ALKERAN- melphalan tablet, film coated
ApoPharma USA, Inc.
----------
ALKERAN®
(melphalan)
Tablets
WARNING
ALKERAN (melphalan) should be administered under the supervision of a qualified physician experienced in the use of cancer chemotherapeutic agents. Severe bone marrow suppression with resulting infection or bleeding may occur. Melphalan is leukemogenic in humans.
Melphalan produces chromosomal aberrations in vitro and in vivo and, therefore, should be considered potentially mutagenic in humans.
DESCRIPTION
ALKERAN (melphalan), also known as L-phenylalanine mustard, phenylalanine mustard, L-PAM, or L-sarcolysin, is a phenylalanine derivative of nitrogen mustard. Melphalan is a bifunctional alkylating agent which is active against selective human neoplastic diseases. It is known chemically as 4-[bis(2-chloroethyl)amino]-L-phenylalanine. The molecular formula is C13H18Cl2N2O2 and the molecular weight is 305.20. The structural formula is:
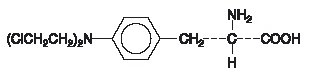
Melphalan is the active L-isomer of the compound and was first synthesized in 1953 by Bergel and Stock; the D-isomer, known as medphalan, is less active against certain animal tumors, and the dose needed to produce effects on chromosomes is larger than that required with the L-isomer. The racemic (DL−) form is known as merphalan or sarcolysin.
Melphalan is practically insoluble in water and has a pKa1 of ~2.5.
ALKERAN (melphalan) is available in tablet form for oral administration. Each film-coated tablet contains 2 mg melphalan and the inactive ingredients colloidal silicon dioxide, crospovidone, hypromellose, macrogol/PEG 400, magnesium stearate, microcrystalline cellulose, and titanium dioxide.
CLINICAL PHARMACOLOGY
Melphalan is an alkylating agent of the bischloroethylamine type. As a result, its cytotoxicity appears to be related to the extent of its interstrand cross-linking with DNA, probably by binding at the N7 position of guanine. Like other bifunctional alkylating agents, it is active against both resting and rapidly dividing tumor cells.
Pharmacokinetics
The absorption of oral melphalan is highly variable with respect to both the time to first appearance of the drug in plasma (range: 0 to 6 hours) and peak plasma concentration (Cmax). The average absolute bioavailability of melphalan is also highly variable (range: 56% to 93%). These results may be due to incomplete intestinal absorption, a variable “first pass” hepatic metabolism, or to rapid hydrolysis. Oral administration of melphalan with a high fat meal may reduce melphalan exposure (AUC) by 36% to 54%.
In 18 patients given a single oral dose of 0.6 mg/kg of ALKERAN, the terminal elimination plasma half-life (t1/2) of parent drug was 1.5 ± 0.83 hours. The 24-hour urinary excretion of parent drug in these patients was 10% ± 4.5%, suggesting that renal clearance is not a major route of elimination of parent drug. In a separate study in 18 patients given single oral doses of 0.2 to 0.25 mg/kg of ALKERAN, Cmax and plasma concentration-time curves (AUC), when dose adjusted to a dose of 14 mg, were (mean ± SD) 212 ± 74 ng/mL and 498 ± 137 ng•hr/mL, respectively. Elimination phase t½ in these patients was approximately 1 hour and the median tmax was 1 hour.
One study using universally labeled 14C-melphalan, found substantially less radioactivity in the urine of patients given the drug by mouth (30% of administered dose in 9 days) than in the urine of those given it intravenously (35% to 65% in 7 days). Following either oral or IV administration, the pattern of label recovery was similar, with the majority being recovered in the first 24 hours. Following oral administration, peak radioactivity occurred in plasma at 2 hours and then disappeared with a half-life of approximately 160 hours. In 1 patient where parent drug (rather than just radiolabel) was determined, the melphalan half-disappearance time was 67 minutes.
The steady-state volume of distribution of melphalan is 0.5 L/kg. Penetration into cerebrospinal fluid (CSF) is low. The average melphalan binding to plasma proteins is highly variable (range: 53% to 92%). Serum albumin is the major binding protein, accounting for approximately 40% to 60% of the plasma protein binding, while α1-acid glycoprotein accounts for about 20% of the plasma protein binding. Approximately 30% of melphalan is (covalently) irreversibly bound to plasma proteins. Interactions with immunoglobulins have been found to be negligible.
