Label: BYOOVIZ- ranibizumab-nuna injection, solution

- NDC Code(s): 64406-019-01, 64406-019-07, 64406-019-08, 64406-019-09
- Packager: BIOGEN INC.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated October 6, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BYOOVIZ safely and effectively. See full prescribing information for BYOOVIZ.
BYOOVIZ™ (ranibizumab-nuna) injection, for intravitreal use
Initial U.S. Approval: 2021
BYOOVIZ (ranibizumab-nuna) is biosimilar* to LUCENTIS® (ranibizumab injection)INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
For ophthalmic intravitreal injection only (2.1)
- Neovascular (Wet) Age-Related Macular Degeneration (AMD) (2.2):
BYOOVIZ 0.5 mg (0.05 mL) is recommended to be administered by intravitreal injection once a month (approximately 28 days).- -
- Although not as effective, patients may be treated with 3 monthly doses followed by less frequent dosing with regular assessment.
- -
- Although not as effective, patients may also be treated with one dose every 3 months after 4 monthly doses. Patients should be assessed regularly.
- Macular Edema Following Retinal Vein Occlusion (RVO) (2.3):
BYOOVIZ 0.5 mg (0.05 mL) is recommended to be administered by intravitreal injection once a month (approximately 28 days). - Myopic Choroidal Neovascularization (mCNV) (2.4):
BYOOVIZ 0.5 mg (0.05 mL) is recommended to be initially administered by intravitreal injection once a month (approximately 28 days) for up to three months. Patients may be retreated if needed.
DOSAGE FORMS AND STRENGTHS
Single-dose glass vial designed to provide 0.05 mL for intravitreal injections: 10 mg/mL solution. (3)
WARNINGS AND PRECAUTIONS
- Endophthalmitis and retinal detachments may occur following intravitreal injections. Patients should be monitored following the injection (5.1).
- Increases in intraocular pressure (IOP) have been noted both pre- and post-intravitreal injection (5.2).
- There is a potential risk of arterial thromboembolic events following intravitreal use of VEGF inhibitors (5.3).
ADVERSE REACTIONS
- The most common adverse reactions (reported more frequently in ranibizumab treated subjects than control subjects) are conjunctival hemorrhage, eye pain, vitreous floaters, and increased IOP (6.2).
To report SUSPECTED ADVERSE REACTIONS, contact Biogen Inc. at 1-877-422-8360 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
- *
- Biosimilar means that the biological product is approved based on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product. Biosimilarity of BYOOVIZ has been demonstrated for the condition(s) of use (e.g., indication(s), dosing regimen(s)), strength(s), dosage form(s), and route(s) of administration described in its Full Prescribing Information.
Revised: 10/2023
- Neovascular (Wet) Age-Related Macular Degeneration (AMD) (2.2):
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
2.3 Macular Edema Following Retinal Vein Occlusion (RVO)
2.4 Myopic Choroidal Neovascularization (mCNV)
2.5 Preparation for Administration
2.6 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
5.2 Increases in Intraocular Pressure
5.3 Thromboembolic Events
6 ADVERSE REACTIONS
6.1 Injection Procedure
6.2 Clinical Trials Experience
6.3 Immunogenicity
6.4 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
14.2 Macular Edema Following Retinal Vein Occlusion (RVO)
14.3 Myopic Choroidal Neovascularization (mCNV)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
BYOOVIZ 0.5 mg (0.05 mL of 10 mg/mL solution) is recommended to be administered by intravitreal injection once a month (approximately 28 days).
Although not as effective, patients may be treated with 3 monthly doses followed by less frequent dosing with regular assessment. In the 9 months after three initial monthly doses, less frequent dosing with 4-5 doses on average is expected to maintain visual acuity while monthly dosing may be expected to result in an additional average 1-2 letter gain. Patients should be assessed regularly [see Clinical Studies (14.1)].
Although not as effective, patients may also be treated with one dose every 3 months after 4 monthly doses. Compared with continued monthly dosing, dosing every 3 months over the next 9 months will lead to an approximate 5-letter (1-line) loss of visual acuity benefit, on average. Patients should be assessed regularly [see Clinical Studies (14.1)].
