FULL PRESCRIBING INFORMATION
WARNING: ABUSE, MISUSE, AND ADDICTION
AZSTARYS has a high potential for abuse and misuse, which can lead to the development of a substance use disorder, including addiction. Misuse and abuse of CNS stimulants, including AZSTARYS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing AZSTARYS, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug. Throughout AZSTARYS treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction [see Warnings and Precautions (5.1) and Drug Abuse and Dependence (9.2)].
1 INDICATIONS AND USAGE
AZSTARYS is indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in patients 6 years of age and older.
2 DOSAGE AND ADMINISTRATION
2.1 Pretreatment Screening
Prior to treating patients with AZSTARYS, assess:
- for the presence of cardiac disease (i.e., perform a careful history, family history of sudden death or ventricular arrhythmia, and physical exam) [see Warnings and Precautions (5.2)].
- the family history and clinically evaluate patients for motor or verbal tics or Tourette’s syndrome before initiating AZSTARYS [see Warnings and Precautions (5.10)].
2.2 Recommended Dosage
Pediatric Patients 6 to 12 years of age
- The recommended starting dosage of AZSTARYS is 39.2 mg serdexmethylphenidate/7.8 mg dexmethylphenidate once daily in the morning.
- The dosage may be increased after one week to a dosage of 52.3 mg serdexmethylphenidate/10.4 mg dexmethylphenidate per day, or decreased after one week to a dosage of 26.1 mg serdexmethylphenidate/5.2 mg dexmethylphenidate per day, depending on response and tolerability.
- Maximum recommended dosage is 52.3 mg serdexmethylphendiate/10.4 mg dexmethyphenidate once daily.
Adults and Pediatric Patients 13 to 17 years of age
- The recommended starting dosage of AZSTARYS is 39.2 mg serdexmethylphenidate/7.8 mg dexmethylphenidate once daily in the morning.
- Increase the dosage after one week to a dosage of 52.3 mg serdexmethylphenidate/10.4 mg dexmethylphenidate per day, depending on response and tolerability.
- Maximum recommended dosage is 52.3 mg serdexmethylphenidate/10.4 mg dexmethylphenidate once daily.
2.3 Administration Instructions
Administer AZSTARYS orally once daily in the morning with or without food [see Clinical Pharmacology (12.3)].
AZSTARYS capsules may be taken whole, or opened and the entire contents sprinkled into 50 mL of water or over 2 tablespoons of applesauce. Consume all the drug/food mixture immediately or within 10 minutes of mixing; do not store for future use [see Clinical Pharmacology (12.3)].
2.4 Switching from Other Methylphenidate Products
If switching from other methylphenidate products, discontinue that treatment, and titrate with AZSTARYS using the titration schedule described above.
Do not substitute AZSTARYS for other methylphenidate products on a milligram-per-milligram basis because these products have different pharmacokinetic profiles from AZSTARYS and may have different methylphenidate base composition [see Description (11), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
AZSTARYS capsules are available as:
- 26.1 mg/5.2 mg (serdexmethylphenidate/dexmethylphenidate) – blue cap/grey body, imprinted with "286" on cap and "KP415" on the body
- 39.2 mg/7.8 mg (serdexmethylphenidate/dexmethylphenidate) – dark blue cap/grey body, imprinted with "429" on cap and "KP415" on the body
- 52.3 mg/10.4 mg (serdexmethylphenidate/dexmethylphenidate) – orange cap/grey body, imprinted with "5612" on cap and "KP415" on the body
4 CONTRAINDICATIONS
AZSTARYS is contraindicated in patients:
- with known hypersensitivity to serdexmethylphenidate, methylphenidate, or other components of AZSTARYS. Bronchospasm, rash, and pruritus have been reported in patients who received AZSTARYS. Hypersensitivity reactions such as angioedema and anaphylactic reactions have been reported in patients treated with other methylphenidate products [see Adverse Reactions (6.2)].
- receiving concomitant treatment with monoamine oxidase inhibitors (MAOIs), or within 14 days following discontinuation of treatment with an MAOI, because of the risk of hypertensive crisis [see Drug Interactions (7.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Abuse, Misuse, and Addiction
AZSTARYS has a high potential for abuse and misuse. The use of AZSTARYS exposes individuals to the risks of abuse and misuse, which can lead to the development of a substance use disorder, including addiction. AZSTARYS can be diverted for non-medical use into illicit channels or distribution [see Drug Abuse and Dependence (9.2)]. Misuse and abuse of CNS stimulants, including AZSTARYS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing AZSTARYS, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their families about these risks and proper disposal of any unused drug. Advise patients to store AZSTARYS in a safe place, preferably locked, and instruct patients to not give AZSTARYS to anyone else. Throughout AZSTARYS treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
5.2 Risks to Patients with Serious Cardiac Disease
Sudden death has been reported in patients with structural cardiac abnormalities or other serious cardiac disease who were treated with CNS stimulants at the recommended ADHD dosage.
Avoid AZSTARYS use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease.
5.3 Increased Blood Pressure and Heart Rate
CNS stimulants cause an increase in blood pressure (mean increase approximately 2 to 4 mmHg) and heart rate (mean increase approximately 3 to 6 beats per minute). Some patients may have larger increases.
Monitor all AZSTARYS-treated patients for hypertension and tachycardia.
5.4 Psychiatric Adverse Reactions
Exacerbation of Pre-Existing Psychosis
CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Induction of a Manic Episode in Patients with Bipolar Disorder
CNS stimulants may induce a manic or mixed mood episode in patients. Prior to initiating AZSTARYS treatment, screen patients for risk factors for developing a manic episode (e.g., comorbid or history of depressive symptoms or a family history of suicide, bipolar disorder, or depression).
New Psychotic or Manic Symptoms
CNS stimulants, at the recommended dosage, may cause psychotic or manic symptoms (e.g., hallucinations, delusional thinking, or mania) in patients without a prior history of psychotic illness or mania. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in approximately 0.1% of CNS stimulant-treated patients, compared to 0% of placebo-treated patients. If such symptoms occur, consider discontinuing AZSTARYS.