Melphalan is eliminated from plasma primarily by chemical hydrolysis to monohydroxymelphalan and dihydroxymelphalan. Aside from these hydrolysis products, no other melphalan metabolites have been observed in humans. Although the contribution of renal elimination to melphalan clearance appears to be low, one pharmacokinetic study showed a significant positive correlation between the elimination rate constant for melphalan and renal function and a significant negative correlation between renal function and the area under the plasma melphalan concentration/time curve.
INDICATIONS AND USAGE
ALKERAN Tablets are indicated for the palliative treatment of multiple myeloma and for the palliation of non-resectable epithelial carcinoma of the ovary.
CONTRAINDICATIONS
ALKERAN should not be used in patients whose disease has demonstrated a prior resistance to this agent. Patients who have demonstrated hypersensitivity to melphalan should not be given the drug.
WARNINGS
ALKERAN should be administered in carefully adjusted dosage by or under the supervision of experienced physicians who are familiar with the drug's actions and the possible complications of its use.
As with other nitrogen mustard drugs, excessive dosage will produce marked bone marrow suppression. Bone marrow suppression is the most significant toxicity associated with ALKERAN in most patients. Therefore, the following tests should be performed at the start of therapy and prior to each subsequent course of ALKERAN: platelet count, hemoglobin, white blood cell count, and differential. Thrombocytopenia and/or leukopenia are indications to withhold further therapy until the blood counts have sufficiently recovered. Frequent blood counts are essential to determine optimal dosage and to avoid toxicity (see PRECAUTIONS: Laboratory Tests). Dose adjustment on the basis of blood counts at the nadir and day of treatment should be considered.
Hypersensitivity reactions, including anaphylaxis, have occurred rarely (see ADVERSE REACTIONS). These reactions have occurred after multiple courses of treatment and have recurred in patients who experienced a hypersensitivity reaction to IV ALKERAN. If a hypersensitivity reaction occurs, oral or IV ALKERAN should not be readministered.
Carcinogenesis
Secondary malignancies, including acute nonlymphocytic leukemia, myeloproliferative syndrome, and carcinoma have been reported in patients with cancer treated with alkylating agents (including melphalan). Some patients also received other chemotherapeutic agents or radiation therapy. Precise quantitation of the risk of acute leukemia, myeloproliferative syndrome, or carcinoma is not possible. Published reports of leukemia in patients who have received melphalan (and other alkylating agents) suggest that the risk of leukemogenesis increases with chronicity of treatment and with cumulative dose. In one study, the 10-year cumulative risk of developing acute leukemia or myeloproliferative syndrome after melphalan therapy was 19.5% for cumulative doses ranging from 730 mg to 9,652 mg. In this same study, as well as in an additional study, the 10-year cumulative risk of developing acute leukemia or myeloproliferative syndrome after melphalan therapy was less than 2% for cumulative doses under 600 mg. This does not mean that there is a cumulative dose below which there is no risk of the induction of secondary malignancy. The potential benefits from melphalan therapy must be weighed on an individual basis against the possible risk of the induction of a second malignancy.
Adequate and well-controlled carcinogenicity studies have not been conducted in animals. However, i.p. administration of melphalan in rats (5.4 to 10.8 mg/m2) and in mice (2.25 to 4.5 mg/m2) 3 times per week for 6 months followed by 12 months post-dose observation produced peritoneal sarcoma and lung tumors, respectively.
Mutagenesis
ALKERAN has been shown to cause chromatid or chromosome damage in humans. Intramuscular administration of ALKERAN at 6 and 60 mg/m2 produced structural aberrations of the chromatid and chromosomes in bone marrow cells of Wistar rats.
Impairment of Fertility
ALKERAN causes suppression of ovarian function in premenopausal women, resulting in amenorrhea in a significant number of patients. Reversible and irreversible testicular suppression have also been reported.
Pregnancy
ALKERAN may cause fetal harm when administered to a pregnant woman. Melphalan was embryolethal and teratogenic in rats following oral (6 to 18 mg/m2/day for 10 days) and intraperitoneal (18 mg/m2) administration. Malformations resulting from melphalan included alterations of the brain (underdevelopment, deformation, meningocele, and encephalocele) and eye (anophthalmia and microphthalmos), reduction of the mandible and tail, as well as hepatocele (exomphaly).