2.3 Macular Edema Following Retinal Vein Occlusion (RVO)
BYOOVIZ 0.5 mg (0.05 mL of 10 mg/mL solution) is recommended to be administered by intravitreal injection once a month (approximately 28 days).
In Studies RVO-1 and RVO-2, patients received monthly injections of ranibizumab for 6 months. In spite of being guided by optical coherence tomography and visual acuity re-treatment criteria, patients who were then not treated at Month 6 experienced on average, a loss of visual acuity at Month 7, whereas patients who were treated at Month 6 did not. Patients should be treated monthly [see Clinical Studies (14.2)].
2.4 Myopic Choroidal Neovascularization (mCNV)
BYOOVIZ 0.5 mg (0.05 mL of 10 mg/mL BYOOVIZ solution) is recommended to be initially administered by intravitreal injection once a month (approximately 28 days) for up to 3 months. Patients may be retreated if needed [see Clinical Studies (14.3)].
2.5 Preparation for Administration
Vial:
Using aseptic technique, all of the BYOOVIZ vial contents are withdrawn through a 5-micron (19- gauge × 1-1/2 inch), sterile filter needle attached to a 1 mL syringe (not included). The filter needle should be discarded after withdrawal of the vial contents and should not be used for intravitreal injection. The filter needle should be replaced with a sterile 30-gauge × ½ inch needle for the intravitreal injection.
Use aseptic technique to carry out the following preparation steps:
- Prepare for intravitreal injection with the following medical devices for single use (not included):
- a 5-micron sterile filter needle (19-gauge × 1-1/2 inch)
- a 1 mL sterile Luer lock syringe (with marking to measure 0.05 mL)
- a sterile injection needle (30-gauge × 1/2-inch)
- Before withdrawal, disinfect the outer part of the rubber stopper of the vial.
- Place a 5-micron filter needle (19-gauge × 1-1/2 inch) onto a 1 mL Luer lock syringe using aseptic technique.
- Push the filter needle into the center of the vial stopper until the needle touches the bottom edge of the vial.
- Withdraw all the liquid from the vial, keeping the vial in an upright position, slightly inclined to ease complete withdrawal.
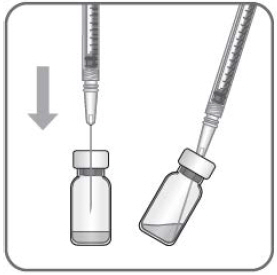
- Ensure that the plunger rod is drawn sufficiently back when emptying the vial in order to completely empty the filter needle.
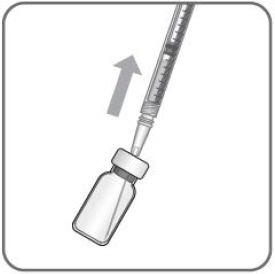
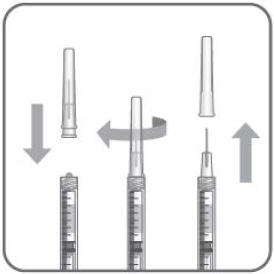
- The filter needle should be discarded after withdrawal of the vial contents and must not be used for the intravitreal injection.
- Attach a 30-gauge × 1/2-inch sterile injection needle firmly onto the syringe by screwing it tightly onto the Luer lock. Carefully remove the needle cap by pulling it straight off. Do not wipe the needle at any time.
- Hold the syringe with the needle pointing up. If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top.
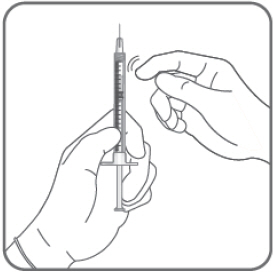
- Hold the syringe at eye level, and carefully push the plunger rod until the plunger tip is aligned with the line that marks 0.05 mL on the syringe.
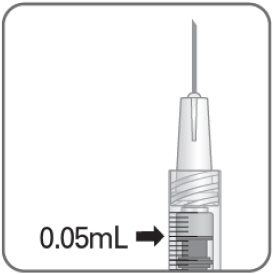
2.6 Administration
The intravitreal injection procedure should be carried out under controlled aseptic conditions, which include the use of sterile gloves, a sterile drape, and a sterile eyelid speculum (or equivalent).
Adequate anesthesia and a broad-spectrum microbicide should be given prior to the injection.
