/neutropenia, ALT/AST increased, and pneumonia) occurred in more than one patient (<1%).
Tables 4and 5summarize the adverse reactions and laboratory abnormalities, respectively, in patients with BRCA--mutated mCRPC in TRITON2.
| Adverse Reaction | Rubraca
N = 115 |
|
|---|---|---|
| Grades 1-4
a
(%) | Grades 3-4
(%) |
|
|
aNCI CTCAE version 4.03. |
||
|
bIncludes platelet count decreased |
||
|
cIncludes blister, blood blister, dermatitis, dermatitis contact, eczema, genital rash, palmar-plantar erythrodysaesthesia syndrome, photosensitivity reaction, psoriasis, rash, rash maculo-papular, rash pruritic, skin exfoliation, skin lesion, urticaria |
||
| General disorders and administration site conditions | ||
| Asthenia/Fatigue | 62 | 9 |
| Gastrointestinal disorders | ||
| Nausea | 52 | 3 |
| Constipation | 27 | 1 |
| Vomiting | 22 | 1 |
| Diarrhea | 20 | 0 |
| Blood and lymphatic system disorders | ||
| Anemia | 43 | 25 |
| Thrombocytopenia b | 25 | 10 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 28 | 2 |
| Skin and subcutaneous tissue disorders | ||
| Rash c | 27 | 2 |
| Investigations | ||
| ALT/AST increased | 33 | 5 |
Other clinically relevant adverse reactions that occurred in less than 20% of patients included dyspnea, dizziness, bleeding, urinary tract infection, dysgeusia, dyspepsia, hypersensitivity (including flushing, asthma, choking sensation, periorbital swelling, swelling face, and wheezing), pneumonia, sepsis, ischemic cardiovascular events, renal failure, venous thromboembolism, and stomatitis.
|
aDenominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available for 111 to 115 patients. |
||
|
bNCI CTCAE version 5.0; decrease in phosphate is graded using NCI CTACE Version 4.03 |
||
|
cGrade 3-4 ALT or AST elevation led to drug interruption in 4 patients, of which 1 had dose reduction upon rechallenge. |
||
| Laboratory Parameter | Rubraca
N = 115 a |
|
| Grades 1-4
b
(%) | Grades 3-4
(%) |
|
| Clinical Chemistry | ||
| Increase in ALT c | 69 | 5 |
| Decrease in phosphate | 68 | 15 |
| Increase in alkaline phosphatase | 44 | 2 |
| Increase in creatinine | 43 | 2 |
| Increase in triglycerides | 42 | 5 |
| Decrease in sodium | 38 | 3 |
| Hematologic | ||
| Decrease in leukocytes | 69 | 5 |
| Decrease in absolute neutrophil count | 62 | 10 |
| Decrease in hemoglobin | 59 | 25 |
| Decrease in lymphocytes | 42 | 17 |
| Decrease in platelets | 40 | 10 |
7 DRUG INTERACTIONS
7.1 Effect of Rubraca on Other Drugs
Certain CYP1A2, CYP3A, CYP2C9, or CYP2C19 Substrates
Concomitant administration of Rubraca with CYP1A2, CYP3A, CYP2C9, or CYP2C19 substrates can increase the systemic exposure of these substrates [see Clinical Pharmacology ( 12.3)] , which may increase the frequency or severity of adverse reactions of these substrates. If concomitant administration is unavoidable between Rubraca and substrates of these enzymes where minimal concentration changes may lead to serious adverse reactions, decrease the substrate dosage in accordance with the approved prescribing information.
If concomitant administration with warfarin (a CYP2C9 substrate) cannot be avoided, consider increasing the frequency of international normalized ratio (INR) monitoring.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, Rubraca can cause fetal harm when administered to pregnant women. There are no available data in pregnant women to inform the drug-associated risk. In an animal reproduction study, administration of rucaparib to pregnant rats during organogenesis resulted in embryo-fetal death at maternal exposures that were 0.04 times the AUC 0-24hin patients receiving the recommended dose of 600 mg twice daily [see Data]. Apprise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
In a dose range-finding embryo-fetal development study, pregnant rats received oral doses of 50, 150, 500, or 1000 mg/kg/day of rucaparib during the period of organogenesis. Post-implantation loss (100% early resorptions) was observed in all animals at doses greater than or equal to 50 mg/kg/day (with maternal systemic exposures approximately 0.04 times the human exposure at the recommended dose based on AUC 0-24h).