There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
PRECAUTIONS
General
In all instances where the use of ALKERAN is considered for chemotherapy, the physician must evaluate the need and usefulness of the drug against the risk of adverse events. ALKERAN should be used with extreme caution in patients whose bone marrow reserve may have been compromised by prior irradiation or chemotherapy, or whose marrow function is recovering from previous cytotoxic therapy. If the leukocyte count falls below 3,000 cells/mcL, or the platelet count below 100,000 cells/mcL, ALKERAN should be discontinued until the peripheral blood cell counts have recovered.
A recommendation as to whether or not dosage reduction should be made routinely in patients with renal insufficiency cannot be made because:
a) There is considerable inherent patient-to-patient variability in the systemic availability of melphalan in patients with normal renal function.
b) Only a small amount of the administered dose appears as parent drug in the urine of patients with normal renal function.
Patients with azotemia should be closely observed, however, in order to make dosage reductions, if required, at the earliest possible time.
Administration of live vaccines to immunocompromised patients should be avoided.
Information for Patients
Patients should be informed that the major toxicities of ALKERAN are related to bone marrow suppression, hypersensitivity reactions, gastrointestinal toxicity, and pulmonary toxicity. The major long-term toxicities are related to infertility and secondary malignancies. Patients should never be allowed to take the drug without close medical supervision and should be advised to consult their physician if they experience skin rash, vasculitis, bleeding, fever, persistent cough, nausea, vomiting, amenorrhea, weight loss, or unusual lumps/masses. Women of childbearing potential should be advised to avoid becoming pregnant.
Laboratory Tests
Periodic complete blood counts with differentials should be performed during the course of treatment with ALKERAN. At least one determination should be obtained prior to each treatment course. Patients should be observed closely for consequences of bone marrow suppression, which include severe infections, bleeding, and symptomatic anemia (see WARNINGS).
Nursing Mothers
It is not known whether this drug is excreted in human milk. ALKERAN should not be given to nursing mothers.
Pediatric Use
The safety and effectiveness of ALKERAN in pediatric patients have not been established.
Geriatric Use
Clinical studies of ALKERAN Tablets did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
Hematologic
The most common side effect is bone marrow suppression leading to leukopenia, thrombocytopenia, and anemia. Although bone marrow suppression frequently occurs, it is usually reversible if melphalan is withdrawn early enough. However, irreversible bone marrow failure has been reported.
Gastrointestinal
Nausea, vomiting, diarrhea, and oral ulceration occur. Hepatic disorders ranging from abnormal liver function tests to clinical manifestations such as hepatitis and jaundice have been reported.
Miscellaneous
Other reported adverse reactions include: pulmonary fibrosis (including fatal outcomes) and interstitial pneumonitis, skin hypersensitivity, maculopapular rashes, vasculitis, alopecia, and hemolytic anemia. Allergic reactions, including urticaria, edema, skin rashes, and rare anaphylaxis, have occurred after multiple courses of treatment. Cardiac arrest has also been reported rarely in association with such reports.
OVERDOSAGE
Overdoses, including doses up to 50 mg/day for 16 days, have been reported. Immediate effects are likely to be vomiting, ulceration of the mouth, diarrhea, and hemorrhage of the gastrointestinal tract. The principal toxic effect is bone marrow suppression. Hematologic parameters should be closely followed for 3 to 6 weeks. An uncontrolled study suggests that administration of autologous bone marrow or hematopoietic growth factors (i.e., sargramostim, filgrastim) may shorten the period of pancytopenia. General supportive measures, together with appropriate blood transfusions and antibiotics, should be instituted as deemed necessary by the physician. This drug is not removed from plasma to any significant degree by hemodialysis.
DOSAGE AND ADMINISTRATION
Multiple Myeloma
The usual oral dose is 6 mg (3 tablets) daily. The entire daily dose may be given at one time. The dose is adjusted, as required, on the basis of blood counts done at approximately weekly intervals. After 2 to 3 weeks of treatment, the drug should be discontinued for up to 4 weeks, during which time the blood count should be followed carefully. When the white blood cell and platelet counts are rising, a maintenance dose of 2 mg daily may be instituted. Because of the patient-to-patient variation in melphalan plasma levels following oral administration of the drug, several investigators have recommended that the dosage of ALKERAN be cautiously escalated until some myelosuppression is observed in order to assure that potentially therapeutic levels of the drug have been reached.