Prior to and 30 minutes following the intravitreal injection, patients should be monitored for elevation in intraocular pressure using tonometry. Monitoring may also consist of a check for perfusion of the optic nerve head immediately after the injection [see Warnings and Precautions (5.2)]. Patients should also be monitored for and instructed to report any symptoms suggestive of endophthalmitis without delay following the injection [see Warnings and Precautions (5.1)].
Each vial should only be used for the treatment of a single eye. If the contralateral eye requires treatment, a new vial should be used and the sterile field, syringe, gloves, drapes, eyelid speculum, filter needle (vial only), and injection needles should be changed before BYOOVIZ is administered to the other eye.
No special dosage modification is required for any of the populations that have been studied (e.g., gender, elderly).
- Prepare for intravitreal injection with the following medical devices for single use (not included):
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
Intravitreal injections, including those with ranibizumab products, have been associated with endophthalmitis and retinal detachments. Proper aseptic injection technique should always be used when administering BYOOVIZ. In addition, patients should be monitored following the injection to permit early treatment should an infection occur [see Dosage and Administration (2.5, 2.6) and Patient Counseling Information (17)].
5.2 Increases in Intraocular Pressure
Increases in intraocular pressure have been noted both pre-injection and post-injection (at 60 minutes) while being treated with ranibizumab products. Monitor intraocular pressure prior to and following intravitreal injection with BYOOVIZ and manage appropriately [see Dosage and Administration (2.6)].
5.3 Thromboembolic Events
Although there was a low rate of arterial thromboembolic events (ATEs) observed in the ranibizumab clinical trials, there is a potential risk of ATEs following intravitreal use of VEGF inhibitors. Arterial thromboembolic events are defined as nonfatal stroke, nonfatal myocardial infarction, or vascular death (including deaths of unknown cause).
Neovascular (Wet) Age-Related Macular Degeneration
The ATE rate in the three controlled neovascular AMD studies (AMD-1, AMD-2, AMD-3) during the first year was 1.9% (17 of 874) in the combined group of patients treated with 0.3 mg or 0.5 mg ranibizumab compared with 1.1% (5 of 441) in patients from the control arms [see Clinical Studies (14.1)]. In the second year of Studies AMD-1 and AMD-2, the ATE rate was 2.6% (19 of 721) in the combined group of ranibizumab-treated patients compared with 2.9% (10 of 344) in patients from the control arms. In Study AMD-4, the ATE rates observed in the 0.5 mg arms during the first and second year were similar to rates observed in Studies AMD-1, AMD-2, and AMD-3.
In a pooled analysis of 2-year controlled studies [AMD-1, AMD-2, and a study of ranibizumab used adjunctively with verteporfin photodynamic therapy (PDT)], the stroke rate (including both ischemic and hemorrhagic stroke) was 2.7% (13 of 484) in patients treated with 0.5 mg ranibizumab compared to 1.1% (5 of 435) in patients in the control arms [odds ratio 2.2 (95% confidence interval (0.8-7.1)].
Macular Edema Following Retinal Vein Occlusion
The ATE rate in the two controlled RVO studies during the first 6 months was 0.8% in both the ranibizumab and control arms of the studies (4 of 525 in the combined group of patients treated with 0.3 mg or 0.5 mg ranibizumab and 2 of 260 in the control arms) [see Clinical Studies (14.2)]. The stroke rate was 0.2% (1 of 525) in the combined group of ranibizumab -treated patients compared to 0.4% (1 of 260) in the control arms.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Endophthalmitis and Retinal Detachments [see Warnings and Precautions (5.1)]
- Increases in Intraocular Pressure [see Warnings and Precautions (5.2)]
- Thromboembolic Events [see Warnings and Precautions (5.3)]
6.1 Injection Procedure
Serious adverse reactions related to the injection procedure have occurred in < 0.1% of intravitreal injections, including endophthalmitis [see Warnings and Precautions (5.1)], rhegmatogenous retinal detachment, and iatrogenic traumatic cataract.
6.2 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of the same or another drug and may not reflect the rates observed in practice.
The data below reflect exposure to 0.5 mg ranibizumab in 440 patients with neovascular AMD in Studies AMD-1, AMD-2, and AMD-3; in 259 patients with macular edema following RVO.