5.5 Priapism
Prolonged and painful erections, sometimes requiring surgical intervention, have been reported with methylphenidate use, in both adult and pediatric male patients. Although priapism was not reported with methylphenidate initiation, it developed after some time on methylphenidate , often subsequent to an increase in dosage. Priapism also occurred during methylphenidate withdrawal (drug holidays or during discontinuation).
AZSTARYS-treated patients who develop abnormally sustained or frequent and painful erections should seek immediate medical attention.
5.6 Peripheral Vasculopathy, including Raynaud's Phenomenon
CNS stimulants used to treat ADHD, including AZSTARYS, are associated with peripheral vasculopathy, including Raynaud's phenomenon. Signs and symptoms are usually intermittent and mild; however, sequelae have included digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud's phenomenon, were observed in post- marketing reports and at the therapeutic dosage of CNS stimulants in all age groups throughout the course of treatment. Signs and symptoms generally improved after dosage reduction or discontinuation of the CNS stimulant.
Careful observation for digital changes is necessary during AZSTARYS treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for AZSTARYS-treated patients who develop signs or symptoms of peripheral vasculopathy.
5.7 Long-Term Suppression of Growth in Pediatric Patients
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients.
In a long-term, open-label safety study with AZSTARYS conducted in pediatric patients 6 to 12 years of age with ADHD, there was a lower than expected increase in height and weight compared to pediatric patients of the same age and sex, on average [see Adverse Reactions (6.1)].
Closely monitor growth (weight and height) in AZSTARYS-treated pediatric patients. Pediatric patients who are not growing or gaining height or weight as expected may need to have their treatment interrupted.
5.8 Acute Angle Closure Glaucoma
There have been reports of angle closure glaucoma associated with methylphenidate treatment.
Although the mechanism is not clear, AZSTARYS-treated patients considered at risk for acute angle closure glaucoma (e.g., patients with significant hyperopia) should be evaluated by an ophthalmologist.
5.9 Increased Intraocular Pressure and Glaucoma
There have been reports of an elevation of intraocular pressure (IOP) associated with methylphenidate treatment [see Adverse Reactions (6.2)].
Prescribe AZSTARYS to patients with open-angle glaucoma or abnormally increased IOP only if the benefit of treatment is considered to outweigh the risk. Closely monitor AZSTARYS-treated patients with a history of abnormally increased IOP or open angle glaucoma.
5.10 Motor and Verbal Tics, and Worsening of Tourette’s Syndrome
CNS stimulants, including methylphenidate, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette’s syndrome has also been reported [see Adverse Reactions (6.2)].
Before initiating AZSTARYS, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor AZSTARYS-treated patients for the emergence or worsening of tics or Tourette’s syndrome, and discontinue treatment if clinically appropriate.
6 ADVERSE REACTIONS
The following are discussed in more detail in other sections of the labeling:
- Abuse, Misuse, and Addiction [see Boxed Warning, Warnings and Precautions (5.1), and Drug Abuse and Dependence (9.2, 9.3)]
- Known hypersensitivity to methylphenidate or other ingredients of AZSTARYS [see Contraindications (4)]
- Hypertensive Crisis with Concomitant Use of Monoamine Oxidase Inhibitors [see Contraindications (4)]
- Risks to Patients with Serious Cardiac Disease [see Warnings and Precautions (5.2)]
- Increased Blood Pressure and Heart Rate [see Warnings and Precautions (5.3)]
- Psychiatric Adverse Reactions [see Warnings and Precautions (5.4)]
- Priapism [see Warnings and Precautions (5.5)]
- Peripheral Vasculopathy, including Raynaud's Phenomenon [see Warnings and Precautions (5.6)]
- Long-Term Suppression of Growth in Pediatric Patients [see Warnings and Precautions (5.7)]
- Acute Angle Closure Glaucoma [see Warnings and Precautions (5.8)]
- Increased Intraocular Pressure and Glaucoma [see Warnings and Precautions (5.9)]
- Motor and Verbal Tics, and Worsening of Tourette’s Syndrome [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse Reactions in Studies with Other Methylphenidate Products in Pediatric Patients and Adults with ADHD
Commonly reported (≥ 5% of the methylphenidate group and at least twice the rate of the placebo group) adverse reactions from placebo-controlled trials of methylphenidate products include: decreased appetite, decreased weight, nausea, abdominal pain, dyspepsia, vomiting, insomnia, anxiety, affect lability, irritability, dizziness, increased blood pressure, and tachycardia.
Adverse Reactions in Studies with AZSTARYS in Pediatric Patients (6 to 12 years) with ADHD
Short-Term Study
A short-term study conducted in pediatric patients 6 to 12 years of age with ADHD was comprised of a 3-week, open-label, dose optimization phase in which all patients received AZSTARYS (n=155), followed by a 1-week, double-blind, controlled phase in which patients were randomized to continue AZSTARYS (n=74) or switch to placebo (n=76). Because of the study design, the reported adverse reaction rates cannot be used to predict the rates that may be expected in clinical practice.
Long-Term Study
A long-term, open-label safety study was conducted in pediatric patients 6 to 12 years of age with ADHD who either completed the short-term study or were de novo patients. This study was comprised of a 3-week dose optimization phase for patients not recently treated with AZSTARYS followed by a 12-month treatment phase for all patients during which 238 patients received open- label AZSTARYS and had evaluable safety data. A total of 154 patients were treated for 12 months. Because of the open-label, uncontrolled design of this study, the reported adverse reaction rates cannot be assessed in terms of a causal relationship to AZSTARYS treatment.
To adjust for normal growth, z-scores were derived (measured in standard deviations [SD]); z- scores normalize for the natural growth of children and adolescents by comparisons to age- and sex-matched population standards. A z-score change less than 0.5 SD is considered not clinically significant.
In this study, the mean increase in weight from baseline to Month 12 was 3.4 kg among study completers. The mean change in z-score from baseline to Month 12 was -0.20, indicating a lower than expected increase in body weight compared to children of the same age and sex, on average. Most of the weight z-score decline occurred in the first 4 months of treatment.