Other dosage regimens have been used by various investigators. Osserman and Takatsuki have used an initial course of 10 mg/day for 7 to 10 days. They report that maximal suppression of the leukocyte and platelet counts occurs within 3 to 5 weeks and recovery within 4 to 8 weeks. Continuous maintenance therapy with 2 mg/day is instituted when the white blood cell count is greater than 4,000 cells/mcL and the platelet count is greater than 100,000 cells/mcL. Dosage is adjusted to between 1 and 3 mg/day depending upon the hematological response. It is desirable to try to maintain a significant degree of bone marrow depression so as to keep the leukocyte count in the range of 3,000 to 3,500 cells/mcL.
Hoogstraten et al have started treatment with 0.15 mg/kg/day for 7 days. This is followed by a rest period of at least 14 days, but it may be as long as 5 to 6 weeks. Maintenance therapy is started when the white blood cell and platelet counts are rising. The maintenance dose is 0.05 mg/kg/day or less and is adjusted according to the blood count.
Available evidence suggests that about one third to one half of the patients with multiple myeloma show a favorable response to oral administration of the drug.
One study by Alexanian et al has shown that the use of ALKERAN in combination with prednisone significantly improves the percentage of patients with multiple myeloma who achieve palliation. One regimen has been to administer courses of ALKERAN at 0.25 mg/kg/day for 4 consecutive days (or, 0.20 mg/kg/day for 5 consecutive days) for a total dose of 1 mg/kg/course. These 4- to 5-day courses are then repeated every 4 to 6 weeks if the granulocyte count and the platelet count have returned to normal levels.
It is to be emphasized that response may be very gradual over many months; it is important that repeated courses or continuous therapy be given since improvement may continue slowly over many months, and the maximum benefit may be missed if treatment is abandoned too soon.
In patients with moderate to severe renal impairment, currently available pharmacokinetic data do not justify an absolute recommendation on dosage reduction to those patients, but it may be prudent to use a reduced dose initially.
HOW SUPPLIED
ALKERAN is supplied as white, film-coated, round, biconvex tablets containing 2 mg melphalan in amber glass bottles with child-resistant closures. One side is engraved with “GX EH3” and the other side is engraved with an “A.”
Bottle of 50 (NDC 52609-0001-5).
Store in a refrigerator, 2° to 8°C (36° to 46°F). Protect from light.
REFERENCES
- 1.
- NIOSH Alert: Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Healthcare Settings. U.S. Department of Health and Human Services, Public Health Service. Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- 2.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- 3.
- American Society of Health-System Pharmacists. (2006) ASHP Guidelines on Handling Hazardous Drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
- 4.
- Polovich M, White JM, Kelleher LO (eds.) 2005. Chemotherapy and Biotherapy Guidelines and Recommendations for Practice. (2nd ed.) Pittsburgh, PA: Oncology Nursing Society.
ALKERAN is a registered trademark of Apotex Inc.
Manufactured by Excella GmbH & Co. KG, 90537 Feucht, Germany for Apotex Inc., Toronto, Ontario, Canada M9L 1T9
Distributed by ApoPharma USA Inc.
Weston, FL 33326
©2016, Apotex Inc. All rights reserved.
May 2017
PRINICIPAL DISPLAY PANEL
NDC 52609-0001-5
ALKERAN®
(melphalan)
Tablets 2 mg
50 Tablets
Each tablet contains 2 mg melphalan.
Rx only
ApoPharma
WARNING: This drug is only to be taken under close medical supervision. Do not take in larger doses or more frequently or for a longer time than specifically directed by the physician. Periodic blood counts are necessary to determine proper dose and to avoid ill effects.
See prescribing information for Dosage and Administration.
Store in a refrigerator, 2o to 8oC (36o to 46oF). Protect from light.
Dispense in glass, tight, light-resistant container as defined in the USP.
Excella GmbH & Co. KG, 90537 Feucht, Germany
7579701-0647

| ALKERAN
melphalan tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - ApoPharma USA, Inc. (962810821) |