Safety data observed in 224 patients with mCNV, as well as Studies AMD-4 and D-3, were consistent with these results. On average, the rates and types of adverse reactions in patients were not significantly affected by dosing regimen.
Ocular Reactions
Table 1 shows frequently reported ocular adverse reactions in ranibizumab-treated patients compared with the control group.
Table 1 Ocular Reactions in the AMD, and RVO Studies Adverse Reaction AMD
2-yearAMD
1-yearRVO
6-monthRanibizumab
0.5 mgControl Ranibizumab
0.5 mgControl Ranibizumab
0.5 mgControl n=379 n=379 n=440 n=441 n=259 n=260 Conjunctival hemorrhage 74% 60% 64% 50% 48% 37% Eye pain 35% 30% 26% 20% 17% 12% Vitreous floaters 27% 8% 19% 5% 7% 2% Intraocular pressure increased 24% 7% 17% 5% 7% 2% Vitreous detachment 21% 19% 15% 15% 4% 2% Intraocular inflammation 18% 8% 13% 7% 1% 3% Cataract 17% 14% 11% 9% 2% 2% Foreign body sensation in eyes 16% 14% 13% 10% 7% 5% Eye irritation 15% 15% 13% 12% 7% 6% Lacrimation increased 14% 12% 8% 8% 2% 3% Blepharitis 12% 8% 8% 5% 0% 1% Dry eye 12% 7% 7% 7% 3% 3% Visual disturbance or vision blurred 18% 15% 13% 10% 5% 3% Eye pruritis 12% 11% 9% 7% 1% 2% Ocular hyperemia 11% 8% 7% 4% 5% 3% Retinal disorder 10% 7% 8% 4% 2% 1% Maculopathy 9% 9% 6% 6% 11% 7% Retinal degeneration 8% 6% 5% 3% 1% 0% Ocular discomfort 7% 4% 5% 2% 2% 2% Conjunctival hyperemia 7% 6% 5% 4% 0% 0% Posterior capsule opacification 7% 4% 2% 2% 0% 1% Injection site hemorrhage 5% 2% 3% 1% 0% 0% Non-Ocular Reactions
Non-ocular adverse reactions with an incidence of ≥ 5% in patients receiving ranibizumab for AMD, and/or RVO and which occurred at a ≥ 1% higher frequency in patients treated with ranibizumab compared to control are shown in Table 2. Though less common, wound healing complications were also observed in some studies.
Table 2 Non-Ocular Reactions in the AMD, and RVO Studies Adverse Reaction AMD
2-yearAMD
1-yearRVO
6-monthRanibizumab
0.5 mgControl Ranibizumab
0.5 mgControl Ranibizumab
0.5 mgControl n=379 n=379 n=440 n=441 n=259 n=260 Nasopharyngitis 16% 13% 8% 9% 5% 4% Anemia 8% 7% 4% 3% 1% 1% Nausea 9% 6% 5% 5% 1% 2% Cough 9% 8% 5% 4% 1% 2% Constipation 5% 7% 3% 4% 0% 1% Seasonal allergy 4% 4% 2% 2% 0% 2% Hypercholesterolemia 5% 5% 3% 2% 1% 1% Influenza 7% 5% 3% 2% 3% 2% Renal failure 1% 1% 0% 0% 0% 0% Upper respiratory tract infection 9% 8% 5% 5% 2% 2% Gastroesophageal reflux disease 4% 6% 3% 4% 1% 0% Headache 12% 9% 6% 5% 3% 3% Edema peripheral 3% 5% 2% 3% 0% 1% Renal failure chronic 0% 1% 0% 0% 0% 0% Neuropathy peripheral 1% 1% 1% 0% 0% 0% Sinusitis 8% 7% 5% 5% 3% 2% Bronchitis 11% 9% 6% 5% 0% 2% Atrial fibrillation 5% 4% 2% 2% 1% 0% Arthralgia 11% 9% 5% 5% 2% 1% Chronic obstructive pulmonary disease 6% 3% 3% 1% 0% 0% Wound healing complications 1% 1% 1% 0% 0% 0% 6.3 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other ranibizumab products may be misleading.
The pre-treatment incidence of immunoreactivity to ranibizumab was 0%-5% across treatment groups. After monthly dosing with ranibizumab for 6 to 24 months, antibodies to ranibizumab were detected in approximately 1%-9% of patients.