8.2 Lactation
Risk Summary
There is no information regarding the presence of rucaparib in human milk, or on its effects on milk production or the breast-fed child. Because of the potential for serious adverse reactions in breast-fed children from Rubraca, advise lactating women not to breastfeed during treatment with Rubraca and for 2 weeks following the last dose.
8.3 Females and Males of Reproductive Potential
Rubraca can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations ( 8.1)] .
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating Rubraca.
Females
Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of Rubraca [see Use in Specific Populations ( 8.1)] .
Males
Based on findings in genetic toxicity and animal reproduction studies, advise male patients with female partners of reproductive potential or who are pregnant to use effective methods of contraception during treatment and for 3 months following last dose of Rubraca. Advise male patients not to donate sperm during therapy and for 3 months following the last dose of Rubraca [see Use in Specific Populations ( 8.1) and Nonclinical Toxicology ( 13.1)].
8.4 Pediatric Use
The safety and effectiveness of Rubraca in pediatric patients have not been established.
8.5 Geriatric Use
Of the 937 patients with ovarian cancer who received Rubraca in clinical trials including ARIEL3, 41% were age 65 or older and 10% were 75 years or older. No major differences in safety were observed between younger and older patients with ovarian cancer.
Of the 209 patients with mCRPC who received Rubraca in TRITON2, 77% were age 65 or older and 33% were 75 years or older. No major differences in safety were observed between younger and older patients with mCRPC.
8.6 Renal Impairment
No dosage modification is recommended for patients with mild to moderate renal impairment (creatinine clearance [CLcr] between 30 and 89 mL/min, as estimated by the Cockcroft-Gault method) [see Clinical Pharmacology ( 12.3)]. Rubraca has not been studied in patients with CLcr < 30 mL/min or patients on dialysis.
8.7 Hepatic Impairment
No dosage modification is recommended for patients with mild to moderate hepatic impairment (total bilirubin ≤ 3 x upper limit of normal [ULN] or AST > ULN) [see Clinical Pharmacology ( 12.3)] . Rubraca has not been studied in patients with severe hepatic impairment (total bilirubin > 3 x ULN and any AST).
11 DESCRIPTION
Rucaparib is an inhibitor of the mammalian polyadenosine 5'-diphosphoribose polymerase (PARP) enzyme. The chemical name is 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one ((1S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1]hept-1-yl)methanesulfonic acid salt. The chemical formula of rucaparib camsylate is C 19H 18FN 3O•C 10H 16O 4S and the relative molecular mass is 555.67 Daltons.
The chemical structure of rucaparib camsylate is shown below:
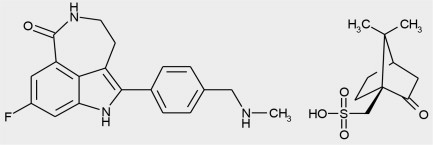
Rucaparib camsylate is a white to pale yellow powder; formulated into a tablet for oral use. Rucaparib shows pH-independent low solubility of approximately 1 mg/mL across the physiological pH range.
Rubraca (rucaparib) tablets contain rucaparib camsylate as the active ingredient. Each 200 mg tablet contains 344 mg rucaparib camsylate equivalent to 200 mg rucaparib free base. Each 250 mg tablet contains 430 mg rucaparib camsylate equivalent to 250 mg rucaparib free base. Each 300 mg tablet contains 516 mg rucaparib camsylate equivalent to 300 mg rucaparib free base.
The inactive ingredients in Rubraca tablets include: microcrystalline cellulose, sodium starch glycolate, colloidal silicon dioxide, and magnesium stearate. The cosmetic blue film coating for 200 mg tablets, cosmetic white film coating for 250 mg tablets, and cosmetic yellow film coating for 300 mg tablets is Opadry II containing polyvinyl alcohol, titanium dioxide, polyethylene glycol/macrogol, and talc. The coating is colorized as blue using brilliant blue aluminum lake and indigo carmine aluminum lake, or yellow using yellow iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Rucaparib is an inhibitor of poly (ADP-ribose) polymerase (PARP) enzymes, including PARP-1, PARP-2, and PARP-3, which play a role in DNA repair. In vitrostudies have shown that rucaparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes resulting in DNA damage, apoptosis, and cancer cell death. Increased rucaparib-induced cytotoxicity and anti-tumor activity was observed in tumor cell lines with deficiencies in BRCA1/2and other DNA repair genes. Rucaparib has been shown to decrease tumor growth in mouse xenograft models of human cancer with or without deficiencies in BRCA.
