DESCRIPTION
CRIXIVAN1 (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, and 400 mg of indinavir (corresponding to 125, 250, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin, titanium dioxide, silicon dioxide, and sodium lauryl sulfate.
The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1-dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula:
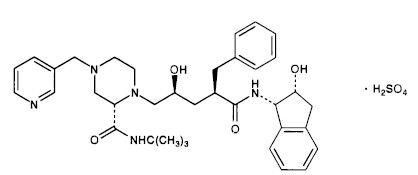
Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4• H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol.
- 1
-
Registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Copyright © 1996, 1997, 1998, 1999, 2004 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved
MICROBIOLOGY
Mechanism of Action:
HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles.
Antiretroviral Activity In Vitro :
The in vitro activity of indinavir was assessed in cell lines of lymphoblastic and monocytic origin and in peripheral blood lymphocytes. HIV-1 variants used to infect the different cell types include laboratory-adapted variants, primary clinical isolates and clinical isolates resistant to nucleoside analogue and nonnucleoside inhibitors of the HIV-1 reverse transcriptase. The IC95 (95% inhibitory concentration) of indinavir in these test systems was in the range of 25 to 100 nM. In drug combination studies with the nucleoside analogues zidovudine and didanosine, indinavir showed synergistic activity in cell culture. The relationship between in vitro susceptibility of HIV-1 to indinavir and inhibition of HIV-1 replication in humans has not been established.
Drug Resistance:
Isolates of HIV-1 with reduced susceptibility to the drug have been recovered from some patients treated with indinavir. Viral resistance was correlated with the accumulation of mutations that resulted in the expression of amino acid substitutions in the viral protease. Eleven amino acid residue positions, (L10l/V/R, K20l/M/R, L24l, M46l/L, l54A/V, L63P, l64V, A71T/V, V82A/F/T, l84V, and L90M), at which substitutions are associated with resistance, have been identified. Resistance was mediated by the co-expression of multiple and variable substitutions at these positions. No single substitution was either necessary or sufficient for measurable resistance (≥4-fold increase in IC95). In general, higher levels of resistance were associated with the co-expression of greater numbers of substitutions, although their individual effects varied and were not additive. At least 3 amino acid substitutions must be present for phenotypic resistance to indinavir to reach measurable levels. In addition, mutations in the p7/ p1 and p1/ p6 gag cleavage sites were observed in some indinavir resistant HIV-1 isolates.
In vitro phenotypic susceptibilities to indinavir were determined for 38 viral isolates from 13 patients who experienced virologic rebounds during indinavir monotherapy. Pre-treatment isolates from five patients exhibited indinavir IC95 values of 50-100 nM. At or following viral RNA rebound (after 12-76 weeks of therapy), IC95 values ranged from 25 to >3000 nM, and the viruses carried 2 to 10 mutations in the protease gene relative to baseline.
Cross-Resistance to Other Antiviral Agents:
Varying degrees of HIV-1 cross-resistance have been observed between indinavir and other HIV-1 protease inhibitors. In studies with ritonavir, saquinavir, and amprenavir, the extent and spectrum of cross-resistance varied with the specific mutational patterns observed. In general, the degree of cross-resistance increased with the accumulation of resistance-associated amino acid substitutions. Within a panel of 29 viral isolates from indinavir-treated patients that exhibited measurable (≥4-fold) phenotypic resistance to indinavir, all were resistant to ritonavir. Of the indinavir resistant HIV-1 isolates, 63% showed resistance to saquinavir and 81% to amprenavir.
CLINICAL PHARMACOLOGY
Pharmacokinetics
Absorption:
Indinavir was rapidly absorbed in the fasted state with a time to peak plasma concentration (Tmax) of 0.8 ± 0.3 hours (mean ± S.D.) (n=11). A greater than dose-proportional increase in indinavir plasma concentrations was observed over the 200-1000 mg dose range. At a dosing regimen of 800 mg every 8 hours, steady-state area under the plasma concentration time curve (AUC) was 30,691 ± 11,407 nM•hour (n=16), peak plasma concentration (Cmax) was 12,617 ± 4037 nM (n=16), and plasma concentration eight hours post dose (trough) was 251 ± 178 nM (n=16).
Effect of Food on Oral Absorption:
Administration of indinavir with a meal high in calories, fat, and protein (784 kcal, 48.6 g fat, 31.3 g protein) resulted in a 77% ± 8% reduction in AUC and an 84% ± 7% reduction in Cmax (n=10). Administration with lighter meals (e.g., a meal of dry toast with jelly, apple juice, and coffee with skim milk and sugar or a meal of corn flakes, skim milk and sugar) resulted in little or no change in AUC, Cmax or trough concentration.
Distribution:
Indinavir was approximately 60% bound to human plasma proteins over a concentration range of 81 nM to 16,300 nM.
Metabolism:
Following a 400-mg dose of 14C-indinavir, 83 ± 1% (n=4) and 19 ± 3% (n=6) of the total radioactivity was recovered in feces and urine, respectively; radioactivity due to parent drug in feces and urine was 19.1% and 9.4%, respectively. Seven metabolites have been identified, one glucuronide conjugate and six oxidative metabolites. In vitro studies indicate that cytochrome P-450 3A4 (CYP3A4) is the major enzyme responsible for formation of the oxidative metabolites.
Elimination:
Less than 20% of indinavir is excreted unchanged in the urine. Mean urinary excretion of unchanged drug was 10.4 ± 4.9% (n=10) and 12.0 ± 4.9% (n=10) following a single 700-mg and 1000-mg dose, respectively. Indinavir was rapidly eliminated with a half-life of 1.8 ± 0.4 hours (n=10). Significant accumulation was not observed after multiple dosing at 800 mg every 8 hours.
Special Populations
Hepatic Insufficiency:
Patients with mild to moderate hepatic insufficiency and clinical evidence of cirrhosis had evidence of decreased metabolism of indinavir resulting in approximately 60% higher mean AUC following a single 400-mg dose (n=12). The half-life of indinavir increased to 2.8 ± 0.5 hours. Indinavir pharmacokinetics have not been studied in patients with severe hepatic insufficiency (see DOSAGE AND ADMINISTRATION, Hepatic Insufficiency).
Renal Insufficiency:
The pharmacokinetics of indinavir have not been studied in patients with renal insufficiency.
Gender:
The effect of gender on the pharmacokinetics of indinavir was evaluated in 10 HIV seropositive women who received CRIXIVAN 800 mg every 8 hours with zidovudine 200 mg every 8 hours and lamivudine 150 mg twice a day for one week. Indinavir pharmacokinetic parameters in these women were compared to those in HIV seropositive men (pooled historical control data). Differences in indinavir exposure, peak concentrations, and trough concentrations between males and females are shown in Table 1 below:
| PK Parameter | % change in PK parameter for females relative to males | 90% Confidence Interval |
| ↓ Indicates a decrease in the PK parameter; ↑ Indicates an increase in the PK parameter. | ||
| AUC0-8h (nM•hr) | ↓13% | (↓32%, ↑12%) |
| Cmax (nM) | ↓13% | (↓32%, ↑10%) |
| C8h (nM) | ↓22% | (↓47%, ↑15%) |
The clinical significance of these gender differences in the pharmacokinetics of indinavir is not known.
Race:
Pharmacokinetics of indinavir appear to be comparable in Caucasians and Blacks based on pharmacokinetic studies including 42 Caucasians (26 HIV-positive) and 16 Blacks (4 HIV-positive).
Pediatric:
The optimal dosing regimen for use of indinavir in pediatric patients has not been established. In HIV-infected pediatric patients (age 4-15 years), a dosage regimen of indinavir capsules, 500 mg/m2 every 8 hours, produced AUC0-8hr of 38,742 ± 24,098 nM•hour (n=34), Cmax of 17,181 ± 9809 nM (n=34), and trough concentrations of 134 ± 91 nM (n=28). The pharmacokinetic profiles of indinavir in pediatric patients were not comparable to profiles previously observed in HIV-infected adults receiving the recommended dose of 800 mg every 8 hours. The AUC and Cmax values were slightly higher and the trough concentrations were considerably lower in pediatric patients. Approximately 50% of the pediatric patients had trough values below 100 nM; whereas, approximately 10% of adult patients had trough levels below 100 nM. The relationship between specific trough values and inhibition of HIV replication has not been established.
Pregnant Patients:
The optimal dosing regimen for use of indinavir in pregnant patients has not been established. A CRIXIVAN dose of 800 mg every 8 hours (with zidovudine 200 mg every 8 hours and lamivudine 150 mg twice a day) has been studied in 16 HIV-infected pregnant patients at 14 to 28 weeks of gestation at enrollment (study PACTG 358). The mean indinavir plasma AUC0-8hr at weeks 30-32 of gestation (n=11) was 9231 nM•hr, which is 74% (95% CI: 50%, 86%) lower than that observed 6 weeks postpartum. Six of these 11 (55%) patients had mean indinavir plasma concentrations 8 hours post-dose (Cmin) below assay threshold of reliable quantification. The pharmacokinetics of indinavir in these 11 patients at 6 weeks postpartum were generally similar to those observed in non-pregnant patients in another study (see PRECAUTIONS, Pregnancy).
Drug Interactions:
(also see CONTRAINDICATIONS, WARNINGS, PRECAUTIONS, Drug Interactions)
Indinavir is an inhibitor of the cytochrome P450 isoform CYP3A4. Coadministration of CRIXIVAN and drugs primarily metabolized by CYP3A4 may result in increased plasma concentrations of the other drug, which could increase or prolong its therapeutic and adverse effects (see CONTRAINDICATIONS and WARNINGS). Based on in vitro data in human liver microsomes, indinavir does not inhibit CYP1A2, CYP2C9, CYP2E1 and CYP2B6. However, indinavir may be a weak inhibitor of CYP2D6.
Indinavir is metabolized by CYP3A4. Drugs that induce CYP3A4 activity would be expected to increase the clearance of indinavir, resulting in lowered plasma concentrations of indinavir. Coadministration of CRIXIVAN and other drugs that inhibit CYP3A4 may decrease the clearance of indinavir and may result in increased plasma concentrations of indinavir.