The mean increase in height from baseline to Month 12 was 4.9 cm among completers. Using the same z-score analysis for height, the mean change in z-score from baseline to Month 12 was - 0.21, indicating a lower than expected increase in height compared to pediatric patients of the same age and sex, on average.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of methylphenidate products. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These adverse reactions are as follows:
Blood and Lymphatic System Disorders: pancytopenia, thrombocytopenia, thrombocytopenic purpura
Cardiac Disorders: angina pectoris, bradycardia, extrasystole, supraventricular tachycardia, ventricular extrasystole, palpitations, increased heart rate
Eye Disorders: diplopia, increased intraocular pressure, mydriasis, visual impairment, blurred vision
General Disorders: chest pain, chest discomfort, hyperpyrexia
Gastrointestinal Disorders: dry mouth
Hepatobiliary disorders: hepatocellular injury, acute hepatic failure
Immune System Disorders: hypersensitivity reactions such as angioedema, anaphylactic reactions, auricular swelling, bullous conditions, exfoliative conditions, urticarias, pruritus NEC, rashes, eruptions, and exanthemas NEC
Investigations: alkaline phosphatase increased, bilirubin increased, hepatic enzyme increased, platelet count decreased, white blood cell count abnormal
Musculoskeletal, Connective Tissue and Bone Disorders: arthralgia, myalgia, muscle twitching, rhabdomyolysis, muscle cramps
Nervous System: convulsion, grand mal convulsion, dyskinesia, serotonin syndrome in combination with serotonergic drugs, nervousness, headache, tremor, drowsiness, vertigo, motor and verbal tics
Psychiatric Disorders: disorientation, libido changes, hallucination, hallucination auditory, hallucination visual, logorrhea, mania, restlessness, agitation
Skin and Subcutaneous Tissue Disorders: alopecia, erythema, hyperhidrosis
Urogenital System: priapism
Vascular Disorders: Raynaud's phenomenon
7 DRUG INTERACTIONS
7.1 Clinically Important Interactions with AZSTARYS
Table 1 presents clinically important drug interactions with AZSTARYS.
| Monoamine Oxidase Inhibitors (MAOIs) | |
| Clinical Impact | Concomitant use of MAOIs and CNS stimulants, including AZSTARYS, can cause hypertensive crisis. Potential outcomes include death, stroke, myocardial infarction, aortic dissection, ophthalmological complications, eclampsia, pulmonary edema, and renal failure [see Contraindications (4)]. |
| Intervention | Do not administer AZSTARYS concomitantly with MAOIs or within 14 days after discontinuing MAOI treatment [see Contraindications (4)] |
| Antihypertensive Drugs | |
| Clinical Impact | AZSTARYS may decrease the effectiveness of drugs used to treat hypertension [see Warnings and Precautions (5.3)]. |
| Intervention | Monitor blood pressure and adjust the dosage of the antihypertensive drug, as needed. |
| Halogenated Anesthetics | |
| Clinical Impact | Concomitant use of halogenated anesthetics and AZSTARYS may increase the risk of sudden blood pressure and heart rate increase during surgery. |
| Intervention | Avoid use of AZSTARYS in patients being treated with anesthetics on the day of surgery. |
| Risperidone | |
| Clinical Impact | Combined use of methylphenidate with risperidone when there is a change, whether an increase or decrease, in dosage of either or both medications, may increase the risk of extrapyramidal symptoms (EPS). |
| Intervention | Monitor for signs of EPS. |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ADHD medications, including AZSTARYS, during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Psychostimulants at 1-866-961-2388.
Risk Summary
There are no available data on AZSTARYS use in pregnant women to evaluate for a drug- associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes; however, AZSTARYS contains dexmethylphenidate and serdexmethylphenidate, a prodrug of dexmethylphenidate. Dexmethylphenidate is the d-threo enantiomer of racemic methylphenidate. Published studies and postmarketing reports on methylphenidate use during pregnancy have not identified a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There may be risks to the fetus associated with the use of CNS stimulants use during pregnancy (see Clinical Considerations). Embryo-fetal development studies in rats showed delayed fetal skeletal ossification at doses up to 3 times the maximum recommended human dose (MRHD) of 40 mg/day dexmethylphenidate hydrochloride given to adults based on plasma levels. A decrease in pup weight in males was observed in a pre- and post-natal development study with oral administration of dexmethylphenidate to rats throughout pregnancy and lactation at doses 3 times the MRHD of 40 mg/day dexmethylphenidate hydrochloride given to adults based on plasma levels (see Data).
No evidence of developmental effects were found in an embryo-fetal development study with oral administration of serdexmethylphenidate to rabbits during organogenesis at doses up to approximately 49 times the MRHD of 52 mg/day serdexmethylphenidate given to adults based on plasma levels (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
CNS stimulants, such as AZSTARYS, can cause vasoconstriction and thereby decrease placental perfusion. No fetal and/or neonatal adverse reactions have been reported with the use of therapeutic doses of methylphenidate during pregnancy; however, premature delivery and low birth weight infants have been reported in amphetamine-dependent mothers.
Data
Animal Data
In embryo-fetal development studies conducted in rats and rabbits, dexmethylphenidate hydrochloride was administered orally at doses of up to 20 and 100 mg/kg/day, respectively, during the period of organogenesis. No evidence of malformations was found in either the rat or rabbit study; however, delayed fetal skeletal ossification was observed at the highest dose level in rats. When dexmethylphenidate hydrochloride was administered to rats throughout pregnancy and lactation at doses of up to 20 mg/kg/day, post-weaning body weight gain was decreased in male offspring at the highest dose, but no other effects on postnatal development were observed. At the highest doses tested, plasma levels [area under the curves (AUCs)] of dexmethylphenidate in pregnant rats and rabbits were approximately 3 and 1 times, respectively, those in adults dosed with 40 mg/day dexmethylphenidate hydrochloride.
Racemic methylphenidate hydrochloride has been shown to cause malformations (increased incidence of fetal spina bifida) in rabbits when given in doses of 200 mg/kg/day throughout organogenesis.
No evidence of developmental effects were found in an embryo-fetal development study with oral administration of serdexmethylphenidate in rabbits during organogenesis at doses of up to 374 mg/kg/day. At the highest dose tested, the plasma level [area under the curve (AUC)] of serdexmethylphenidate in pregnant rabbits was approximately 49 times that in adults dosed with 52 mg/day serdexmethylphenidate.