The clinical significance of immunoreactivity to ranibizumab products are unclear at this time. Among neovascular AMD patients with the highest levels of immunoreactivity, some were noted to have iritis or vitritis.
Intraocular inflammation was not observed in patients with RVO patients with the highest levels of immunoreactivity.
6.4 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ranibizumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Ocular: Tear of retinal pigment epithelium among patients with neovascular AMD
-
7 DRUG INTERACTIONS
Drug interaction studies have not been conducted with ranibizumab products.
Ranibizumab intravitreal injection has been used adjunctively with Photodynamic Therapy (PDT). Twelve of 105 (11%) patients with neovascular AMD developed serious intraocular inflammation; in 10 of the 12 patients, this occurred when ranibizumab was administered 7 days (± 2 days) after PDT.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of ranibizumab products administered in pregnant women.
Administration of ranibizumab to pregnant monkeys throughout the period of organogenesis resulted in a low incidence of skeletal abnormalities at intravitreal doses 13-times the predicted human exposure (based on maximal serum trough levels [Cmax]) after a single eye treatment at the recommended clinical dose. No skeletal abnormalities were observed at serum trough levels equivalent to the predicted human exposure after a single eye treatment at the recommended clinical dose [see Animal Data].
Animal reproduction studies are not always predictive of human response, and it is not known whether ranibizumab products can cause fetal harm when administered to a pregnant woman. Based on the anti-VEGF mechanism of action for ranibizumab products [see Clinical Pharmacology (12.1)], treatment with ranibizumab products may pose a risk to human embryofetal development.
BYOOVIZ should be given to a pregnant woman only if clearly needed.
Data
Animal Data
An embryo-fetal developmental toxicity study was performed on pregnant cynomolgus monkeys. Pregnant animals received intravitreal injections of ranibizumab every 14 days starting on Day 20 of gestation, until Day 62 at doses of 0, 0.125, and 1 mg/eye. Skeletal abnormalities including incomplete and/or irregular ossification of bones in the skull, vertebral column, and hindlimbs and shortened supernumerary ribs were seen at a low incidence in fetuses from animals treated with 1 mg/eye of ranibizumab. The 1 mg/eye dose resulted in trough serum ranibizumab levels up to 13 times higher than predicted Cmax levels with single eye treatment in humans. No skeletal abnormalities were seen at the lower dose of 0.125 mg/eye, a dose which resulted in trough exposures equivalent to single eye treatment in humans. No effect on the weight or structure of the placenta, maternal toxicity, or embryotoxicity was observed.
8.2 Lactation
Risk Summary
There are no data available on the presence of ranibizumab products in human milk, the effects of ranibizumab products on the breastfed infant or the effects of ranibizumab products on milk production/excretion.
Because many drugs are excreted in human milk, and because the potential for absorption and harm to infant growth and development exists, caution should be exercised when BYOOVIZ is administered to a nursing woman.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BYOOVIZ and any potential adverse effects on the breastfed child from BYOOVIZ.
8.3 Females and Males of Reproductive Potential
Infertility
No studies on the effects of ranibizumab products on fertility have been conducted and it is not known whether ranibizumab products can affect reproduction capacity. Based on the anti-VEGF mechanism of action for ranibizumab products, treatment with ranibizumab products may pose a risk to reproductive capacity.
8.4 Pediatric Use
The safety and effectiveness of ranibizumab products in pediatric patients have not been established.
8.5 Geriatric Use
In the clinical studies, approximately 76% (2449 of 3227) of patients randomized to treatment with ranibizumab were ≥ 65 years of age and approximately 51% (1644 of 3227) were ≥ 75 years of age [see Clinical Studies (14)]. No notable differences in efficacy or safety were seen with increasing age in these studies. Age did not have a significant effect on systemic exposure.
- 10 OVERDOSAGE
-
11 DESCRIPTION
BYOOVIZ (ranibizumab-nuna) is a recombinant humanized IgG1 kappa isotype monoclonal antibody fragment designed for intraocular use. Ranibizumab-nuna binds to and inhibits the biologic activity of human vascular endothelial growth factor A (VEGF-A). Ranibizumab-nuna, which lacks an Fc region, has a molecular weight of approximately 48 kilodaltons and is produced by an E. coli expression system in a nutrient medium containing the antibiotic tetracycline. Tetracycline is not detectable in the final product.