Drug interaction studies were performed with CRIXIVAN and other drugs likely to be coadministered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of coadministration of CRIXIVAN on the AUC, Cmax and Cmin are summarized in Table 2 (effect of other drugs on indinavir) and Table 3 (effect of indinavir on other drugs). For information regarding clinical recommendations, see Table 9 in PRECAUTIONS.
| Coadministered drug | Dose of Coadministered drug (mg) | Dose of CRIXIVAN (mg) | n | Ratio (with/without coadministered drug) of Indinavir Pharmacokinetic Parameters (90% CI); No Effect = 1.00 |
||
| Cmax | AUC | Cmin | ||||
| All interaction studies conducted in healthy, HIV-negative adult subjects, unless otherwise indicated. | ||||||
| Cimetidine | 600 twice daily, 6 days | 400 single dose | 12 | 1.07 (0.77, 1.49) | 0.98 (0.81, 1.19) | 0.82 (0.69, 0.99) |
| Clarithromycin | 500 q12h, 7 days | 800 three times daily, 7 days | 10 | 1.08 (0.85, 1.38) | 1.19 (1.00, 1.42) | 1.57 (1.16, 2.12) |
| Delavirdine | 400 three times daily | 400 three times daily, 7 days | 28 | 0.64*
(0.48, 0.86) | No significant change* | 2.18*
(1.16, 4.12) |
| Delavirdine | 400 three times daily | 600 three times daily, 7 days | 28 | No significant change | 1.53*
(1.07, 2.20) | 3.98*
(2.04, 7.78) |
| Efavirenz† | 600 once daily, 10 days | 1000 three times daily, 10 days | 20 | |||
| After morning dose | No significant change* | 0.67*
(0.61, 0.74) | 0.61*
(0.49, 0.76) |
|||
| After afternoon dose | No significant change* | 0.63*
(0.54, 0.74) | 0.48*
(0.43, 0.53) |
|||
| After evening dose | 0.71*
(0.57, 0.89) | 0.54*
(0.46, 0.63) | 0.43*
(0.37, 0.50) |
|||
| Fluconazole† | 400 once daily, 8 days | 1000 three times daily, 7 days | 11 | 0.87 (0.72, 1.05) | 0.76 (0.59, 0.98) | 0.90 (0.72, 1.12) |
| Grapefruit Juice | 8 oz. | 400 single dose | 10 | 0.65 (0.53, 0.79) | 0.73 (0.60, 0.87) | 0.90 (0.71, 1.15) |
| Isoniazid | 300 once daily in the morning, 8 days | 800 three times daily, 7 days | 11 | 0.95 (0.88, 1.03) | 0.99 (0.87, 1.13) | 0.89 (0.75, 1.06) |
| Itraconazole | 200 twice daily, 7 days | 600 three times daily, 7 days | 12 | 0.78*
(0.69, 0.88) | 0.99*
(0.91, 1.06) | 1.49*
(1.28, 1.74) |
| Ketoconazole | 400 once daily, 7 days | 600 three times daily, 7 days | 12 | 0.69*
(0.61, 0.78) | 0.80*
(0.74, 0.87) | 1.29*
(1.11, 1.51) |
| 400 once daily, 7 days | 400 three times daily, 7 days | 12 | 0.42*
(0.37, 0.47) | 0.44*
(0.41, 0.48) | 0.73*
(0.62, 0.85) |
|
| Methadone | 20-60 once daily in the morning, 8 days | 800 three times daily, 8 days | 10 | See text below for discussion of interaction. | ||
| Quinidine | 200 single dose | 400 single dose | 10 | 0.96 (0.79, 1.18) | 1.07 (0.89, 1.28) | 0.93 (0.73, 1.19) |
| Rifabutin | 150 once daily in the morning, 10 days | 800 three times daily, 10 days | 14 | 0.80 (0.72, 0.89) | 0.68 (0.60, 0.76) | 0.60 (0.51, 0.72) |
| Rifabutin | 300 once daily in the morning, 10 days | 800 three times daily, 10 days | 10 | 0.75 (0.61, 0.91) | 0.66 (0.56, 0.77) | 0.61 (0.50, 0.75) |
| Rifampin | 600 once daily in the morning, 8 days | 800 three times daily, 7 days | 12 | 0.13 (0.08, 0.22) | 0.08 (0.06, 0.11) | Not Done |
| Ritonavir | 100 twice daily, 14 days | 800 twice daily, 14 days | 10, 16‡ | See text below for discussion of interaction. | ||
| Ritonavir | 200 twice daily, 14 days | 800 twice daily,14 days | 9, 16‡ | See text below for discussion of interaction. | ||
| Sildenafil | 25 single dose | 800 three times daily | 6 | See text below for discussion of interaction. | ||
| St. John's wort (Hypericum perforatum, standardized to 0.3 % hypericin) | 300 three times daily with meals, 14 days | 800 three times daily | 8 | Not Available | 0.46 (0.34, 0.58)§ | 0.19 (0.06, 0.33)§ |
| Stavudine (d4T)† | 40 twice daily, 7 days | 800 three times daily, 7 days | 11 | 0.95 (0.80, 1.11) | 0.95 (0.80, 1.12) | 1.13 (0.83, 1.53) |
| Trimethoprim/ Sulfamethoxazole | 800 Trimethoprim/ 160 Sulfamethoxazole q12h, 7 days | 400 four times daily, 7 days | 12 | 1.12 (0.87, 1.46) | 0.98 (0.81, 1.18) | 0.83 (0.72, 0.95) |
| Zidovudine† | 200 three times daily, 7 days | 1000 three times daily, 7 days | 12 | 1.06 (0.91, 1.25) | 1.05 (0.86, 1.28) | 1.02 (0.77, 1.35) |
| Zidovudine/ Lamivudine (3TC)† | 200/150 three times daily, 7 days | 800 three times daily, 7 days | 6, 9¶ | 1.05 (0.83, 1.33) | 1.04 (0.67, 1.61) | 0.98 (0.56, 1.73) |
| Coadministered drug | Dose of Coadministered drug (mg) | Dose of CRIXIVAN (mg) | n | Ratio (with/without CRIXIVAN) of Coadministered Drug Pharmacokinetic Parameters (90% CI); No Effect = 1.00 |
||
| Cmax | AUC | Cmin | ||||
| All interaction studies conducted in healthy, HIV-negative adult subjects, unless otherwise indicated. | ||||||
| Clarithromycin | 500 twice daily, 7 days | 800 three times daily, 7 days | 12 | 1.19 (1.02, 1.39) | 1.47 (1.30, 1.65) | 1.97 (1.58, 2.46) n=11 |
| Efavirenz | 200 once daily, 14 days | 800 three times daily, 14 days | 20 | No significant change | No significant change | -- |
| Ethinyl Estradiol (ORTHO-NOVUM 1/35)* | 35 mcg, 8 days | 800 three times daily, 8 days | 18 | 1.02 (0.96, 1.09) | 1.22 (1.15, 1.30) | 1.37 (1.24, 1.51) |
| Isoniazid | 300 once daily in the morning, 8 days | 800 three times daily, 8 days | 11 | 1.34 (1.12, 1.60) | 1.12 (1.03, 1.22) | 1.00 (0.92, 1.08) |
| Methadone† | 20-60 once daily in the morning, 8 days | 800 three times daily, 8 days | 12 | 0.93 (0.84, 1.03) | 0.96 (0.86, 1.06) | 1.06 (0.94, 1.19) |
| Norethindrone (ORTHO-NOVUM 1/35)* | 1 mcg, 8 days | 800 three times daily, 8 days | 18 | 1.05 (0.95, 1.16) | 1.26 (1.20, 1.31) | 1.44 (1.32, 1.57) |
| Rifabutin •150 mg once daily in the morning, 11 days + indinavir compared to 300 mg once daily in the morning, 11 days alone | 150 once daily in the morning, 10 days 300 once daily in the morning, 10 days | 800 three times daily, 10 days 800 three times daily, 10 days | 14 10 | 1.29 (1.05, 1.59) 2.34 (1.64, 3.35) | 1.54 (1.33, 1.79) 2.73 (1.99, 3.77) | 1.99 (1.71, 2.31) n=13 3.44 (2.65, 4.46) n=9 |
| Ritonavir | 100 twice daily, 14 days | 800 twice daily, 14 days | 10, 4‡ | 1.61 (1.13, 2.29) | 1.72 (1.20, 2.48) | 1.62 (0.93, 2.85) |
| 200 twice daily, 14 days | 800 twice daily, 14 days | 9, 5‡ | 1.19 (0.85, 1.66) | 1.96 (1.39, 2.76) | 4.71 (2.66, 8.33) n=9, 4 |
|
| Saquinavir | ||||||
| Hard gel formulation | 600 single dose | 800 three times daily, 2 days | 6 | 4.7 (2.7, 8.1) | 6.0 (4.0, 9.1) | 2.9 (1.7, 4.7)§ |
| Soft gel formulation | 800 single dose | 800 three times daily, 2 days | 6 | 6.5 (4.7, 9.1) | 7.2 (4.3, 11.9) | 5.5 (2.2, 14.1)§ |
| Soft gel formulation | 1200 single dose | 800 three times daily, 2 days | 6 | 4.0 (2.7, 5.9) | 4.6 (3.2, 6.7) | 5.5 (3.7, 8.3)§ |
| Sildenafil | 25 single dose | 800 three times daily | 6 | See text below for discussion of interaction. | ||
| Stavudine¶ | 40 twice daily, 7 days | 800 three times daily, 7 days | 13 | 0.86 (0.73, 1.03) | 1.21 (1.09, 1.33) | Not Done |
| Theophylline | 250 single dose (on Days 1 and 7) | 800 three times daily, 6 days (Days 2 to 7) | 12, 4‡ | 0.88 (0.76, 1.03) | 1.14 (1.04, 1.24) | 1.13 (0.86, 1.49) n=7, 3 |
| Trimethoprim/ Sulfamethoxazole | ||||||
| Trimethoprim | 800 Trimethoprim/ 160 Sulfamethoxazole q12h, 7 days | 400 q6h, 7 days | 12 | 1.18 (1.05, 1.32) | 1.18 (1.05, 1.33) | 1.18 (1.00, 1.39) |
| Trimethoprim/ Sulfamethoxazole | ||||||
| Sulfamethoxazole | 800 Trimethoprim/ 160 Sulfamethoxazole q12h, 7 days | 400 q6h, 7 days | 12 | 1.01 (0.95, 1.08) | 1.05 (1.01, 1.09) | 1.05 (0.97, 1.14) |
| Vardenafil | 10 single dose | 800 three times daily | 18 | See text below for discussion of interaction. | ||
| Zidovudine¶ | 200 three times daily, 7 days | 1000 three times daily, 7 days | 12 | 0.89 (0.73, 1.09) | 1.17 (1.07, 1.29) | 1.51 (0.71, 3.20) n=4 |
| Zidovudine/ Lamivudine¶ | ||||||
| Zidovudine | 200/150 three times daily, 7 days | 800 three times daily, 7 days | 6, 7‡ | 1.23 (0.74, 2.03) | 1.39 (1.02, 1.89) | 1.08 (0.77, 1.50) n=5, 5 |
| Zidovudine/ Lamivudine¶ | ||||||
| Lamivudine | 200/150 three times daily, 7 days | 800 three times daily, 7 days | 6, 7‡ | 0.73 (0.52, 1.02) | 0.91 (0.66, 1.26) | 0.88 (0.59, 1.33) |
Delavirdine: Delavirdine inhibits the metabolism of indinavir such that coadministration of 400-mg or 600-mg indinavir three times daily with 400-mg delavirdine three times daily alters indinavir AUC, Cmax and Cmin (see Table 2). Indinavir had no effect on delavirdine pharmacokinetics (see DOSAGE AND ADMINISTRATION, Concomitant Therapy, Delavirdine), based on a comparison to historical delavirdine pharmacokinetic data.