8.2 Lactation
Risk Summary
There are no available data on the presence of serdexmethylphenidate in human milk, effects on the breastfed infant, or effects on milk production. Dexmethylphenidate is the d-threo enantiomer of racemic methylphenidate. Limited published literature, based on milk sampling from seven mothers reports that methylphenidate is present in human milk, which resulted in infant doses of 0.16% to 0.7% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 1.1 and 2.7. There are no reports of adverse effects on the breastfed infant and no effects on milk production. Long-term neurodevelopmental effects on infants from stimulant exposure are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for AZSTARYS and any potential adverse effects on the breastfed infant from AZSTARYS or from the underlying maternal condition.
Clinical Considerations
Monitor breastfeeding infants for adverse reactions, such as agitation, anorexia, and reduced weight gain.
8.4 Pediatric Use
The safety and effectiveness of AZSTARYS have been established in pediatric patients ages 6 to 17 years of age for the treatment of ADHD. Use of AZSTARYS in patients 6 to 12 years of age is supported by a randomized, double-blind, placebo-controlled, parallel group trial in 155 pediatric patients with ADHD and a 12-month open-label long term safety trial in 238 patients [see Adverse Reactions (6.1), Clinical Studies (14)]. Use of AZSTARYS in pediatric patients 13 to 17 years of age is supported by additional pharmacokinetics analysis showing similar plasma concentration-time profiles of dexmethylphenidate in adolescents and adults after administration of the same dose of AZSTARYS [see Clinical Studies (14)]. The long-term efficacy of methylphenidate in pediatric patients has not been established.
The safety and effectiveness of AZSTARYS in pediatric patients less than 6 years have not been established.
Long Term Suppression of Growth
Growth should be monitored during treatment with stimulants, including AZSTARYS. Pediatric patients who are not growing or gaining weight as expected may need to have their treatment interrupted [see Warnings and Precautions (5.7) and Adverse Reactions (6.1)].
Juvenile Animal Toxicity Data
Rats treated with racemic methylphenidate early in the postnatal period through sexual maturation demonstrated a decrease in spontaneous locomotor activity in adulthood. A deficit in acquisition of a specific learning task was observed in females only. The doses at which these findings were observed are at least 3 times the MRHD of 40 mg/day dexmethylphenidate hydrochloride given to children on a mg/m2 basis.
In a study conducted in young rats, racemic methylphenidate hydrochloride was administered orally at doses of up to 100 mg/kg/day for 9 weeks, starting early in the postnatal period (postnatal Day 7) and continuing through sexual maturity (postnatal Week 10). When these animals were tested as adults (postnatal Weeks 13 to 14), decreased spontaneous locomotor activity was observed in males and females previously treated with 50 mg/kg/day racemic methylphenidate hydrochloride [approximately 3 times the maximum recommended human dose (MRHD) of 40 mg of dexmethylphenidate hydrochloride given to children on a mg/m2 basis] or greater, and a deficit in the acquisition of a specific learning task was seen in females exposed to the highest dose (6 times the MRHD of 40 mg of dexmethylphenidate hydrochloride given to children on a mg/m2 basis). The no effect level for juvenile neurobehavioral development in rats was 5 mg/kg/day racemic methylphenidate hydrochloride (less than the MRHD of 40 mg of dexmethylphenidate hydrochloride given to children on a mg/m2 basis). The clinical significance of the long-term behavioral effects observed in rats is unknown.
Serdexmethylphenidate was administered orally to juvenile rabbits at doses up to 280 mg/kg/day (approximately 50 times the MRHD of 52 mg/day serdexmethylphenidate given to children on a mg/m2 basis), respectively, for 6 months, starting at postnatal Day 28 and continuing through sexual maturity (postnatal Day 196). No adverse findings were observed at the highest dose of serdexmethylphenidate.
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
AZSTARYS contains dexmethylphenidate hydrochloride, a Schedule II controlled substance, and serdexmethylphenidate, a Schedule IV controlled substance.
9.2 Abuse
AZSTARYS has a high potential for abuse and misuse which can lead to the development of a substance use disorder, including addiction [see Warnings and Precautions (5.1)]. AZSTARYS can be diverted for non-medical use into illicit channels or distribution.
Abuse is the intentional non-therapeutic use of a drug, even once, to achieve a desired psychological or physiological effect. Misuse is the intentional use, for therapeutic purposes, of a drug by an individual in a way other than prescribed by a health care provider or for whom it was not prescribed. Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that may include a strong desire to take the drug, difficulties in controlling drug use (e.g., continuing drug use despite harmful consequences, giving a higher priority to drug use than other activities and obligations), and possible tolerance or physical dependence.
Misuse and abuse of the combination of dexmethylphenidate and serdexmethylphenidate may cause increased heart rate, respiratory rate, or blood pressure; sweating; dilated pupils; hyperactivity; restlessness; insomnia; decreased appetite; loss of coordination; tremors; flushed skin; vomiting; and/or abdominal pain. Anxiety, psychosis, hostility, aggression, and suicidal or homicidal ideation have also been observed with CNS stimulants abuse and/or misuse. Misuse and abuse of CNS stimulants, including AZSTARYS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
9.3 Dependence
Physical Dependence
AZSTARYS may produce physical dependence. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
Withdrawal signs and symptoms after abrupt discontinuation or dose reduction following prolonged use of CNS stimulants including AZSTARYS include dysphoric mood; depression; fatigue; vivid, unpleasant dreams; insomnia or hypersomnia; increased appetite; and psychomotor retardation or agitation.
Tolerance
AZSTARYS may produce tolerance. Tolerance is a physiological state characterized by a reduced response to a drug after repeated administration (i.e., a higher dose of a drug is required to produce the same effect that was once obtained at a lower dose).
10 OVERDOSAGE
Clinical Effects of Overdose
Overdose of CNS stimulants is characterized by the following sympathomimetic effects:
- Cardiovascular effects including tachyarrhythmias, and hypertension or hypotension. Vasospasm, myocardial infarction, or aortic dissection may precipitate sudden cardiac death. Takotsubo cardiomyopathy may develop.
- CNS effects including psychomotor agitation, confusion, and hallucinations. Serotonin syndrome, seizures, cerebral vascular accidents, and coma may occur.