BYOOVIZ (ranibizumab-nuna) injection is a sterile, clear to slightly opalescent and colorless to pale yellow solution in a single-dose glass vial for intravitreal use. BYOOVIZ is supplied as a preservative-free, sterile solution in a single-dose container designed to deliver 0.05 mL of 10 mg/mL BYOOVIZ (0.5 mg dose vial) aqueous solution with 10 mM histidine HCl, 10% α,α-trehalose dihydrate, 0.01% polysorbate 20, pH 5.5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ranibizumab products bind to the receptor binding site of active forms of VEGF-A, including the biologically active, cleaved form of this molecule, VEGF110. VEGF-A has been shown to cause neovascularization and leakage in models of ocular angiogenesis and vascular occlusion and is thought to contribute to pathophysiology of neovascular AMD, mCNV, and macular edema following RVO. The binding of ranibizumab products to VEGF-A prevents the interaction of VEGF-A with its receptors (VEGFR1 and VEGFR2) on the surface of endothelial cells, reducing endothelial cell proliferation, vascular leakage, and new blood vessel formation.
12.2 Pharmacodynamics
Increased retinal thickness (i.e., center point thickness (CPT) or central foveal thickness (CFT)), as assessed by optical coherence tomography (OCT) is associated with neovascular AMD, mCNV, and macular edema following RVO. Leakage from choroidal neovascularization (CNV) as assessed by fluorescein angiography (FA) is associated with neovascular AMD and mCNV.
Neovascular (Wet) Age-Related Macular Degeneration
In Study AMD-3, CPT was assessed by time domain (TD)-OCT in 118 of 184 patients. TD-OCT measurements were collected at baseline, Months 1, 2, 3, 5, 8, and 12. In patients treated with ranibizumab, CPT decreased, on average, more than in the sham group from baseline through Month 12. CPT decreased by Month 1 and decreased further at Month 3, on average. In this study, CPT data did not provide information useful in influencing treatment decisions [see Clinical Studies (14.1)].
In Study AMD-4, CFT was assessed by spectral domain (SD)-OCT in all patients; on average, CFT reductions were observed beginning at Day 7 following the first ranibizumab injection through Month 24. CFT data did not provide information capable of predicting final visual acuity results [see Clinical Studies (14.1)].
In patients treated with ranibizumab, the area of CNV leakage, on average, decreased by Month 3 as assessed by FA. The area of CNV leakage for an individual patient was not correlated with visual acuity.
Macular Edema Following Retinal Vein Occlusion
On average, CPT reductions were observed in Studies RVO-1 and RVO-2 beginning at Day 7 following the first ranibizumab injection through Month 6. CPT was not evaluated as a means to guide treatment decisions [see Clinical Studies (14.2)].
Myopic Choroidal Neovascularization
On average CFT reductions were observed as early as Month 1, and were greater in the ranibizumab groups compared to PDT [see Clinical Studies (14.3)].
12.3 Pharmacokinetics
In patients with neovascular AMD, following monthly intravitreal administration of 0.5 mg ranibizumab, mean (±SD) maximum ranibizumab serum concentrations were 1.7 (± 1.1) ng/mL. These concentrations were below the concentration range of ranibizumab (11 to 27 ng/mL) that was necessary to inhibit the biological activity of VEGF-A by 50%, as measured in an in vitro cellular proliferation assay (based on human umbilical vein endothelial cells (HUVEC)). No significant change from baseline was observed in the mean plasma VEGF concentrations following three monthly 0.5 mg intravitreal injections. The maximum observed serum concentration was dose proportional over the dose range of 0.05 to 2 mg/eye. Serum ranibizumab concentrations in RVO patients were similar to those observed in neovascular AMD patients.
Based on a population pharmacokinetic analysis of patients with neovascular AMD, maximum serum concentrations are predicted to be reached at approximately 1 day after monthly intravitreal administration of ranibizumab 0.5 mg/eye. Based on the disappearance of ranibizumab from serum, the estimated average vitreous elimination half-life was approximately 9 days. Steady-state minimum concentration is predicted to be 0.22 ng/mL with a monthly dosing regimen. In humans, serum ranibizumab concentrations are predicted to be approximately 90,000-fold lower than vitreal concentrations.