Methadone: Administration of indinavir (800 mg every 8 hours) with methadone (20 mg to 60 mg daily) for one week in subjects on methadone maintenance resulted in no change in methadone AUC. Based on a comparison to historical data, there was little or no change in indinavir AUC.
Ritonavir: Compared to historical data in patients who received indinavir 800 mg every 8 hours alone, twice-daily coadministration to volunteers of indinavir 800 mg and ritonavir with food for two weeks resulted in a 2.7-fold increase of indinavir AUC24h, a 1.6-fold increase in indinavir Cmax, and an 11-fold increase in indinavir Cmin for a 100-mg ritonavir dose and a 3.6-fold increase of indinavir AUC24h, a 1.8- fold increase in indinavir Cmax, and a 24-fold increase in indinavir Cmin for a 200-mg ritonavir dose. In the same study, twice-daily coadministration of indinavir (800 mg) and ritonavir (100 or 200 mg) resulted in ritonavir AUC24h increases versus the same doses of ritonavir alone (see Table 3).
Sildenafil: The results of one published study in HIV-infected men (n=6) indicated that coadministration of indinavir (800 mg every 8 hours chronically) with a single 25-mg dose of sildenafil resulted in an 11% increase in average AUC0-8hr of indinavir and a 48% increase in average indinavir peak concentration (Cmax) compared to 800 mg every 8 hours alone. Average sildenafil AUC was increased by 340% following coadministration of sildenafil and indinavir compared to historical data following administration of sildenafil alone (see CONTRAINDICATIONS, WARNINGS, Drug Interactions and PRECAUTIONS, Drug Interactions).
Vardenafil: Indinavir (800 mg every 8 hours) coadministered with a single 10-mg dose of vardenafil resulted in a 16-fold increase in vardenafil AUC, a 7-fold increase in vardenafil Cmax, and a 2-fold increase in vardenafil half-life (see WARNINGS, Drug Interactions and PRECAUTIONS, Drug Interactions).
INDICATIONS AND USAGE
CRIXIVAN in combination with antiretroviral agents is indicated for the treatment of HIV infection.
This indication is based on two clinical trials of approximately 1 year duration that demonstrated: 1) a reduction in the risk of AIDS-defining illnesses or death; 2) a prolonged suppression of HIV RNA.
Description of Studies
In all clinical studies, with the exception of ACTG 320, the AMPLICOR HIV MONITOR assay was used to determine the level of circulating HIV RNA in serum. This is an experimental use of the assay. HIV RNA results should not be directly compared to results from other trials using different HIV RNA assays or using other sample sources.
Study ACTG 320 was a multicenter, randomized, double-blind clinical endpoint trial to compare the effect of CRIXIVAN in combination with zidovudine and lamivudine with that of zidovudine plus lamivudine on the progression to an AIDS-defining illness (ADI) or death. Patients were protease inhibitor and lamivudine naive and zidovudine experienced, with CD4 cell counts of ≤200 cells/mm3. The study enrolled 1156 HIV-infected patients (17% female, 28% Black, 18% Hispanic, mean age 39 years). The mean baseline CD4 cell count was 87 cells/mm3. The mean baseline HIV RNA was 4.95 log10 copies/mL (89,035 copies/mL). The study was terminated after a planned interim analysis, resulting in a median follow-up of 38 weeks and a maximum follow-up of 52 weeks. Results are shown in Table 4 and Figures 1 & 2.
| Number (%) of Patients with AIDS-defining Illness or Death | |||
| Endpoint | IDV+ZDV+L*
(n=577) | ZDV+L (n=579) |
|
| HIV Progression or Death | 35 (6.1) | 63 (10.9) | |
| Death† | 10 (1.7) | 19 (3.3) | |
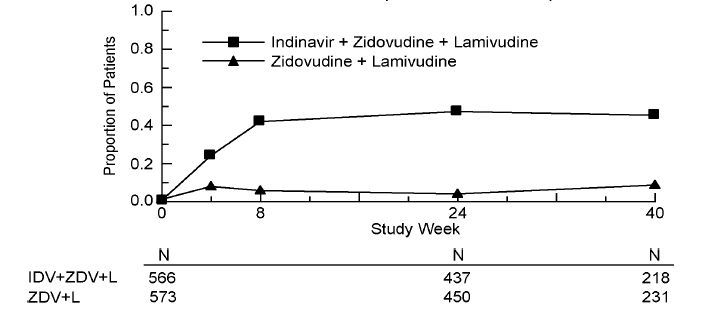
Study ACTG 320: Figure 1 - Indinavir Protocol ACTG 320 Zidovudine Experienced Plasma Viral RNA - Proportions Below 400 copies/mL
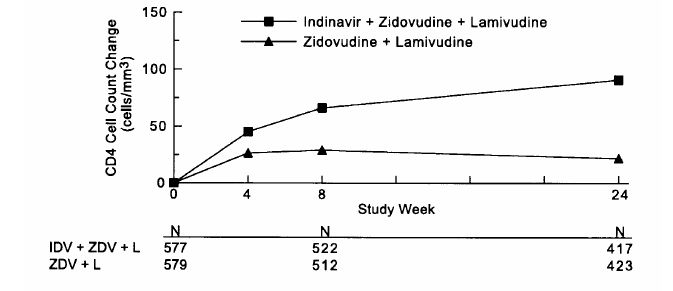
Study ACTG 320: Figure 2 - ACTG 320 Zidovudine Experienced CD4 Cell Counts - Mean Change from Baseline
Study 028, a double-blind, multicenter, randomized, clinical endpoint trial conducted in Brazil, compared the effects of CRIXIVAN plus zidovudine with those of CRIXIVAN alone or zidovudine alone on the progression to an ADI or death, and on surrogate marker responses. All patients were antiretroviral naive with CD4 cell counts of 50 to 250 cells/mm3. The study enrolled 996 HIV-1 seropositive patients [28% female, 11% Black, 1% Asian/Other, median age 33 years, mean baseline CD4 cell count of 152 cells/mm3, mean serum viral RNA of 4.44 log10 copies/mL (27,824 copies/mL)]. Treatment regimens containing zidovudine were modified in a blinded manner with the optional addition of lamivudine (median time: week 40). The median length of follow-up was 56 weeks with a maximum of 97 weeks. The study was terminated after a planned interim analysis, resulting in a median follow-up of 56 weeks and a maximum follow-up of 97 weeks. Results are shown in Table 5 and Figures 3 and 4.
| Number (%) of Patients with AIDS-defining Illness or Death | |||
| Endpoint | IDV+ZDV (n=332) | IDV (n=332) | ZDV (n=332) |
|
|||
| HIV Progression or Death | 21 (6.3) | 27 (8.1) | 62 (18.7) |
| Death* | 8 (2.4) | 5 (1.5) | 11 (3.3) |
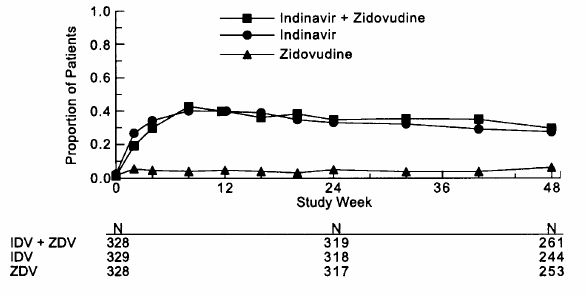
Study 028: Figure 3 - Indinavir Protocol 028 Zidovudine Naive Viral RNA - Proportions Below 500 copies/mL in Serum
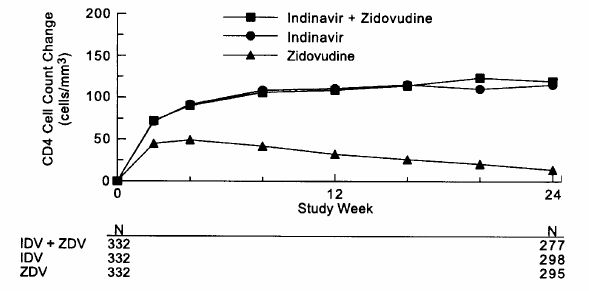
Study 028: Figure 4 - Indinavir Protocol 028 Zidovudine Naive CD4 Cell Counts - Mean Change from Baseline
Study 035 was a multicenter, randomized trial in 97 HIV-1 seropositive patients who were zidovudine-experienced (median exposure 30 months), protease-inhibitor- and lamivudine-naive, with mean baseline CD4 count 175 cells/mm3 and mean baseline serum viral RNA 4.62 log10 copies/mL (41,230 copies/mL). Comparisons included CRIXIVAN plus zidovudine plus lamivudine vs. CRIXIVAN alone vs. zidovudine plus lamivudine. After at least 24 weeks of randomized, double-blind therapy, patients were switched to open-label CRIXIVAN plus lamivudine plus zidovudine. Mean changes in log10 viral RNA in serum, the proportions of patients with viral RNA below 500 copies/mL in serum, and mean changes in CD4 cell counts, during 24 weeks of randomized, double-blinded therapy are summarized in Figures 5, 6, and 7, respectively. A limited number of patients remained on randomized, double-blind treatment for longer periods; based on this extended treatment experience, it appears that a greater number of subjects randomized to CRIXIVAN plus zidovudine plus lamivudine demonstrated HIV RNA levels below 500 copies/mL during one year of therapy as compared to those in other treatment groups.