- Life-threatening hyperthermia (temperatures greater than 104°F) and rhabdomyolysis may develop.
Overdose Management
Consider the possibility of multiple drug ingestion. The pharmacokinetic profile of AZSTARYS should be considered when treating patients with overdose. Because methylphenidate has a large volume of distribution and is rapidly metabolized, dialysis is not useful. Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
11 DESCRIPTION
AZSTARYS (serdexmethylphenidate and dexmethylphenidate) capsules contain dexmethylphenidate, a CNS stimulant, and serdexmethylphenidate, a prodrug of dexmethylphenidate.
AZSTARYS capsules are intended for oral administration and each capsule contains a fixed molar ratio of 30% dexmethylphenidate and 70% serdexmethylphenidate.
AZSTARYS contains 26.1/5.2, 39.2/7.8, or 52.3/10.4 mg of serdexmethylphenidate/ dexmethylphenidate (equivalent to 28/6, 42/9, or 56/12 mg of serdexmethylphenidate chloride/ dexmethylphenidate hydrochloride, respectively. The combined molar dose of serdexmethylphenidate and dexmethylphenidate in each dosage strength of AZSTARYS is equivalent to 20, 30, or 40 mg dexmethylphenidate hydrochloride, respectively (equivalent to 17.3, 25.9 or 34.6 mg dexmethylphenidate free base, respectively).
The chemical name of serdexmethylphenidate chloride is 3-(((1S)-1-carboxy-2-hydroxyethyl)carbamoyl)-1-((((2R)-2-(2-(1R)-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridinium chloride. Its molecular formula is C25H30N3O8+•Cl-, and its structural formula is:
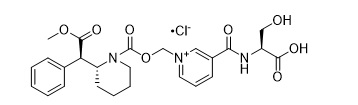
Serdexmethylphenidate chloride is a white to off-white crystalline powder. Its solutions are acid to litmus. It is freely soluble in water, soluble in methanol, and slightly soluble in alcohol and acetone. Its molecular weight is 535.98 g/mol.
Dexmethylphenidate is the d-threo enantiomer of racemic d,l-methylphenidate hydrochloride. The chemical name of dexmethylphenidate hydrochloride is methyl (R)-2-phenyl-2-((R)-piperidin-2-yl)acetate hydrochloride. Its molecular formula is C14H19NO2•HCl, and its structural formula is:
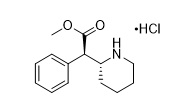
Dexmethylphenidate hydrochloride is a white to off-white powder. Its solutions are acid to litmus. It is freely soluble in water and in methanol, soluble in alcohol, and slightly soluble in chloroform and in acetone. Its molecular weight is 269.77 g/mol.
Inactive ingredients: colloidal silicon dioxide, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, and talc.
Each strength capsule also contains colorant ingredients in the capsule shell as follows:
- 26.1/5.2 mg: Back Iron Oxide, FD&C Blue No. 1, Titanium Dioxide
- 39.2/7.8 mg: Black Iron Oxide, FD&C Blue No. 1, FD&C Red No. 40, Titanium Dioxide
- 52.3/10.4 mg: Black Iron Oxide, FD&C Red No. 40, FD&C Yellow No. 6, Titanium Dioxide
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Serdexmethylphenidate is a prodrug of dexmethylphenidate. Dexmethylphenidate HCl is a central nervous system (CNS) stimulant. The mode of therapeutic action in ADHD is not known.
12.2 Pharmacodynamics
Dexmethylphenidate
Dexmethylphenidate is the more pharmacologically active d-enantiomer of racemic d,l-methylphenidate. Methylphenidate blocks the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. In vitro studies with serdexmethylphenidate showed little or no binding of the prodrug to monoaminergic reuptake transporters.
Cardiac Electrophysiology
The effect of serdexmethylphenidate on QTc interval was evaluated in a randomized, double-blind, placebo-controlled, human abuse potential study (intranasal administration) in 46 healthy subjects. At a mean concentration 40 times the Cmax for the highest dose of AZSTARYS (52.3/10.4 mg base equivalent), serdexmethylphenidate does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Serdexmethylphenidate is a prodrug of dexmethylphenidate. Following a single dose administration of 52.3 mg/10.4 mg AZSTARYS and 40 mg of an dexmethylphenidate hydrochloride extended-release (ER) capsule in healthy volunteers under fasted conditions:
- The mean peak plasma concentration (Cmax) of dexmethylphenidate was 14.0 ng/mL and 28.2 ng/mL, respectively;
- The mean area under concentration curve (AUC) of dexmethylphenidate was 186 hour*ng/mL and 248 hour*ng/mL, respectively.
The plasma PK profiles of dexmethylphenidate following administration of AZSTARYS or dexmethylphenidate hydrochloride extended-release (ER) capsule are presented in Figure 1.
Figure 1: Mean Dexmethylphenidate Plasma Concentration-Time Profiles After A Single Dose Administration of AZSTARYS or Dexmethylphenidate Hydrochloride Extended-Release (ER) Capsule in Healthy Adults Under Fasted Conditions
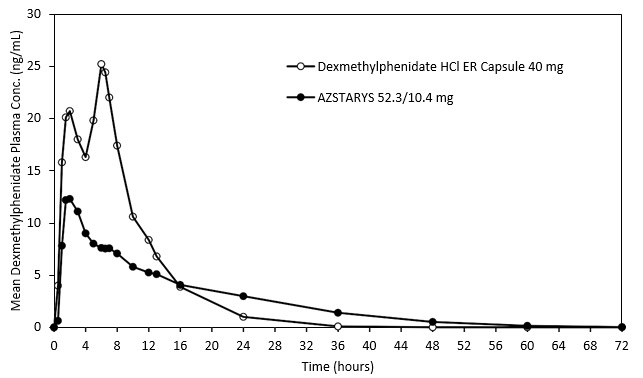
Approximate linear PK was demonstrated for dexmethylphenidate following single dose administration of AZSTARYS in the dose range of 26.1 mg/5.2 mg to 52.3 mg/10.4 mg. Steady state of dexmethylphenidate was approached after the third once-daily dose. At steady-state, dexmethylphenidate mean exposures (Cmax and AUC0-24h) were approximately 37% higher relative to a single-dose administration of AZSTARYS. No accumulation of serdexmethylphenidate was observed after once-daily administration of AZSTARYS. The mean relative exposure of serdexmethylphenidate to dexmethylphenidate based on molar concentrations for Cmax, Cmin, and AUC0-24hr was about 101%, 8.5%, and 55.7%, respectively, following multiple once-daily oral dosing under fasted conditions.