In pharmacokinetic covariate analyses, 48% (520/1091) of patients had renal impairment (35% mild, 11% moderate, and 2% severe). Because the increases in plasma ranibizumab exposures in these patients are not considered clinically significant, no dosage adjustment is needed based on renal impairment status.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to determine the carcinogenic potential of ranibizumab products. Based on the anti-VEGF mechanism of action of ranibizumab products, treatment with ranibizumab products may pose a risk to reproductive capacity [see Females and Males of Reproductive Potential (8.3)].
-
14 CLINICAL STUDIES
Unless otherwise noted, visual acuity was measured at a distance of 4 meters.
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The safety and efficacy of ranibizumab were assessed in three randomized, double-masked, sham- or active-controlled studies in patients with neovascular AMD. A total of 1323 patients (ranibizumab 879, control 444) were enrolled in the three studies.
Studies AMD-1 and AMD-2
In Study AMD-1, patients with minimally classic or occult (without classic) CNV lesions received monthly ranibizumab 0.3 mg or 0.5 mg intravitreal injections or monthly sham injections. Data are available through Month 24. Patients treated with ranibizumab in Study AMD-1 received a mean of 22 total treatments out of a possible 24 from Day 0 to Month 24.
In Study AMD-2, patients with predominantly classic CNV lesions received one of the following: 1) monthly ranibizumab 0.3 mg intravitreal injections and sham PDT; 2) monthly ranibizumab 0.5 mg intravitreal injections and sham PDT; or 3) sham intravitreal injections and active PDT. Sham PDT (or active PDT) was given with the initial ranibizumab (or sham) intravitreal injection and every 3 months thereafter if FA showed persistence or recurrence of leakage. Data are available through Month 24. Patients treated with ranibizumab in Study AMD-2 received a mean of 21 total treatments out of a possible 24 from Day 0 through Month 24.
In both studies, the primary efficacy endpoint was the proportion of patients who maintained vision, defined as losing fewer than 15 letters of visual acuity at 12 months compared with baseline. Almost all ranibizumab-treated patients (approximately 95%) maintained their visual acuity. Among ranibizumab-treated patients, 31% to 37% experienced a clinically significant improvement in vision, defined as gaining 15 or more letters at 12 months. The size of the lesion did not significantly affect the results. Detailed results are shown in Table 3, Table 4, and Figure 1 below.
Table 3 Visual Acuity Outcomes at Month 12 and Month 24 in Study AMD-1 Outcome Measure Month Sham
n=229Ranibizumab
0.5 mg n=230Estimated Difference (95% CI)* - *
- Adjusted estimate based on the stratified model; p < 0.01
Loss of <15 letters in visual acuity (%) 12 60% 91% 30%
(23%, 37%)24 56% 89% 33%
(26%, 41%)Gain of ≥15 letters in visual acuity (%) 12 6% 31% 25%
(18%, 31%)24 4% 30% 25%
(18%, 31%)Mean change in visual acuity (letters) (SD) 12 -11.0 (17.9) +6.3 (14.1) 17.1
(14.2, 20.0)24 -15.0 (19.7) +5.5 (15.9) 20.1
(16.9, 23.4)Table 4 Visual Acuity Outcomes at Month 12 and Month 24 in Study AMD-2 Outcome Measure Month PDT
n=141Ranibizumab
0.5 mg n=139Estimated Difference (95% CI)* - *
- Adjusted estimate based on the stratified model; p < 0.01
Loss of <15 letters in visual acuity (%) 12 66% 98% 32%
(24%, 40%)24 65% 93% 28%