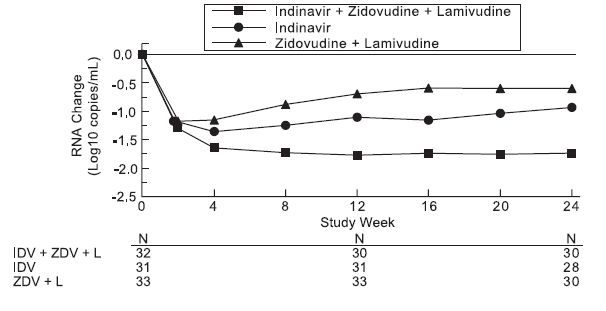
Study 035: Figure 5 - Indinavir Protocol 035 Zidovudine Experienced Viral RNA - Mean Log10 Change from Baseline in Serum
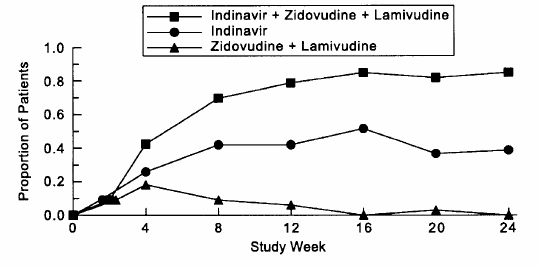
Study 035: Figure 6 - Indinavir Protocol 035 Zidovudine Experienced Viral RNA - Proportions Below 500 Copies/mL in Serum
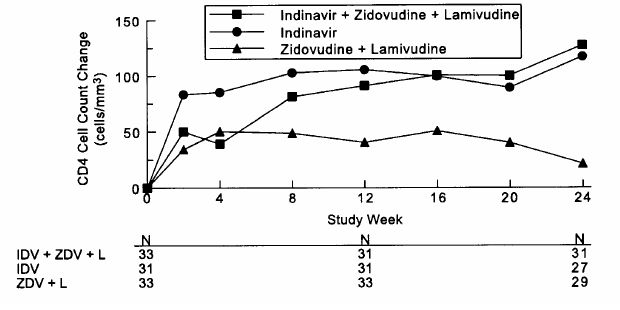
Study 035: Figure 7 - Indinavir Protocol 035 Zidovudine Experienced CD4 Cell Counts - Mean Change from Baseline
Genotypic Resistance in Clinical Studies
Study 006 (10/15/93-10/12/94) was a dose-ranging study in which patients were initially treated with CRIXIVAN at a dose of <2.4 g/day followed by 2.4 g/day. Study 019 (6/23/94-4/10/95) was a randomized comparison of CRIXIVAN 600 mg every 6 hours, CRIXIVAN plus zidovudine, and zidovudine alone. Table 6 shows the incidence of genotypic resistance at 24 weeks in these studies.
| Treatment Group | Resistance to IDV n/N* | Resistance to ZDV n/N* |
|
||
| IDV | — | — |
| <2.4 g/day | 31/37 (84%) | — |
| 2.4 g/day | 9/21 (43%) | 1/17 (6%) |
| IDV/ZDV | 4/22 (18%) | 1/22 (5%) |
| ZDV | 1/18 (6%) | 11/17 (65%) |
CONTRAINDICATIONS
CRIXIVAN is contraindicated in patients with clinically significant hypersensitivity to any of its components.
Inhibition of CYP3A4 by CRIXIVAN can result in elevated plasma concentrations of the following drugs, potentially causing serious or life-threatening reactions:
| Drug Class | Drugs Within Class That Are Contraindicated With CRIXIVAN |
|
|
| Alpha 1-adrenoreceptor antagonist | alfuzosin |
| Antiarrhythmics | amiodarone |
| Ergot derivatives | dihydroergotamine, ergonovine, ergotamine, methylergonovine |
| GI motility agents | cisapride |
| Neuroleptics | pimozide |
| PDE5 Inhibitors | Revatio* (sildenafil) [for treatment of pulmonary aterial hypertension] |
| Sedative/hypnotics | oral midazolam, triazolam, alprazolam |
WARNINGS
ALERT: Find out about medicines that should NOT be taken with CRIXIVAN. This statement is included on the product’s bottle label.
Nephrolithiasis/Urolithiasis
Nephrolithiasis/urolithiasis has occurred with CRIXIVAN therapy. The cumulative frequency of nephrolithiasis is substantially higher in pediatric patients (29%) than in adult patients (12.4%; range across individual trials: 4.7% to 34.4%). The cumulative frequency of nephrolithiasis events increases with increasing exposure to CRIXIVAN; however, the risk over time remains relatively constant. In some cases, nephrolithiasis/urolithiasis has been associated with renal insufficiency or acute renal failure, pyelonephritis with or without bacteremia. If signs or symptoms of nephrolithiasis/urolithiasis occur, (including flank pain, with or without hematuria or microscopic hematuria), temporary interruption (e.g., 1-3 days) or discontinuation of therapy may be considered. Adequate hydration is recommended in all patients treated with CRIXIVAN. (See ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION, Nephrolithiasis/Urolithiasis.)
Hemolytic Anemia
Acute hemolytic anemia, including cases resulting in death, has been reported in patients treated with CRIXIVAN. Once a diagnosis is apparent, appropriate measures for the treatment of hemolytic anemia should be instituted, including discontinuation of CRIXIVAN.
Hepatitis
Hepatitis including cases resulting in hepatic failure and death has been reported in patients treated with CRIXIVAN. Because the majority of these patients had confounding medical conditions and/or were receiving concomitant therapy(ies), a causal relationship between CRIXIVAN and these events has not been established.
Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus and hyperglycemia have been reported during post-marketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
Drug Interactions
Concomitant use of CRIXIVAN with lovastatin, simvastatin, or rosuvastatin is not recommended. Caution should be exercised if HIV protease inhibitors, including CRIXIVAN, are used concurrently with atorvastatin. The interaction of CRIXIVAN with pravastatin and fluvastatin is not known. The risk of myopathy including rhabdomyolysis may be increased when HIV protease inhibitors, including CRIXIVAN, are used in combination with these statin drugs (see PRECAUTIONS, Drug Interactions).
Midazolam is extensively metabolized by CYP3A4. Co-administration with CRIXIVAN with or without ritonavir may cause a large increase in the concentration of this benzodiazepine. No drug interaction study has been performed for the co-administration of CRIXIVAN with benzodiazepines. Based on data from other CYP3A4 inhibitors, plasma concentrations of midazolam are expected to be significantly higher when midazolam is given orally. Therefore CRIXIVAN should not be co-administered with orally administered midazolam (see CONTRAINDICATIONS), whereas caution should be used with co-administration of CRIXIVAN and parenteral midazolam. Data from concomitant use of parenteral midazolam with other protease inhibitors suggest a possible 3-4 fold increase in midazolam plasma levels. If CRIXIVAN with or without ritonavir is co-administered with parenteral midazolam, it should be done in a setting which ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dosage reduction for midazolam should be considered, especially if more than a single dose of midazolam is administered.
Particular caution should be used when prescribing sildenafil, tadalafil, or vardenafil in patients receiving indinavir. Coadministration of CRIXIVAN with these medications is expected to substantially increase plasma concentrations of sildenafil, tadalafil, and vardenafil and may result in an increase in adverse events, including hypotension, visual changes, and priapism, which have been associated with sildenafil, tadalafil, and vardenafil (see CONTRAINDICATIONS and PRECAUTIONS, Drug Interactions and Information for Patients, and the manufacturer’s complete prescribing information for sildenafil, tadalafil, or vardenafil).
Concomitant use of CRIXIVAN and St. John’s wort (Hypericum perforatum) or products containing St. John’s wort is not recommended. Coadministration of CRIXIVAN and St. John’s wort has been shown to substantially decrease indinavir concentrations (see CLINICAL PHARMACOLOGY, Drug Interactions) and may lead to loss of virologic response and possible resistance to CRIXIVAN or to the class of protease inhibitors.
PRECAUTIONS
General
Indirect hyperbilirubinemia has occurred frequently during treatment with CRIXIVAN and has infrequently been associated with increases in serum transaminases (see also ADVERSE REACTIONS, Clinical Trials and Post-Marketing Experience). It is not known whether CRIXIVAN will exacerbate the physiologic hyperbilirubinemia seen in neonates. (See Pregnancy.)
Tubulointerstitial Nephritis
Reports of tubulointerstitial nephritis with medullary calcification and cortical atrophy have been observed in patients with asymptomatic severe leukocyturia (>100 cells/ high power field). Patients with asymptomatic severe leukocyturia should be followed closely and monitored frequently with urinalyses. Further diagnostic evaluation may be warranted, and discontinuation of CRIXIVAN should be considered in all patients with severe leukocyturia.
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy (CART), including CRIXIVAN. During the initial phase of treatment, patients responding to antiretroviral therapy whose immune system responds to CART may develop an inflammatory response to indolent or residual opportunistic infections (such as MAI, CMV, PCP, or TB), which may necessitate further evaluation and treatment.
Coexisting Conditions
Patients with hemophilia: There have been reports of spontaneous bleeding in patients with hemophilia A and B treated with protease inhibitors. In some patients, additional factor VIII was required. In many of the reported cases, treatment with protease inhibitors was continued or restarted. A causal relationship between protease inhibitor therapy and these episodes has not been established. (See ADVERSE REACTIONS, Post-Marketing Experience.)
Patients with hepatic insufficiency due to cirrhosis: In these patients, the dosage of CRIXIVAN should be lowered because of decreased metabolism of CRIXIVAN (see DOSAGE AND ADMINISTRATION).
Patients with renal insufficiency: Patients with renal insufficiency have not been studied.
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance” have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Information for Patients
A statement to patients and health care providers is included on the product’s bottle label. ALERT: Find out about medicines that should NOT be taken with CRIXIVAN. A Patient Package Insert (PPI) for CRIXIVAN is available for patient information.
CRIXIVAN is not a cure for HIV infection and patients may continue to develop opportunistic infections and other complications associated with HIV disease. The long-term effects of CRIXIVAN are unknown at this time. CRIXIVAN has not been shown to reduce the risk of transmission of HIV to others through sexual contact or blood contamination.