12.2 Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of rucaparib has not fully been characterized.
Cardiac Electrophysiology
A positive concentration-QTc relationship was observed in patients who were administered continuous dosages of Rubraca ranging from 40 mg once daily (0.03 times the approved recommended dosage) to 840 mg twice daily (1.4 times the approved recommended dosage). The mean (90% confidence interval [CI]) QTcF increase from baseline at steady state of Rubraca 600 mg twice daily was 14.9 msec (11.1, 18.7 msec).
12.3 Pharmacokinetics
The AUC and C maxof rucaparib demonstrated linear pharmacokinetics over a dose range from 240 mg to 840 mg twice daily (0.4 times to 1.4 times the approved recommended dosage). The mean (coefficient of variation [CV]) steady-state rucaparib C maxis 1,940 ng/mL (54%) and AUC 0-12his 16,900 h×ng/mL (54%) at the approved recommended dosage. The mean AUC accumulation ratio is 3.5 to 6.2 fold.
Absorption
The median T max(min, max) at the steady state is 1.9 hours (0, 5.98) at the approved recommended dosage. The mean (min, max) absolute bioavailability is 36% (30%, 45%).
Effect of Food
Following a high-fat meal (approximately 800-1000 calories, including approximately 250 calories from carbohydrates, approximately 500-600 calories from fat, approximately 150 calories from protein), the C maxwas increased by 20%, AUC 0-24hwas increased by 38%, and the T maxwas delayed by 2.5 hours, as compared to fasted conditions [see Dosage and Administration ( 2.2)] .
Distribution
The mean apparent volume of distribution is 2300 L (21%).
Rucaparib is 70% bound to human plasma proteins in vitro. Rucaparib preferentially distributed to red blood cells with a blood-to-plasma concentration ratio of 1.8.
Elimination
The mean apparent total clearance at steady state is 44.2 L/h (45%) and the mean terminal elimination half-life is 26 (39%) hours.
Metabolism
In vitro, rucaparib is primarily metabolized by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. In addition to CYP-based oxidation, rucaparib also undergoes N-demethylation, N-methylation, and glucuronidation.
Excretion
Following a single oral dose of radiolabeled rucaparib, unchanged rucaparib accounted for 64% of the radioactivity. Rucaparib accounted for 45% and 95% of radioactivity in urine and feces, respectively.
Specific Populations
Age (20 to 86 years), race (White, Black, and Asian), sex, body weight (41 to 171 kg), mild to moderate renal impairment (CLcr ≥ 30 mL/min), mild hepatic impairment (total bilirubin < ULN and AST > ULN or total bilirubin 1 to 1.5 x ULN and any AST), and CYP2D6 or CYP1A2 genotype polymorphisms did not have a clinically meaningful effect on the pharmacokinetics of rucaparib. The effect of severe renal impairment (CLcr 15 to 29 mL/min), end-stage renal disease (CLcr < 15 mL/min), or severe hepatic impairment (total bilirubin > 3 x ULN and any AST) has not been studied.
Hepatic Impairment
Moderate hepatic impairment (total bilirubin > 1.5 to 3 x ULN and any AST) increased rucaparib AUC by 45%, but had no effect on C maxcompared to patients with normal hepatic function.
Effect of Other Drugs on Rucaparib
Concomitant administration of Rubraca with a proton pump inhibitor had no clinically meaningful effect on steady-state concentrations of rucaparib.
Effect of Rucaparib on Other Drugs
Concomitant administration of Rubraca with rosuvastatin (BCRP substrate) had no clinically meaningful effect on the concentrations of rosuvastatin.
Coadministration of Rubraca with the following substrates increased the C maxof each coadministered substrate by ≤ 1.1-fold and increased the AUC of each substrate as follows:
- Caffeine (CYP1A2): by 2.6-fold
- Midazolam (CYP3A4): by 1.4-fold
- Warfarin (CYP2C9): by 1.5-fold
- Omeprazole (CYP2C19): by 1.6-fold
- Digoxin (P-glycoprotein): by 1.2-fold
Concomitant administration of Rubraca with an oral contraceptive containing ethinylestradiol and levonorgestrel (CYP3A substrates): increased ethinylestradiol AUC by 1.4-fold and levonorgestrel AUC by 1.6-fold, but did not have a clinically meaningful effect on their C max.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Rucaparib inhibited CYP2C8 and CYP2D6 and induced CYP1A2.