Absorption
Cross-study calculation estimated an absolute oral bioavailability for serdexmethylphenidate of less than 3%. The median time to reach Cmax of serdexmethylphenidate and dexmethylphenidate (Tmax) is about 2 hours following a single dose administration of AZSTARYS under fasted conditions. Following oral administration of serdexmethylphenidate single moiety alone, dexmethylphenidate Tmax is reached at about 8 hours post dose.
Effect of Food
No clinically meaningful differences in the exposure of dexmethylphenidate were observed when administered after an overnight fast, with a high-fat, high-caloric meal, or sprinkled onto applesauce or water. The median time to peak plasma concentration (Tmax) was lengthened from 2 to 4-4.5 hours in the presence of food.
Distribution
Plasma protein binding of serdexmethylphenidate and dexmethylphenidate is approximately 56% and 47%, respectively, at 5 µM (about 60-fold higher than the therapeutic concentrations at the highest recommended dose). The mean apparent volume of distribution for serdexmethylphenidate was about 29.3 L/kg after AZSTARYS administration. Dexmethylphenidate shows a mean volume of distribution of 2.65 L/kg after intravenous administration.
Elimination
The mean plasma terminal elimination half-life of serdexmethylphenidate and dexmethylphenidate in healthy adult subjects was about 5.7 hours and 11.7 hours, respectively, following a single dose of 52.3 mg/10.4 mg AZSTARYS. The mean apparent clearance for serdexmethylphenidate was about 3.6 L/hr/kg after AZSTARYS administration. Dexmethylphenidate was eliminated with a mean clearance of 0.40 L/hr/kg after intravenous administration.
Metabolism
Serdexmethylphenidate is a prodrug of dexmethylphenidate and is likely converted to dexmethylphenidate mainly in the lower gastrointestinal tract. Enzymes involved in the conversion process are not identified.
Dexmethylphenidate is metabolized primarily via de-esterification to d-α-phenyl-piperidine acetic acid (also known as d-ritalinic acid). Ritalinic acid has little or no pharmacological activity. There is no in vivo interconversion to the l-threo-enantiomer observed.
Excretion
After oral dosing of radiolabeled serdexmethylphenidate in humans, about 62% and 37% of the radioactivity was recovered in urine and feces, respectively. Metabolite ritalinic acid accounted for approximately 63% of the total recovered dose in urine and feces. About 0.4% and 11% of the dose was excreted as unchanged serdexmethylphenidate in the urine and feces, respectively.
After oral dosing of radiolabeled racemic methylphenidate in humans, about 90% of the radioactivity was recovered in urine. The main urinary metabolite of racemic d,l-methylphenidate was d,l-ritalinic acid, accountable for approximately 80% of the dose. Urinary excretion of unchanged methylphenidate accounted for 0.5% of an intravenous dose.
Specific Populations
Sex
No significant pharmacokinetic differences based on sex have been observed for AZSTARYS.
Race
There is insufficient experience with the use of AZSTARYS to detect ethnic variations in pharmacokinetics.
Age
The shapes of the plasma concentration time profiles for dexmethylphenidate were similar in pediatric patients (6 to 17 years of age) with ADHD and healthy adults. After the same dose administration of AZSTARYS, dexmethylphenidate exposure in pediatric patients (13 to 17 years of age) and adults was about half of that in pediatric patients 6 to 12 years of age. Plasma concentrations of dexmethylphenidate when adjusted for dose and body weight were similar across all age groups.
Renal Impairment
There is no experience with the use of AZSTARYS in patients with renal impairment. Since renal clearance is not an important route of serdexmethylphenidate or methylphenidate elimination, renal impairment is expected to have little effect on the pharmacokinetics of AZSTARYS.
Hepatic Impairment
There is no experience with the use of AZSTARYS in patients with hepatic impairment.
Drug Interaction Studies
Clinical Studies
CYP2D6 substrate: No clinically significant differences in desipramine (CYP2D6 substrate) were observed when co-administered with methylphenidate.
In Vitro Studies
Alcohol: No clinically significant differences in the rate or amount of release of either serdexmethylphenidate or methylphenidate were observed with alcohol concentrations of 5% and 40%.
Cytochrome P450 (CYP) enzymes: Serdexmethylphenidate and methylphenidate do not appear to be substrates, inducers or inhibitors of CYP1A2, 2C8, 2C9, 2C19, 2D6, 2E1 or 3A.
Transporters: Serdexmethylphenidate does not appear to be a substrate or inhibitor of P-gp, BCRP, OATP1B1/3, OAT1/3, OCT2, or MATE1/2-K.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime studies to evaluate the carcinogenic potential of serdexmethylphenidate have not been conducted.
Lifetime carcinogenicity studies have not been carried out with dexmethylphenidate hydrochloride.
In a lifetime carcinogenicity study carried out in B6C3F1 mice, racemic methylphenidate caused an increase in hepatocellular adenomas, and in males only, an increase in hepatoblastomas was seen at a daily dose of approximately 60 mg/kg/day. This dose is approximately 4 times the MRHD of 40 mg of dexmethylphenidate hydrochloride on a mg/m2 basis. Hepatoblastoma is a relatively rare rodent malignant tumor type. There was no increase in total malignant hepatic tumors. The mouse strain used is sensitive to the development of hepatic tumors, and the significance of these results to humans is unknown.
Racemic methylphenidate hydrochloride did not cause any increase in tumors in a lifetime carcinogenicity study carried out in F344 rats; the highest dose used was approximately 45 mg/kg/day, which is approximately 5 times the MRHD of 40 mg of dexmethylphenidate hydrochloride on a mg/m2 basis.
In a 24-week carcinogenicity study with racemic methylphenidate in the transgenic mouse strain p53+/-, which is sensitive to genotoxic carcinogens, there was no evidence of carcinogenicity. Male and female mice were fed diets containing the same concentrations as in the lifetime carcinogenicity study; the high-dose group was exposed to 60 to 74 mg/kg/day of racemic methylphenidate hydrochloride.