Patients should be advised to remain under the care of a physician when using CRIXIVAN and should not modify or discontinue treatment without first consulting the physician. Therefore, if a dose is missed, patients should take the next dose at the regularly scheduled time and should not double this dose. Therapy with CRIXIVAN should be initiated and maintained at the recommended dosage.
CRIXIVAN may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John’s wort.
For optimal absorption, CRIXIVAN should be administered without food but with water 1 hour before or 2 hours after a meal. Alternatively, CRIXIVAN may be administered with other liquids such as skim milk, juice, coffee, or tea, or with a light meal, e.g., dry toast with jelly, juice, and coffee with skim milk and sugar; or corn flakes, skim milk and sugar (see CLINICAL PHARMACOLOGY, Effect of Food on Oral Absorption and DOSAGE AND ADMINISTRATION). Ingestion of CRIXIVAN with a meal high in calories, fat, and protein reduces the absorption of indinavir.
Patients receiving a phosphodiesterase type 5 (PDE5) inhibitor (sildenafil, tadalafil, or vardenafil) should be advised that they may be at an increased risk of PDE5 inhibitor-associated adverse events including hypotension, visual changes, and priapism, and should promptly report any symptoms to their doctors ( see CONTRAINDICATIONS and WARNINGS, Drug Interactions).
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time.
CRIXIVAN Capsules are sensitive to moisture. Patients should be informed that CRIXIVAN should be stored and used in the original container and the desiccant should remain in the bottle.
Drug Interactions
Indinavir is an inhibitor of the cytochrome P450 isoform CYP3A4. Coadministration of CRIXIVAN and drugs primarily metabolized by CYP3A4 may result in increased plasma concentrations of the other drug, which could increase or prolong its therapeutic and adverse effects (see CONTRAINDICATIONS and WARNINGS).
Indinavir is metabolized by CYP3A4. Drugs that induce CYP3A4 activity would be expected to increase the clearance of indinavir, resulting in lowered plasma concentrations of indinavir. Coadministration of CRIXIVAN and other drugs that inhibit CYP3A4 may decrease the clearance of indinavir and may result in increased plasma concentrations of indinavir.
| Drug Class: Drug Name | Clinical Comment |
|
|
| Alpha 1-adrenoreceptor antagonist: alfuzosin | Potentially increased alfuzosin concentrations can result in hypotension. |
| Antiarrhythmics: amiodarone | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| Antimycobacterial: rifampin | May lead to loss of virologic response and possible resistance to CRIXIVAN or to the class of protease inhibitors or other coadministered antiretroviral agents. |
| Ergot derivatives: dihydroergotamine, ergonovine, ergotamine, methylergonovine | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| GI motility agents: cisapride | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| Herbal products: St. John's wort (Hypericum perforatum) | May lead to loss of virologic response and possible resistance to CRIXIVAN or to the class of protease inhibitors. |
| HMG-CoA Reductase inhibitors: lovastatin, simvastatin, rosuvastatin | Potential for serious reactions such as risk of myopathy including rhabdomyolysis. |
| Neuroleptic: pimozide | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| PDE5 inhibitor: Revatio* (sildenafil) [for treatment of pulmonary arterial hypertension] | A safe and effective dose has not been established when used with CRIXIVAN. There is increased potential for sildenafil-associated adverse events (which include visual disturbances, hypotension, prolonged erection, and syncope). |
| Protease inhibitor: atazanavir | Both CRIXIVAN and atazanavir are associated with indirect (unconjugated) hyperbilirubinemia. Combinations of these drugs have not been studied and coadministration of CRIXIVAN and atazanavir is not recommended. |
| Sedative/hypnotics: Oral midazolam, triazolam, alprazolam | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as prolonged or increased sedation or respiratory depression. |
| Drug Name | Effect | Clinical Comment |
| HIV Antiviral Agents | ||
| Note: ↑= increase; ↓ = decrease | ||
| Delavirdine | ↑ indinavir concentration | Dose reduction of CRIXIVAN to 600 mg every 8 hours should be considered when taking delavirdine 400 mg three times a day. |
| Didanosine | Indinavir and didanosine formulations containing buffer should be administered at least one hour apart on an empty stomach. | |
| Efavirenz | ↓ indinavir concentration | The optimal dose of indinavir, when given in combination with efavirenz, is not known. Increasing the indinavir dose to 1000 mg every 8 hours does not compensate for the increased indinavir metabolism due to efavirenz. |
| Nelfinavir | ↑ indinavir concentration | The appropriate doses for this combination, with respect to efficacy and safety, have not been established. |
| Nevirapine | ↓ indinavir concentration | Indinavir concentrations may be decreased in the presence of nevirapine. The appropriate doses for this combination, with respect to efficacy and safety, have not been established. |
| Ritonavir | ↑ indinavir concentration ↑ ritonavir concentration | The appropriate doses for this combination, with respect to efficacy and safety, have not been established. Preliminary clinical data suggest that the incidence of nephrolithiasis is higher in patients receiving indinavir in combination with ritonavir than those receiving CRIXIVAN 800 mg q8h. |
| Saquinavir | ↑ saquinavir concentration | The appropriate doses for this combination, with respect to efficacy and safety, have not been established. |
| Other Agents | ||
| Antiarrhythmics: bepridil, lidocaine(systemic) and quinidine | ↑ antiarrhythmic agents concentration | Caution is warranted and therapeutic concentration monitoring is recommended for antiarrhythmics when coadministered with CRIXIVAN. |
| Anticonvulsants: carbamazepine, phenobarbital, phenytoin | ↓ indinavir concentration | Use with caution. CRIXIVAN may not be effective due to decreased indinavir concentrations in patients taking these agents concomitantly. |
| Antidepressant: Trazodone | ↑ trazodone concentration | Concomitant use of trazodone and CRIXIVAN may increase plasma concentrations of trazodone. Adverse events of nausea, dizziness, hypotension and syncope have been observed following coadministration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as CRIXIVAN, the combination should be used with caution and a lower dose of trazodone should be considered. |
| Anti-gout: Colchicine | ↑ colchicine concentration | Patients with renal or hepatic impairment should not be given colchicine with CRIXIVAN. Treatment of gout flares: Co-administration of colchicine in patients on CRIXIVAN: 0.6 mg (1 tablet) x 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days. Prophylaxis of gout flares: Co-administration of colchicine in patients on CRIXIVAN: If the original colchicine regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day. If the original colchicine regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day. Treatment of familial Mediterranean fever (FMF):Co-administration of colchicine in patients on CRIXIVAN: Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day). |
| Calcium Channel Blockers, Dihydropyridine: e.g., felodipine, nifedipine, nicardipine | ↑ dihydropyridine calcium channel blockers concentration | Caution is warranted and clinical monitoring of patients is recommended. |
| Clarithromycin | ↑ clarithromycin concentration ↑ indinavir concentration | The appropriate doses for this combination, with respect to efficacy and safety, have not been established. |
| Endothelin receptor antagonist: Bosentan | ↑ bosentan concentration | Co-administration of bosentan in patients on CRIXIVAN or co-administration of CRIXIVAN in patients on bosentan: Start at or adjust bosentan to 62.5 mg once daily or every other day based upon individual tolerability. |
| HMG-CoA Reductase Inhibitors: atorvastatin, pravastatin, fluvastatin | ↑ atorvastatin concentration pravastatin, fluvastatin-interaction not studied | Use the lowest possible dose of atorvastatin with careful monitoring. If no alternative treatment is available, use with careful monitoring. |
| Immunosuppressants: cyclosporine, tacrolimus, sirolimus | ↑ immunosuppressant agents concentration | Plasma concentrations may be increased by CRIXIVAN. |
| Inhaled beta agonist: Salmeterol | ↑ salmeterol | Concurrent administration of salmeterol with CRIXIVAN is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia. |
| Inhaled/nasal steroid: Fluticasone | ↑ fluticasone concentration | Concomitant use of fluticasone propionate and CRIXIVAN may increase plasma concentrations of fluticasone propionate. Use with caution. Consider alternatives to fluticasone propionate, particularly for long-term use. Fluticasone use is not recommended in situations where CRIXIVAN is coadministered with a potent CYP3A4 inhibitor such as ritonavir unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effects. |
| Itraconazole | ↑ indinavir concentration | Dose reduction of CRIXIVAN to 600 mg every 8 hours is recommended when administering itraconazole concurrently. |
| Ketoconazole | ↑ indinavir concentration | Dose reduction of CRIXIVAN to 600 mg every 8 hours should be considered. |
| Midazolam (parenteral administration) | ↑ midazolam concentration | Concomitant use of parenteral midazolam with CRIXIVAN may increase plasma concentrations of midazolam. Coadministration should be done in a setting which ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dosage reduction for midazolam should be considered, especially if more than a single dose of midazolam is administered. Coadministration of oral midazolam with CRIXIVAN is CONTRAINDICATED (see Table 8). |
| Rifabutin | ↓ indinavir concentration ↑ rifabutin concentration | Dose reduction of rifabutin to half the standard dose and a dose increase of CRIXIVAN to 1000 mg every 8 hours are recommended when rifabutin and CRIXIVAN are coadministered. |
| Sildenafil | ↑ sildenafil concentration (only the use of sildenafil at doses used for treatment of erectile dysfunction has been studied with CRIXIVAN) | May result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, syncope, visual disturbances, and priapism. Use of sildenafil for pulmonary arterial hypertension (PAH): Use of Revatio* (sildenafil) is contraindicated when used for the treatment of pulmonary arterial hypertension (PAH) [see CONTRAINDICATIONS]. Use of sildenafil for erectile dysfunction: Sildenafil dose should not exceed a maximum of 25 mg in a 48-hour period in patients receiving concomitant CRIXIVAN therapy. Use with increased monitoring for adverse events. |
| Tadalafil | ↑ tadalafil concentration | May result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, visual disturbances, and priapism. Use of tadalafil for pulmonary arterial hypertension (PAH): The following dose adjustments are recommended for use of Adcirca† (tadalafil) with CRIXIVAN: Co-administration of Adcirca in patients on CRIXIVAN or co-administration of CRIXIVAN in patients on Adcirca: Start at or adjust Adcirca to 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Use of tadalafil for erectile dysfunction: Tadalafil dose should not exceed a maximum of 10 mg in a 72-hour period in patients receiving concomitant CRIXIVAN therapy. Use with increased monitoring for adverse events. |
| Vardenafil | ↑ vardenafil concentration | Vardenafil dose should not exceed a maximum of 2.5 mg in a 24-hour period in patients receiving concomitant indinavir therapy. |
| Venlafaxine | ↓ indinavir concentration | In a study of 9 healthy volunteers, venlafaxine administered under steady-state conditions at 150 mg/day resulted in a 28% decrease in the AUC of a single 800 mg oral dose of indinavir and a 36% decrease in indinavir Cmax. Indinavir did not affect the pharmacokinetics of venlafaxine and ODV. The clinical significance of this finding is unknown. |
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were conducted in mice and rats. In mice, no increased incidence of any tumor type was observed. The highest dose tested in rats was 640 mg/kg/day; at this dose a statistically significant increased incidence of thyroid adenomas was seen only in male rats. At that dose, daily systemic exposure in rats was approximately 1.3 times higher than daily systemic exposure in humans. No evidence of mutagenicity or genotoxicity was observed in in vitro microbial mutagenesis (Ames) tests, in vitro alkaline elution assays for DNA breakage, in vitro and in vivo chromosomal aberration studies, and in vitro mammalian cell mutagenesis assays. No treatment-related effects on mating, fertility, or embryo survival were seen in female rats and no treatment-related effects on mating performance were seen in male rats at doses providing systemic exposure comparable to or slightly higher than that with the clinical dose. In addition, no treatment-related effects were observed in fecundity or fertility of untreated females mated to treated males.