UDP-glucuronosyltransferase (UGT) Enzymes: Rucaparib inhibited UGT1A1.
Transporter Systems: Rucaparib is a substrate of P-gp and BCRP. Rucaparib is not a substrate of OATP1B1, OATP1B3, OAT1, OAT3, or OCT2.
Rucaparib inhibited OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, OCT1, OCT2, and MRP4. Rucaparib did not inhibit MRP2, MRP3, or BSEP.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with rucaparib.
Rucaparib was clastogenic in an in vitrochromosomal aberration assay in cultured human lymphocytes. The clastogenic response in mitotically-stimulated cells was anticipated based on the mechanism of action of rucaparib and indicates potential genotoxicity in humans. Rucaparib was not mutagenic in a bacterial reverse mutation (Ames) test.
Fertility studies with rucaparib have not been conducted. In 3-month repeat-dose general toxicology studies, rucaparib had no effects on male and female reproductive organs at doses up to 100 mg/kg/day and 20 mg/kg/day in rats and dogs, respectively. These dose levels resulted in systemic exposures of approximately 0.3 and 0.09 times the human exposure (AUC 0-24h), respectively, at the recommended dose.
14 CLINICAL STUDIES
14.1 Maintenance Treatment of BRCA-mutated Recurrent Ovarian Cancer
The efficacy of Rubraca was investigated in ARIEL3 (NCT01968213), a double-blind, multicenter clinical trial in which 564 patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who were in response to platinum-based chemotherapy were randomized (2:1) to receive Rubraca tablets 600 mg orally twice daily (n=375) or placebo (n=189). Treatment was continued until disease progression or unacceptable toxicity. All patients had achieved a response (complete or partial) to their most recent platinum-based chemotherapy. Randomization was stratified by best response to last platinum (complete or partial), time to progression following the penultimate platinum therapy (6 to ≤ 12 months and > 12 months), and tumor biomarker status.
Tumor tissue samples were tested using a clinical trial assay (CTA) (N=564) and an investigational Foundation Medicine tissue test (n=518). Of the samples evaluated with both tests, tumor BRCA(t BRCA) mutant status was confirmed for 99% (177/178) of t BRCA-positive patients determined by the CTA. Blood samples for 94% (186/196) of the t BRCApatients were evaluated using a central blood germline BRCAtest. Based on these results, 70% (130/186) of the t BRCApatients had a germline BRCAmutation and 30% (56/186) had a somatic BRCAmutation. The efficacy results are based on the t BRCA(germline or somatic) subgroup.
The major efficacy outcome was investigator-assessed progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (v1.1). Overall survival (OS) was an additional outcome measure.
Of the 564 enrolled patients, 196 patients (35%) had a t BRCAmutation. Among the patients who had a tBRCAmutation, the median age was 58 years (range: 42 to 81) for patients receiving Rubraca and 59 years (range: 36 to 84) for those on placebo; the majority were White (84%); and 100% had an ECOG performance status of 0 or 1. All patients had received at least two prior platinum-based chemotherapies (range: 2 to 5). A total of 33% of patients were in complete response (CR) to their most recent therapy. The progression-free interval to penultimate platinum was 6-12 months in 41% of patients and > 12 months in 59%. Prior bevacizumab therapy was reported for 22% of patients who received Rubraca and 17% of patients who received placebo. Measurable disease was present at baseline in 32% of patients.
ARIEL3 demonstrated a statistically significant improvement in PFS for patients randomized to Rubraca as compared with placebo in patients who had a t BRCAmutation. Results from a blinded independent radiology review were consistent.