Mutagenesis
Serdexmethylphenidate was not mutagenic in the in vitro Ames reverse mutation assay, in the in vitro mammalian cell micronucleus assay using human peripheral blood lymphocytes, in the in vivo rat bone barrow micronucleus assay, or in the in vivo rat alkaline comet assay.
Dexmethylphenidate was not mutagenic in the in vitro Ames reverse mutation assay, in the in vitro mouse lymphoma cell forward mutation assay, or in the in vivo mouse bone marrow micronucleus test. In an in vitro assay using cultured Chinese Hamster Ovary (CHO) cells treated with racemic methylphenidate, sister chromatid exchanges and chromosome aberrations were increased, indicative of a weak clastogenic response.
Impairment of Fertility
Racemic methylphenidate hydrochloride did not impair fertility in male or female mice that were fed diets containing the drug in an 18-week continuous breeding study. The study was conducted at doses of up to 160 mg/kg/day, approximately 10-times the MRHD of 40 mg of dexmethylphenidate hydrochloride on a mg/m2 basis.
14 CLINICAL STUDIES
Pediatric Patients 6 to 12 years of age with ADHD
The efficacy of AZSTARYS for the treatment of ADHD in pediatric patients 6 to 12 years of age was evaluated in a randomized, double-blind, placebo-controlled, parallel group, analog classroom study (Study 1; NCT# 03292952). That study was conducted in 150 pediatric patients 6 to 12 years of age who met Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) criteria for a primary diagnosis of ADHD (combined, inattentive, or hyperactive/impulsive presentation) confirmed by the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID).
Following washout of previous ADHD medication, subjects entered an open-label dose- optimization period (3 weeks) with an initial dosage of 39.2 mg/7.8 mg once daily in the morning. The dose could be titrated on a weekly basis to either 26.1 mg/5.2 mg, 39.2 mg/7.8 mg, or 52.3 mg/10.4 mg, until an optimal dose or the maximum dosage of 52.3 mg/10.4 mg/day was reached. At the end of optimization period, subjects were randomly assigned into a 1-week parallel group treatment period to receive either the individually optimized dose of AZSTARYS (mean dose of 45.6 mg/9.0 mg) or placebo.
At the end of the 1-week treatment period, raters evaluated the attention and behavior of the subjects in a laboratory classroom setting over a period of 13 hours using the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) rating scale. SKAMP is a validated 13-item teacher-rated scale that assesses manifestations of ADHD in a classroom setting. On this day, the dose was administered in the morning immediately after breakfast.
The primary efficacy endpoint was the mean change from baseline (pre-dose at randomization visit) of the SKAMP-Combined scores averaged across the test day (not including baseline score), with assessments conducted at 0.5, 1, 2, 4, 8, 10, 12, and 13 hours post-dose.
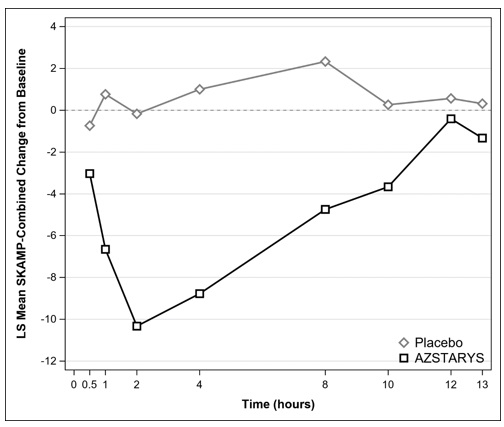
The mean change from baseline in the SKAMP-Combined scores, averaged across the test day, was statistically significantly lower (indicating improvement) with AZSTARYS compared to placebo (Table 2).
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval. * Baseline score assessed at pre-dose on the practice classroom day/randomization visit after 2 days of active drug washout. † Classroom day least-squares mean change from baseline over hours 0.5. 1, 2, 4, 8, 10, 12, and 13. ‡ Difference (active drug minus placebo) in least-squares mean change from baseline. |
|||||
| Study Number | Treatment Group | N | Mean Baseline Score* (SD) | LS Mean Change from Baseline† (SE) | Placebo-subtracted Difference‡ (95% CI) |
| Study 1 | AZSTARYS (26.1 /5.2, 39.2/7.8, 52.3/10.4 mg/day) | 74 | 17.9 (9.2) | -4.87 (0.62) | -5.4 (-7.1, -3.7) |
| Placebo | 76 | 17.9 (10.4) | 0.54 (0.70) | ||
Figure 2: LS Mean Change in SKAMP-Combined Score from Baseline after Treatment with AZSTARYS or Placebo during Classroom Day in Pediatric Patients (6 to 12 years) with ADHD
Adults and Pediatric Patients 13 to 17 years of age with ADHD
The efficacy of 52.3 mg/10.4 mg AZSTARYS in adults and pediatric patients 13 to 17 years of age was established by pharmacokinetic bridging between AZSTARYS (52.3 mg/10.4 mg) and dexmethylphenidate hydrochloride extended-release capsules [see Clinical Pharmacology (12.3)].
16 HOW SUPPLIED/STORAGE AND HANDLING
AZSTARYS (serdexmethylphenidate/dexmethylphenidate) capsules are available as follows:
-
26.1 mg/5.2 mg Capsules – blue cap/grey body, imprinted with "286" on cap and "KP415" on the body
Bottles of 100 ................................................................... NDC 65038-0286-99 -
39.2 mg/7.8 mg Capsules – dark blue cap/grey body, imprinted with "429" on cap and "KP415" on the body
Bottles of 100 ................................................................... NDC 65038-0429-99 -
52.3 mg/10.4 mg Capsules – orange cap/grey body, imprinted with "5612" on cap and "KP415" on the body
Bottles of 100 ................................................................... NDC 65038-0561-99
Storage
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F). [See USP Controlled Room Temperature]. Protect from moisture.
Dispense in tight container (USP).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Abuse, Misuse, and Addiction
Educate patients and their families about the risks of abuse, misuse, and addiction of AZSTARYS, which can lead to overdose and death, and proper disposal of any unused drug [see Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2), Overdosage (10)]. Advise patients to store AZSTARYS in a safe place, preferably locked, and instruct patients to not give AZSTARYS to anyone else.