Pregnancy
Pregnancy Category C:
Developmental toxicity studies were performed in rabbits (at doses up to 240 mg/kg/day), dogs (at doses up to 80 mg/kg/day), and rats (at doses up to 640 mg/kg/day). The highest doses in these studies produced systemic exposures in these species comparable to or slightly greater than human exposure. No treatment-related external, visceral, or skeletal changes were observed in rabbits or dogs. No treatment-related external or visceral changes were observed in rats. Treatment-related increases over controls in the incidence of supernumerary ribs (at exposures at or below those in humans) and of cervical ribs (at exposures comparable to or slightly greater than those in humans) were seen in rats. In all three species, no treatment-related effects on embryonic/fetal survival or fetal weights were observed.
In rabbits, at a maternal dose of 240 mg/kg/day, no drug was detected in fetal plasma 1 hour after dosing. Fetal plasma drug levels 2 hours after dosing were approximately 3% of maternal plasma drug levels. In dogs, at a maternal dose of 80 mg/kg/day, fetal plasma drug levels were approximately 50% of maternal plasma drug levels both 1 and 2 hours after dosing. In rats, at maternal doses of 40 and 640 mg/kg/day, fetal plasma drug levels were approximately 10 to 15% and 10 to 20% of maternal plasma drug levels 1 and 2 hours after dosing, respectively.
Indinavir was administered to Rhesus monkeys during the third trimester of pregnancy (at doses up to 160 mg/kg twice daily) and to neonatal Rhesus monkeys (at doses up to 160 mg/kg twice daily). When administered to neonates, indinavir caused an exacerbation of the transient physiologic hyperbilirubinemia seen in this species after birth; serum bilirubin values were approximately fourfold above controls at 160 mg/kg twice daily. A similar exacerbation did not occur in neonates after in utero exposure to indinavir during the third trimester of pregnancy. In Rhesus monkeys, fetal plasma drug levels were approximately 1 to 2% of maternal plasma drug levels approximately 1 hour after maternal dosing at 40, 80, or 160 mg/kg twice daily.
Hyperbilirubinemia has occurred during treatment with CRIXIVAN (see PRECAUTIONS and ADVERSE REACTIONS). It is unknown whether CRIXIVAN administered to the mother in the perinatal period will exacerbate physiologic hyperbilirubinemia in neonates.
There are no adequate and well-controlled studies in pregnant patients. CRIXIVAN should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
A CRIXIVAN dose of 800 mg every 8 hours (with zidovudine 200 mg every 8 hours and lamivudine 150 mg twice a day) has been studied in 16 HIV-infected pregnant patients at 14 to 28 weeks of gestation at enrollment (study PACTG 358). Given the substantially lower antepartum exposures observed and the limited data in this patient population, indinavir use is not recommended in HIV-infected pregnant patients (see CLINICAL PHARMACOLOGY, Pregnant Patients).
Nursing Mothers
Studies in lactating rats have demonstrated that indinavir is excreted in milk. Although it is not known whether CRIXIVAN is excreted in human milk, there exists the potential for adverse effects from indinavir in nursing infants. Mothers should be instructed to discontinue nursing if they are receiving CRIXIVAN. This is consistent with the recommendation by the U.S. Public Health Service Centers for Disease Control and Prevention that HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV.
Pediatric Use
The optimal dosing regimen for use of indinavir in pediatric patients has not been established. A dose of 500 mg/m2 every eight hours has been studied in uncontrolled studies of 70 children, 3 to 18 years of age. The pharmacokinetic profiles of indinavir at this dose were not comparable to profiles previously observed in adults receiving the recommended dose (see CLINICAL PHARMACOLOGY, Pediatric). Although viral suppression was observed in some of the 32 children who were followed on this regimen through 24 weeks, a substantially higher rate of nephrolithiasis was reported when compared to adult historical data (see WARNINGS, Nephrolithiasis/Urolithiasis). Physicians considering the use of indinavir in pediatric patients without other protease inhibitor options should be aware of the limited data available in this population and the increased risk of nephrolithiasis.
Geriatric Use
Clinical studies of CRIXIVAN did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal or cardiac function and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
Clinical Trials in Adults
Nephrolithiasis/urolithiasis, including flank pain with or without hematuria (including microscopic hematuria), has been reported in approximately 12.4% (301/2429; range across individual trials: 4.7% to 34.4%) of patients receiving CRIXIVAN at the recommended dose in clinical trials with a median follow-up of 47 weeks (range: 1 day to 242 weeks; 2238 patient-years follow-up). The cumulative frequency of nephrolithiasis events increases with duration of exposure to CRIXIVAN; however, the risk over time remains relatively constant. Of the patients treated with CRIXIVAN who developed nephrolithiasis/urolithiasis in clinical trials during the double-blind phase, 2.8% (7/246) were reported to develop hydronephrosis and 4.5% (11/246) underwent stent placement. Following the acute episode, 4.9% (12/246) of patients discontinued therapy. (See WARNINGS and DOSAGE AND ADMINISTRATION, Nephrolithiasis/Urolithiasis.)
Asymptomatic hyperbilirubinemia (total bilirubin ≥2.5 mg/dL), reported predominantly as elevated indirect bilirubin, has occurred in approximately 14% of patients treated with CRIXIVAN. In <1% this was associated with elevations in ALT or AST.
Hyperbilirubinemia and nephrolithiasis/urolithiasis occurred more frequently at doses exceeding 2.4 g/day compared to doses ≤2.4 g/day.
Clinical adverse experiences reported in ≥2% of patients treated with CRIXIVAN alone, CRIXIVAN in combination with zidovudine or zidovudine plus lamivudine, zidovudine alone, or zidovudine plus lamivudine are presented in Table 10.
| Study 028 Considered Drug-Related and of Moderate or Severe Intensity | Study ACTG 320 of Unknown Drug Relationship and of Severe or Life-threatening Intensity |
|||||
| CRIXIVAN | CRIXIVAN plus Zidovudine | Zidovudine | CRIXIVAN plus Zidovudine plus Lamivudine | Zidovudine plus Lamivudine |
||
| Adverse Experience | Percent (n=332) | Percent (n=332) | Percent (n=332) | Percent (n=571) | Percent (n=575) |
|
|
||||||
| Body as a Whole | ||||||
| Abdominal pain | 16.6 | 16.0 | 12.0 | 1.9 | 0.7 | |
| Asthenia/fatigue | 2.1 | 4.2 | 3.6 | 2.4 | 4.5 | |
| Fever | 1.5 | 1.5 | 2.1 | 3.8 | 3.0 | |
| Malaise | 2.1 | 2.7 | 1.8 | 0 | 0 | |
| Digestive System | ||||||
| Nausea | 11.7 | 31.9 | 19.6 | 2.8 | 1.4 | |
| Diarrhea | 3.3 | 3.0 | 2.4 | 0.9 | 1.2 | |
| Vomiting | 8.4 | 17.8 | 9.0 | 1.4 | 1.4 | |
| Acid regurgitation | 2.7 | 5.4 | 1.8 | 0.4 | 0 | |
| Anorexia | 2.7 | 5.4 | 3.0 | 0.5 | 0.2 | |
| Appetite increase | 2.1 | 1.5 | 1.2 | 0 | 0 | |
| Dyspepsia | 1.5 | 2.7 | 0.9 | 0 | 0 | |
| Jaundice | 1.5 | 2.1 | 0.3 | 0 | 0 | |
| Hemic and Lymphatic System | ||||||
| Anemia | 0.6 | 1.2 | 2.1 | 2.4 | 3.5 | |
| Musculoskeletal System | ||||||
| Back pain | 8.4 | 4.5 | 1.5 | 0.9 | 0.7 | |
| Nervous System/Psychiatric | ||||||
| Headache | 5.4 | 9.6 | 6.0 | 2.4 | 2.8 | |
| Dizziness | 3.0 | 3.9 | 0.9 | 0.5 | 0.7 | |
| Somnolence | 2.4 | 3.3 | 3.3 | 0 | 0 | |
| Skin and Skin Appendage | ||||||
| Pruritus | 4.2 | 2.4 | 1.8 | 0.5 | 0 | |
| Rash | 1.2 | 0.6 | 2.4 | 1.1 | 0.5 | |
| Respiratory System | ||||||
| Cough | 1.5 | 0.3 | 0.6 | 1.6 | 1.0 | |
| Difficulty breathing/ dyspnea/ shortness of breath | 0 | 0.6 | 0.3 | 1.8 | 1.0 | |
| Urogenital System | ||||||
| Nephrolithiasis/urolithiasis* | 8.7 | 7.8 | 2.1 | 2.6 | 0.3 | |
| Dysuria | 1.5 | 2.4 | 0.3 | 0.4 | 0.2 | |
| Special Senses | ||||||
| Taste perversion | 2.7 | 8.4 | 1.2 | 0.2 | 0 | |
In Phase I and II controlled trials, the following adverse events were reported significantly more frequently by those randomized to the arms containing CRIXIVAN than by those randomized to nucleoside analogues: rash, upper respiratory infection, dry skin, pharyngitis, taste perversion.