Efficacy results for patients with a t BRCAmutation are summarized in Table 6and Figure 1.
|
at BRCAincludes all patients with a deleterious germline or somatic BRCAmutation, as determined by the CTA. |
||
| Rubraca
N=130 | Placebo
N=66 |
|
| Progression Free Survival | ||
| Number of events, n (%) | 67 (52%) | 56 (85%) |
| Median in months (95% CI) | 16.6 (13.4-22.9) | 5.4 (3.4-6.7) |
| HR (95% CI) | 0.23 (0.16, 0.34) | |
| p-value | < 0.0001 | |
Figure 1. Kaplan-Meier Curves of Progression-Free Survival in ARIEL3 as Assessed by Investigator: tBRCAGroup
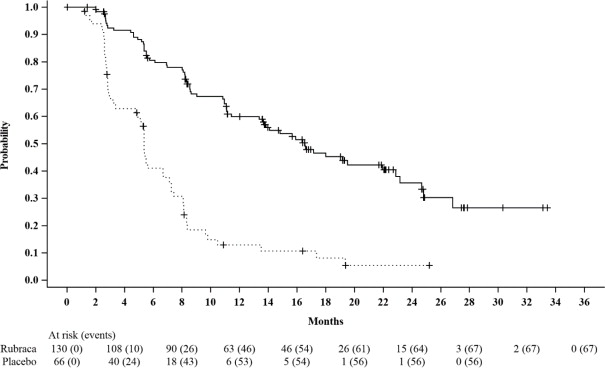
A final OS analysis was conducted after 130 events were observed. Exploratory OS results showed a HR of 0.83 (95% CI: 0.58, 1.19) in the t BRCAsubgroup with a median OS of 45.9 months (95% CI: 37.7, 59.6) for patients treated with Rubraca and 47.8 months (95% CI: 43.2, 55.8) for patients on placebo.
14.2 BRCA-mutated Metastatic Castration-Resistant Prostate Cancer
The efficacy of Rubraca was investigated in TRITON2 (NCT02952534), an ongoing multi-center, single arm clinical trial in patients with BRCA-mutated mCRPC who had been treated with androgen receptor-directed therapy and taxane-based chemotherapy. There were 115 patients with either germline or somatic BRCAmutations enrolled in TRITON2, of whom 62 patients had measurable disease at baseline by independent radiology review (IRR). Patients received Rubraca 600 mg orally twice daily until disease progression or unacceptable toxicity. Patients also received concomitant GnRH analog or had prior bilateral orchiectomy. Objective response rate (ORR) and duration of response (DOR) were assessed in patients with measurable disease by blinded IRR and by the investigator according to modified RECIST v1.1/ Prostate Cancer Working Group 3 (PCWG3) criteria.
For the 62 patients with measurable disease at baseline, the median age was 73 years (range 52 to 88); the majority were White (73%) and 10% were Black; and 98% of patients had an ECOG performance status of 0 or 1. All patients had received at least one prior androgen receptor-directed therapy, 34% had received 2 prior androgen receptor-directed therapies and 2% had received 3 prior androgen receptor-directed therapies, and all patients also received prior taxane chemotherapy. Eighteen percent of patients had lung and 21% had liver metastases at baseline. Twenty-four percent had metastases to lymph nodes alone. Forty percent had 10 or more bone lesions at baseline.
All 62 patients had a deleterious somatic or germline BRCAmutation detected from either central plasma (26%), central tissue (32%), or local (42%) testing. Of the 62 patients, 66% had a somatic BRCAmutation, 34% had a germline BRCAmutation, 85% had a BRCA2mutation, and 15% had a BRCA1mutation.
The major efficacy outcomes of the study were confirmed ORR by IRR using modified RECIST v1.1/PCWG3 criteria and DOR. Efficacy results of TRITON2 are provided in Table 7. The ORR by IRR was similar in patients with germline versus somatic BRCAmutation.