Risks to Patients with Serious Cardiac Disease
Advise patients that there are potential risks to patients with serious cardiac disease, including sudden death, with AZSTARYS use. Instruct patients to contact a healthcare provider immediately if they develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease [see Warnings and Precautions (5.2)].
Increased Blood Pressure and Heart Rate
Advise patients and their caregivers that AZSTARYS can elevate blood pressure and heart rate [see Warnings and Precautions (5.3)].
Psychiatric Adverse Reactions
Advise patients and their caregivers that AZSTARYS, at recommended doses, can cause psychotic or manic symptoms, even in patients without a prior history or psychotic symptoms or mania [see Warnings and Precautions (5.4)].
Priapism
Advise patients and their caregivers of the possibility of painful or prolonged penile erections (priapism). Instruct the patient to seek immediate medical attention in the event of priapism [see Warnings and Precautions (5.5)].
Circulation Problems in Fingers and Toes [Peripheral vasculopathy, including Raynaud's phenomenon]
- Instruct patients about the risk of peripheral vasculopathy, including Raynaud's phenomenon, and associated signs and symptoms: fingers or toes may feel numb, cool, painful, and/or may change color from pale, to blue, to red.
- Instruct patients to report to their physician any new numbness, pain, skin color change, or sensitivity to temperature in fingers or toes.
- Instruct patients to call their physician immediately with any signs of unexplained wounds appearing on fingers or toes while taking AZSTARYS.
- Further clinical evaluation (e.g., rheumatology referral) may be appropriate for certain patients [see Warnings and Precautions (5.6)].
Long-Term Suppression of Growth in Pediatric Patients
Advise patients and their caregivers that AZSTARYS can cause slowing of growth and weight loss [see Warnings and Precautions (5.7)].
Increased Intraocular Pressure (IOP) and Glaucoma
Advise patients that IOP and glaucoma may occur during treatment with AZSTARYS [see Warnings and Precautions (5.9)].
Motor and Verbal Tics, and Worsening of Tourette’s Syndrome
Advise patients that motor and verbal tics and worsening of Tourette’s Syndrome may occur during treatment with AZSTARYS. Instruct patients to notify their healthcare provider if emergence of new tics or worsening of tics or Tourette’s syndrome occurs [see Warnings and Precautions (5.10)].
Administration Instructions
Advise patients and their caregivers to administer AZSTARYS capsules whole or opened and sprinkled over applesauce or added to water. If sprinkled, advise patients and their caregivers to consume all the drug/food mixture immediately or within 10 minutes of mixing and not to store for future use [see Dosage and Administration (2.3)].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in females exposed to AZSTARYS during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise nursing mother to monitor infants exposed to AZSTARYS through breastmilk for agitation, poor feeding, and reduced weight gain [see Use in Specific Populations (8.2)].
Distributed by:
Corium, LLC
11 Farnsworth St., 4th Floor
Boston, MA 02210

| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 10/2023 | ||
| Medication Guide AZSTARYS™ (az star’ is) (serdexmethylphenidate and dexmethylphenidate) capsules, CII |
|||
|
What is the most important information I should know about AZSTARYS? |
|||
AZSTARYS can cause serious side effects, including:
|
|||
| What is AZSTARYS? | |||
| AZSTARYS is a central nervous system stimulant prescription medicine used for the treatment of Attention-Deficit Hyperactivity Disorder (ADHD) in people 6 years of age and older. AZSTARYS may help increase attention and decrease impulsiveness and hyperactivity in people 6 years of age and older with ADHD. | |||
| It is not known if AZSTARYS is safe and effective in children younger than 6 years of age. | |||
| AZSTARYS is a federally controlled substance (CII) because it contains dexmethylphenidate that can be a target for people who abuse prescription medicines or street drugs. Keep AZSTARYS in a safe place to protect it from theft. Never give AZSTARYS to anyone else because it may cause death or harm them. Selling or giving away AZSTARYS may harm others and is against the law. | |||
Do not take AZSTARYS if you or your child are:
|
|||
Before taking AZSTARYS, tell your healthcare provider about all medical conditions, including if you or your child:
|
|||
| Tell your healthcare provider about all of the medicines that you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | |||
| AZSTARYS and some medicines may interact with each other and cause serious side effects. Sometimes the doses of other medicines will need to be adjusted during treatment with AZSTARYS. Your healthcare provider will decide whether AZSTARYS can be taken with other medicines. | |||
| Especially tell your healthcare provider if you or your child takes: blood pressure medicines (anti-hypertensive) | |||
| Know the medicines that you or your child take. Keep a list of the medicines with you to show your healthcare provider and pharmacist. Do not start any new medicine during treatment with AZSTARYS without talking to your healthcare provider first. | |||
How should AZSTARYS be taken?
|
|||
| What are possible side effects of AZSTARYS? | |||
AZSTARYS can cause serious side effects, including:
|
|||
| The most common side effects of AZSTARYS include: | |||
|
| ||
| These are not all the possible side effects of AZSTARYS. | |||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
How should I store AZSTARYS?
|
|||
| Keep AZSTARYS and all medicines out of the reach of children. | |||
| General information about the safe and effective use of AZSTARYS. | |||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use AZSTARYS for a condition for which it was not prescribed. Do not give AZSTARYS to other people, even if they have the same symptoms that you have. It may harm them, and it is against the law. You can ask your healthcare provider or pharmacist for information about AZSTARYS that was written for healthcare professionals. | |||
| What are the ingredients in AZSTARYS? | |||
| Active ingredients: serdexmethylphenidate and dexmethylphenidate | |||
| Inactive ingredients: colloidal silicon dioxide, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, talc, black iron oxide, titanium dioxide, FD&C Blue No. 1 (26/5.2 mg and 39/7.8 mg), FD&C Red No. 40 (39/7.8 mg and 52/10.4 mg), FD&C Yellow No. 6 (52/10.4 mg) | |||
| Distributed by Corium, LLC, 11 Farnsworth St., 4th Floor , Boston, MA 02210 | |||
| For more information call 1-800-910-8432 or go to www.AZSTARYS.com | |||