Selected laboratory abnormalities of severe or life-threatening intensity reported in patients treated with CRIXIVAN alone, CRIXIVAN in combination with zidovudine or zidovudine plus lamivudine, zidovudine alone, or zidovudine plus lamivudine are presented in Table 11.
| Study 028 | Study ACTG 320 | ||||
| CRIXIVAN | CRIXIVAN plus Zidovudine | Zidovudine | CRIXIVAN plus Zidovudine plus Lamivudine | Zidovudine plus Lamivudine |
|
| Percent (n=329) | Percent (n=320) | Percent (n=330) | Percent (n=571) | Percent (n=575) |
|
|
|||||
| Hematology | |||||
| Decreased hemoglobin <7.0 g/dL | 0.6 | 0.9 | 3.3 | 2.4 | 3.5 |
| Decreased platelet count <50 THS/mm3 | 0.9 | 0.9 | 1.8 | 0.2 | 0.9 |
| Decreased neutrophils <0.75 THS/mm3 | 2.4 | 2.2 | 6.7 | 5.1 | 14.6 |
| Blood chemistry | |||||
| Increased ALT >500% ULN* | 4.9 | 4.1 | 3.0 | 2.6 | 2.6 |
| Increased AST >500% ULN | 3.7 | 2.8 | 2.7 | 3.3 | 2.8 |
| Total serum bilirubin >250% ULN | 11.9 | 9.7 | 0.6 | 6.1 | 1.4 |
| Increased serum amylase >200% ULN | 2.1 | 1.9 | 1.8 | 0.9 | 0.3 |
| Increased glucose >250 mg/dL | 0.9 | 0.9 | 0.6 | 1.6 | 1.9 |
| Increased creatinine >300% ULN | 0 | 0 | 0.6 | 0.2 | 0 |
Post-Marketing Experience
Body As A Whole: redistribution/accumulation of body fat (see PRECAUTIONS, Fat Redistribution).
Cardiovascular System: cardiovascular disorders including myocardial infarction and angina pectoris; cerebrovascular disorder.
Digestive System: liver function abnormalities; hepatitis including reports of hepatic failure (see WARNINGS); pancreatitis; jaundice; abdominal distention; dyspepsia.
Hematologic: increased spontaneous bleeding in patients with hemophilia (see PRECAUTIONS); acute hemolytic anemia (see WARNINGS).
Endocrine/Metabolic: new onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, hyperglycemia (see WARNINGS).
Hypersensitivity: anaphylactoid reactions; urticaria; vasculitis.
Musculoskeletal System: arthralgia.
Nervous System/Psychiatric: oral paresthesia; depression.
Skin and Skin Appendage: rash including erythema multiforme and Stevens-Johnson syndrome; hyperpigmentation; alopecia; ingrown toenails and/or paronychia; pruritus.
Urogenital System: nephrolithiasis/urolithiasis, in some cases resulting in renal insufficiency or acute renal failure, pyelonephritis with or without bacteremia (see WARNINGS); interstitial nephritis sometimes with indinavir crystal deposits; in some patients, the interstitial nephritis did not resolve following discontinuation of CRIXIVAN; renal insufficiency; renal failure; leukocyturia (see PRECAUTIONS), crystalluria; dysuria.
OVERDOSAGE
There have been more than 60 reports of acute or chronic human overdosage (up to 23 times the recommended total daily dose of 2400 mg) with CRIXIVAN. The most commonly reported symptoms were renal (e.g., nephrolithiasis/urolithiasis, flank pain, hematuria) and gastrointestinal (e.g., nausea, vomiting, diarrhea).
It is not known whether CRIXIVAN is dialyzable by peritoneal or hemodialysis.
DOSAGE AND ADMINISTRATION
The recommended dosage of CRIXIVAN is 800 mg (usually two 400-mg capsules) orally every 8 hours.
CRIXIVAN must be taken at intervals of 8 hours. For optimal absorption, CRIXIVAN should be administered without food but with water 1 hour before or 2 hours after a meal. Alternatively, CRIXIVAN may be administered with other liquids such as skim milk, juice, coffee, or tea, or with a light meal, e.g., dry toast with jelly, juice, and coffee with skim milk and sugar; or corn flakes, skim milk and sugar. (See CLINICAL PHARMACOLOGY, Effect of Food on Oral Absorption.)
To ensure adequate hydration, it is recommended that adults drink at least 1.5 liters (approximately 48 ounces) of liquids during the course of 24 hours.
Concomitant Therapy
(See CLINICAL PHARMACOLOGY, Drug Interactions, and/or PRECAUTIONS, Drug Interactions.)
Delavirdine
Dose reduction of CRIXIVAN to 600 mg every 8 hours should be considered when administering delavirdine 400 mg three times a day.
Didanosine
If indinavir and didanosine are administered concomitantly, they should be administered at least one hour apart on an empty stomach (consult the manufacturer's product circular for didanosine).
Itraconazole
Dose reduction of CRIXIVAN to 600 mg every 8 hours is recommended when administering itraconazole 200 mg twice daily concurrently.
HOW SUPPLIED
CRIXIVAN Capsules are supplied as follows:
No. 3758 — 400 mg capsules: semi-translucent white capsules coded "CRIXIVAN™ 400 mg" in green. Available as:
NDC 21695-366-18 unit-dose packages of 18
Storage
Bottles: Store in a tightly-closed container at room temperature, 15-30°C (59-86°F). Protect from moisture.
CRIXIVAN Capsules are sensitive to moisture. CRIXIVAN should be dispensed and stored in the original container. The desiccant should remain in the original bottle.
Unit-Dose Packages: Store at room temperature, 15-30°C (59-86°F). Protect from moisture.
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USA
U.S. Patent Nos.: 5,413,999; 6,645,961
Issued April 2010
Printed in USA
9640612
Repackaged by:
Rebel Distributors Corp
Thousand Oaks, CA 91320
PATIENT PACKAGE INSERT
CRIXIVAN®2 (indinavir sulfate) Capsules
Patient Information about
CRIXIVAN (KRIK-sih-van)
for HIV (Human Immunodeficiency Virus) Infection
Generic name: indinavir (in-DIH-nuh-veer) sulfate
ALERT: Find out about medicines that should NOT be taken with CRIXIVAN. Please also read the section “MEDICINES YOU SHOULD NOT TAKE WITH CRIXIVAN”.
Please read this information before you start taking CRIXIVAN. Also, read the leaflet each time you renew your prescription, just in case anything has changed. Remember, this leaflet does not take the place of careful discussions with your doctor. You and your doctor should discuss CRIXIVAN when you start taking your medication and at regular checkups. You should remain under a doctor’s care when using CRIXIVAN and should not change or stop treatment without first talking with your doctor.
- 2
-
Registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Copyright © 1996, 1999 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved
What is CRIXIVAN?
CRIXIVAN is an oral capsule used for the treatment of HIV (Human Immunodeficiency Virus). HIV is the virus that causes AIDS (acquired immune deficiency syndrome). CRIXIVAN is a type of HIV drug called a protease (PRO-tee-ase) inhibitor.
How does CRIXIVAN work?
CRIXIVAN is a protease inhibitor that fights HIV. CRIXIVAN can help reduce your chances of getting illnesses associated with HIV. CRIXIVAN can also help lower the amount of HIV in your body (called “viral load”) and raise your CD4 (T) cell count. CRIXIVAN may not have these effects in all patients.
CRIXIVAN is usually prescribed with other anti-HIV drugs such as ZDV (also called AZT), 3TC, ddI, ddC, or d4T. CRIXIVAN works differently from these other anti-HIV drugs. Talk with your doctor about how you should take CRIXIVAN.
How should I take CRIXIVAN?
There are six important things you must do to help you benefit from CRIXIVAN:
-
Take CRIXIVAN capsules every day as prescribed by your doctor. Continue taking CRIXIVAN unless your doctor tells you to stop. Take the exact amount of CRIXIVAN that your doctor tells you to take, right from the very start. To help make sure you will benefit from CRIXIVAN, you must not skip doses or take “drug holidays”. If you don’t take CRIXIVAN as prescribed, the activity of CRIXIVAN may be reduced (due to resistance).
-
Take CRIXIVAN capsules every 8 hours around the clock, every day. It may be easier to remember to take CRIXIVAN if you take it at the same time every day. If you have questions about when to take CRIXIVAN, your doctor or health care provider can help you decide what schedule works for you.
-
If you miss a dose by more than 2 hours, wait and then take the next dose at the regularly scheduled time. However, if you miss a dose by less than 2 hours, take your missed dose immediately. Then take your next dose at the regularly scheduled time. Do not take more or less than your prescribed dose of CRIXIVAN at any one time.
-
Take CRIXIVAN with water. You can also take CRIXIVAN with other beverages such as skim or non-fat milk, juice, coffee, or tea.
-
Ideally, take each dose of CRIXIVAN without food but with water at least one hour before or two hours after a meal. Or you can take CRIXIVAN with a light meal. Examples of light meals include:
dry toast with jelly, juice, and coffee (with skim or non-fat milk and sugar if you want)
cornflakes with skim or non-fat milk and sugar
Do not take CRIXIVAN at the same time as any meals that are high in calories, fat, and protein (for example — a bacon and egg breakfast). When taken at the same time as CRIXIVAN, these foods can interfere with CRIXIVAN being absorbed into your bloodstream and may lessen its effect.
- It is critical to drink plenty of fluids while taking CRIXIVAN. Adults should drink at least six 8-ounce glasses of liquids (preferably water) throughout the day, every day. Your health care provider will give you further instructions on the amount of fluid that you should drink. CRIXIVAN can cause kidney stones. Having enough fluids in your body should help reduce the chances of forming a kidney stone. Call your doctor or other health care provider if you develop kidney pains (middle to lower stomach or back pain) or blood in the urine.
Does CRIXIVAN cure HIV or AIDS?
CRIXIVAN is not a cure for HIV or AIDS. People taking CRIXIVAN may still develop infections or other conditions associated with HIV. Because of this, it is very important for you to remain under the care of a doctor. Although CRIXIVAN is not a cure for HIV or AIDS, CRIXIVAN can help reduce your chances of getting illnesses, including death, associated with HIV. CRIXIVAN may not have these effects in all patients.
Does CRIXIVAN reduce the risk of passing HIV to others?
CRIXIVAN has not been shown to reduce the risk of passing HIV to others through sexual contact or blood contamination.