|
NE = not evaluable |
|
|
aDefined per modified RECIST v1.1 criteria and with no confirmed bone progression per PCWG3. |
|
|
bThe range for the DOR was 1.7-24+ months. Fifteen of the 27 (56%) patients with a confirmed objective response had a DOR of ≥ 6 months. |
|
| Rubraca
(N = 62) |
|
| Confirmed Objective Response Rate (95% CI) a | 44% (31, 57) |
| Median DOR in months (95% CI) b | NE (6.4, NE) |
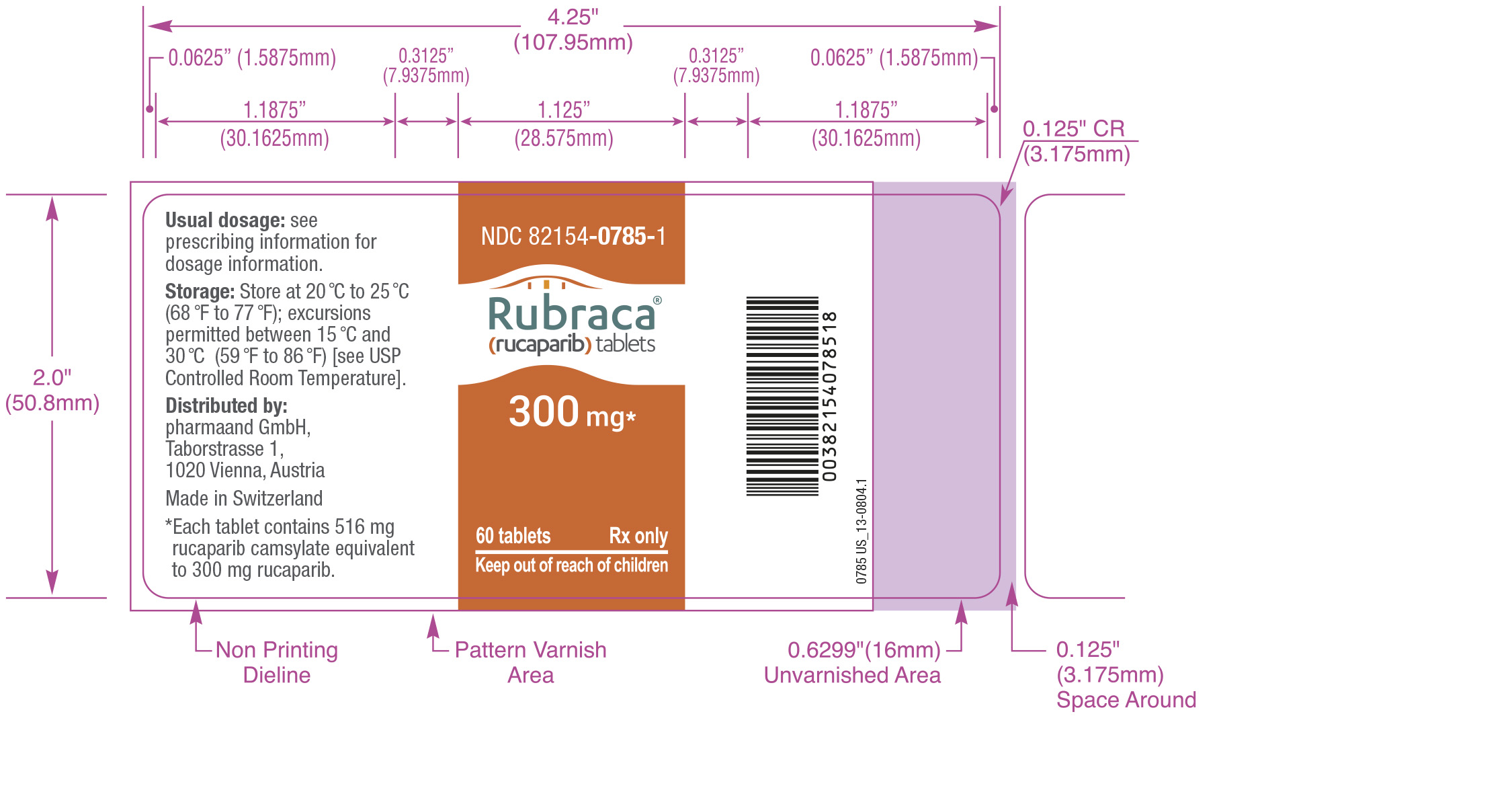
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Rubraca is available as 200 mg, 250 mg, and 300 mg tablets.
200 mg Tablets:
- Blue, round, and debossed with “C2” on one side
- Supplied in bottles of 60 tablets (NDC:82154-0783-1)
250 mg Tablets:
- White, diamond, and debossed with “C25” on one side
- Supplied in bottles of 60 tablets (NDC:82154-0784-1)
300 mg Tablets:
- Yellow, oval, and debossed with “C3” on one side
- Supplied in bottles of 60 tablets (NDC:82154-0785-1)
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling ( Patient Information).
MDS/AML:Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. These may be signs of hematological toxicity or a more serious uncommon bone marrow problem called ‘myelodysplastic syndrome' (MDS) or ‘acute myeloid leukemia' (AML) which have been reported in patients treated with Rubraca [see Warnings and Precautions ( 5.1)] .