Who should not take CRIXIVAN?
Do not take CRIXIVAN if you have had a serious allergic reaction to CRIXIVAN or any of its components.
What other medical problems or conditions should I discuss with my doctor?
Talk to your doctor if:
- You are pregnant or if you become pregnant while you are taking CRIXIVAN. We do not yet know how CRIXIVAN affects pregnant women or their developing babies.
- You are breast-feeding. You should stop breast-feeding if you are taking CRIXIVAN.
Also talk to your doctor if you have:
- Problems with your liver, especially if you have mild or moderate liver disease caused by cirrhosis
- Problems with your kidneys
- Diabetes
- Hemophilia
- High cholesterol and you are taking cholesterol-lowering medicines called “statins”.
Tell your doctor about any medicines you are taking or plan to take, including non-prescription medicines, herbal products including St. John’s wort (Hypericum perforatum ), or dietary supplements.
Can CRIXIVAN be taken with other medications?3
MEDICINES YOU SHOULD NOT TAKE WITH CRIXIVAN
| Oral VERSED® (midazolam) | HALCION® (triazolam) |
| ORAP® (pimozide) | XANAX® (alprazolam) |
| PROPULSID® (cisapride) | REVATIO® (sildenafil for the treatment of pulmonary arterial hypertension) |
| CORDARONE® (amiodarone) | UROXATRAL® (alfuzosin) |
| HISMANAL® (astemizole) | Ergot medications (e.g., Wigraine®, Cafergot®, D.H.E. 45®, Migranal®, Ergotrate®, and Methergine®) |
Taking CRIXIVAN with the above medications could result in serious or life-threatening problems (such as irregular heartbeat or excessive sleepiness).
In addition, you should not take CRIXIVAN with the following:
- Rifampin, known as RIFADIN®, RIFAMATE®, RIFATER®, or RIMACTANE®.
- It is not recommended to take CRIXIVAN with the cholesterol-lowering drugs MEVACOR®4 (lovastatin), ZOCOR®5 (simvastatin), or CRESTOR® (rosuvastatin) because of possible drug interactions. There is also an increased risk of drug interactions between CRIXIVAN and LIPITOR® (atorvastatin); talk to your doctor before you take any of these cholesterol-reducing drugs with CRIXIVAN.
- Taking CRIXIVAN with REYATAZ® (atazanavir) is not recommended because they can both sometimes cause increased levels of bilirubin in the blood.
- Taking CRIXIVAN with St. John’s wort (Hypericum perforatum), an herbal product sold as a dietary supplement, or products containing St. John’s wort is not recommended. Taking St. John’s wort has been shown to decrease CRIXIVAN levels and may lead to increased viral load and possible resistance to CRIXIVAN or cross resistance to other antiretroviral drugs.
Before you take VIAGRA® (sildenafil), CIALIS® (tadalafil), or LEVITRA® (vardenafil) with CRIXIVAN, talk to your doctor about possible drug interactions and side effects. If you take any of these medicines together with CRIXIVAN, you may be at increased risk of side effects such as low blood pressure, visual changes, and penile erection lasting more than 4 hours, which have been associated with sildenafil, tadalafil, and vardenafil. If an erection lasts longer than 4 hours, you should seek immediate medical assistance to avoid permanent damage to your penis. Your doctor can explain these symptoms to you.
MEDICINES YOU CAN TAKE WITH CRIXIVAN
| RETROVIR® (zidovudine, ZDV also called AZT) | EPIVIR™ (lamivudine, 3TC) |
| ZERIT® (stavudine, d4T) | isoniazid (INH) |
| BACTRIM®/SEPTRA® (trimethoprim/sulfamethoxazole) | DIFLUCAN® (fluconazole) |
| BIAXIN® (clarithromycin) | ORTHO-NOVUM 1/35® (oral contraceptive) |
| TAGAMET® (cimetidine) | Methadone |
|
VIDEX® (didanosine, ddI) — If you take CRIXIVAN with VIDEX, take them at least one hour apart. |
|
|
MYCOBUTIN® (rifabutin) — If you take CRIXIVAN with MYCOBUTIN, your doctor may adjust both the dose of MYCOBUTIN and the dose of CRIXIVAN. |
|
|
NIZORAL® (ketoconazole) — If you take CRIXIVAN with NIZORAL, your doctor may adjust the dose of CRIXIVAN. |
|
|
RESCRIPTOR® (delavirdine) — If you take CRIXIVAN with RESCRIPTOR, your doctor may adjust the dose of CRIXIVAN. |
|
|
SPORANOX® (itraconazole) — If you take CRIXIVAN with SPORANOX, your doctor may adjust the dose of CRIXIVAN. |
|
|
SUSTIVA™ (efavirenz) — If you take CRIXIVAN with SUSTIVA, check with your doctor. |
|
| Intravenous VERSED® (midazolam) — If you take CRIXIVAN with Intravenous VERSED®, your doctor may adjust the dose of VERSED®. | |
Talk to your doctor about any medications you are taking.
Calcium Channel Blockers: Tell your doctor if you are taking calcium channel blockers (e.g., amlodipine, felodipine).
Antiarrhythmics: Tell your doctor if you are taking antiarrhythmics (e.g., quinidine).
Anticonvulsants: Tell your doctor if you are taking anticonvulsants (e.g., phenobarbital, phenytoin, or carbamazepine).
Steroids: Tell your doctor if you are taking steroids (e.g., dexamethasone).
- 3
-
The brands listed are the registered trademarks of their respective owners and are not trademarks of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
- 4
-
Registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Copyright © 1996, 1999 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved - 5
-
Registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Copyright © 1996, 1999 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved
What are the possible side effects of CRIXIVAN?
Like all prescription drugs, CRIXIVAN can cause side effects. The following is not a complete list of side effects reported with CRIXIVAN when taken either alone or with other anti-HIV drugs. Do not rely on this leaflet alone for information about side effects. Your doctor can discuss with you a more complete list of side effects.
Some patients treated with CRIXIVAN developed kidney stones. In some of these patients this led to more severe kidney problems, including kidney failure or inflammation of the kidneys or kidney infection which sometimes spread to the blood. Drinking at least six 8-ounce glasses of liquids (preferably water) each day should help reduce the chances of forming a kidney stone (see How should I take CRIXIVAN?). Call your doctor or other health care provider if you develop kidney pains (middle to lower stomach or back pain) or blood in the urine.
Some patients treated with CRIXIVAN have had rapid breakdown of red blood cells (hemolytic anemia) which in some cases was severe or resulted in death.
Some patients treated with CRIXIVAN have had liver problems including liver failure and death. Some patients had other illnesses or were taking other drugs. It is uncertain if CRIXIVAN caused these liver problems.
Diabetes and high blood sugar (hyperglycemia) have occurred in patients taking protease inhibitors. In some of these patients, this led to ketoacidosis, a serious condition caused by poorly controlled blood sugar. Some patients had diabetes before starting protease inhibitors, others did not. Some patients required adjustments to their diabetes medication. Others needed new diabetes medication.
In some patients with hemophilia, increased bleeding has been reported.
Severe muscle pain and weakness have occurred in patients taking protease inhibitors, including CRIXIVAN, together with some of the cholesterol-lowering medicines called “statins”. Call your doctor if you develop severe muscle pain or weakness.
Changes in body fat have been seen in some patients taking antiretroviral therapy. These changes may include increased amount of fat in the upper back and neck (“buffalo hump”), breast, and around the trunk. Loss of fat from the legs, arms and face may also happen. The cause and long term health effects of these conditions are not known at this time.
In some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from opportunistic infections may occur when combination antiretroviral treatment is started.
Clinical Studies
Increases in bilirubin (one laboratory test of liver function) have been reported in approximately 14% of patients. Usually, this finding has not been associated with liver problems. However, on rare occasions, a person may develop yellowing of the skin and/or eyes.
Side effects occurring in 2% or more of patients included: abdominal pain, fatigue or weakness, low red blood cell count, flank pain, painful urination, feeling unwell, nausea, upset stomach, diarrhea, vomiting, acid regurgitation, increased or decreased appetite, back pain, headache, dizziness, taste changes, rash, itchy skin, yellowing of the skin and/or eyes, upper respiratory infection, dry skin, and sore throat.
Swollen kidneys due to blocked urine flow occurred rarely.
Marketing Experience
Other side effects reported since CRIXIVAN has been marketed include: allergic reactions; severe skin reactions; yellowing of the skin and/or eyes; heart problems including heart attack; stroke; abdominal swelling; indigestion; inflammation of the kidneys; decreased kidney function; inflammation of the pancreas; joint pain; depression; itching; hives; change in skin color; hair loss; ingrown toenails with or without infection; crystals in the urine; painful urination; numbness of the mouth and increased cholesterol.
Tell your doctor promptly about these or any other unusual symptoms. If the condition persists or worsens, seek medical attention.
How should I store CRIXIVAN capsules?
- Keep CRIXIVAN capsules in the bottle they came in and at room temperature (59°-86°F).
- Keep CRIXIVAN capsules dry by leaving the small desiccant in the bottle. Keep the bottle closed.
This medication was prescribed for your particular condition. Do not use it for any other condition or give it to anybody else. Keep CRIXIVAN and all medicines out of the reach of children. If you suspect that more than the prescribed dose of this medicine has been taken, contact your local poison control center or emergency room immediately.
This leaflet provides a summary of information about CRIXIVAN. If you have any questions or concerns about either CRIXIVAN or HIV, talk to your doctor.
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Whitehouse Station, NJ 08889, USA
U.S. Patent Nos.: 5,413,999; 6,645,961
Issued April 2010
9640612
PRINCIPAL DISPLAY PANEL - Bottle Label - 400 mg
Crixivan® 400 mg
(Indinavir Sulfate) Capsules
NDC 21695-366-18
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA
Repackaged by:
Rebel Distributors Corp
Thousand Oaks, CA 91320
Each capsule contains indinavir 400 mg (corresponding to indinavir sulfate 500 mg).
Store at room temperature, 15-30°C (59-86ºF). Keep container tightly closed. Protect from moisture.
Bottle contains desiccant.
USUAL DOSAGE: See accompanying circular.
ALERT
Find out about medicines that should NOT be taken with CRIXIVAN.
Note to Pharmacist: Do not cover ALERT box with Pharmacy label.
18 Capsules
Rx only
9966801
90 | No.3758