Embryo-Fetal Toxicity:Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations ( 8.1)] . Advise females of reproductive potential to use effective contraception during treatment and for 6 months after receiving the last dose of Rubraca. Advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 3 months after receiving the last dose of Rubraca. Advise male patients not to donate sperm during therapy and for 3 months following the last dose of Rubraca [see Warnings and Precautions ( 5.2) and Use in Specific Populations ( 8.1, 8.3)] .
Photosensitivity:Advise patients to use appropriate sun protection due to the increased susceptibility to sunburn while taking Rubraca [see Adverse Drug Reactions ( 6.1)] .
Lactation:Advise females not to breastfeed during treatment and for 2 weeks after the last dose of Rubraca [see Use in Specific Populations ( 8.2)] .
Dosing Instructions:Instruct patients to take Rubraca orally twice daily with or without food. Doses should be taken approximately 12 hours apart. Advise patients that if a dose of Rubraca is missed or if the patient vomits after taking a dose of Rubraca, patients should not take an extra dose, but take the next dose at the regular time [see Dosage and Administration ( 2.2)] .
Manufactured for:
pharmaand GmbH
Taborstrasse 1
1020 Vienna
Austria
Distributed by:
Summit SD, LLC
255 Northwest Victoria Drive, Suite A
Lee’s Summit, MO 64086
Rubraca is a registered trademark of pharma&.
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: September 2023 |
|
| PATIENT INFORMATION
Rubraca®(roo-brah'-kah) (rucaparib) tablets |
||
|
What is the most important information I should know about Rubraca? Rubraca may cause serious side effects including:Bone marrow problems called Myelodysplastic Syndrome (MDS) or a type of cancer of the blood called Acute Myeloid Leukemia (AML).Some people who have cancer and who have received previous treatment with chemotherapy or certain other medicines for their cancer have developed MDS or AML during or after treatment with Rubraca. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with Rubraca. Symptoms of low blood cell counts are common during treatment with Rubraca, but can be a sign of serious problems, including MDS or AML. Tell your healthcare provider if you have any of the following symptoms during treatment with Rubraca: |
||
|
|
|
Your healthcare provider will do blood tests to check your blood cell counts:
|
||
| What is Rubraca?
Rubraca is a prescription medicine used in adults for:
|
||
Before you take Rubraca, tell your healthcare provider about all of your medical conditions, including if you:
|
||
How should I take Rubraca?
|
||
| What should I avoid while taking Rubraca?
Avoid spending time in sunlight. Rubraca can make your skin sensitive to the sun (photosensitivity). You may sunburn more easily during treatment with Rubraca. You should wear a hat and clothes that cover your skin and use sunscreen to help protect against sunburn if you have to be in the sunlight. |
||
|
What are the possible side effects of Rubraca? Rubraca may cause serious side effects. The most common side effects of Rubraca in people with ovarian cancer include: |
||
|
|
|
| The most common side effects of Rubraca in people with prostate cancer include: | ||
|
|
|
| These are not all of the possible side effects of Rubraca. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store Rubraca?
|
||
| General information about the safe and effective use of Rubraca.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use Rubraca for a condition for which it was not prescribed. Do not give it to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about Rubraca. |
||
| What are the ingredients in Rubraca?
Active ingredient:rucaparib Inactive ingredients:microcrystalline cellulose, sodium starch glycolate, colloidal silicon dioxide, and magnesium stearate. The film coating contains polyvinyl alcohol, titanium dioxide, polyethylene glycol/macrogol, and talc. The blue film coating contains brilliant blue aluminum lake and indigo carmine aluminum lake. The yellow film coating contains yellow iron oxide. Manufactured for: pharmaand GmbH, Taborstrasse 1, 1020 Vienna, Austria Distributed by: Summit SD LLC, 255 Northwest Victoria Drive, Suite A, Lee’s Summit, MO 64086 For more information call tel: 1-800-506-8501 |
||
Principal Display Panel - Rubraca tablets 200 mg Bottle Label
NDC 82154-0783-1
Rubraca®
(rucaparib)tablets
200 mg*
60 tablets
Each tablet contains 344 mg rucaparib camsylate equivalent to 200 mg rucaparib
Rx only
Keep out of reach of children