(19%, 37%)Gain of ≥15 letters in visual acuity (%) 12 11% 37% 26%
(17%, 36%)24 9% 37% 29%
(20%, 39%)Mean change in visual acuity (letters) (SD) 12 -8.5 (17.8) +11.0 (15.8) 19.8
(15.9, 23.7)24 -9.1 (18.7) +10.9 (17.3) 20
(16.0, 24.4)- *
- Visual acuity was measured at a distance of 2 meters
Figure 1
Mean Change in Visual Acuity* from Baseline to Month 24 in Study AMD-1 and Study AMD-2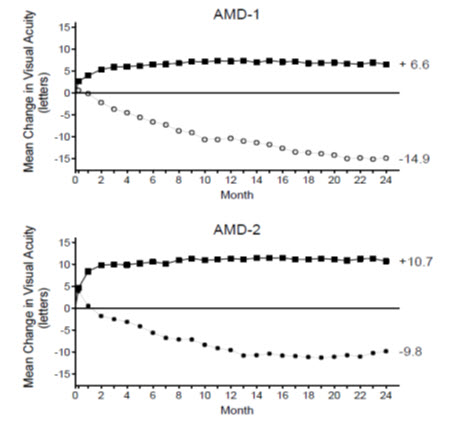
AMD-1 AMD-2 
Ranibizumab 0.5 mg (n=240) 
Ranibizumab 0.5 mg (n=139) 
Sham (n=238) 
Verteporfin PDT (n=143) Patients in the group treated with ranibizumab had minimal observable CNV lesion growth, on average. At Month 12, the mean change in the total area of the CNV lesion was 0.1-0.3 disc areas (DA) for ranibizumab versus 2.3-2.6 DA for the control arms. At Month 24, the mean change in the total area of the CNV lesion was 0.3-0.4 DA for ranibizumab versus 2.9-3.1 DA for the control arms.
Study AMD-3
Study AMD-3 was a randomized, double-masked, sham-controlled, 2-year study designed to assess the safety and efficacy of ranibizumab in patients with neovascular AMD (with or without a classic CNV component). Data are available through Month 12. Patients received ranibizumab 0.3 mg or 0.5 mg intravitreal injections or sham injections once a month for three consecutive doses, followed by a dose administered once every 3 months for 9 months. A total of 184 patients were enrolled in this study (ranibizumab 0.3 mg, 60; ranibizumab 0.5 mg, 61; sham, 63); 171 (93%) completed 12 months of this study. Patients treated with ranibizumab in Study AMD-3 received a mean of six total treatments out of a possible 6 from Day 0 through Month 12.
In Study AMD-3, the primary efficacy endpoint was the mean change in visual acuity at 12 months compared with baseline (see Figure 2). After an initial increase in visual acuity (following monthly dosing), on average, patients dosed once every 3 months with ranibizumab lost visual acuity, returning to baseline at Month 12. In Study AMD-3, almost all ranibizumab-treated patients (90%) lost fewer than 15 letters of visual acuity at Month 12.
Figure 2
Mean Change in Visual Acuity from Baseline to Month 12 in Study AMD-3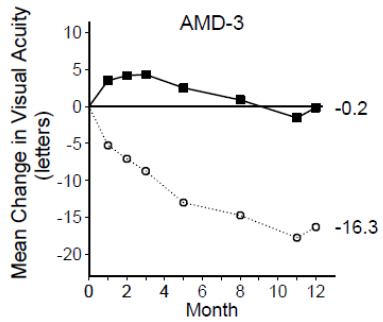

Ranibizumab 0.5 mg (n=61) 
Sham (n=63) Study AMD-4
Study AMD-4 was a randomized, double-masked, active treatment-controlled, two-year study designed to assess the safety and efficacy of ranibizumab 0.5 mg administered monthly or less frequently than monthly in patients with neovascular AMD. Patients randomized to the ranibizumab 0.5 mg less frequent dosing arm received three monthly doses followed by monthly assessments where patients were eligible to receive ranibizumab injections guided by pre-specified re-treatment criteria. A total of 550 patients were enrolled in the two 0.5 mg treatment groups with 467 (85%) completing through Month 24. Data are available through Month 24.
Clinical results at Month 24 remain similar to that observed at Month 12.
From Month 3 through Month 24, visual acuity decreased by 0.3 letters in the 0.5 mg less frequent dosing arm and increased by 0.7 letters in the 0.5 mg monthly arm (see Figure 3). Over this 21-month period, patients in the 0.5 mg less frequent dosing and the 0.5 mg monthly arms averaged 10.3 and 18.5 injections, respectively. The distribution of injections received in the less frequent dosing arm is shown in Figure 4.
Figure 3
Mean Change in Visual Acuity from Baseline to Month 24 in Study AMD-4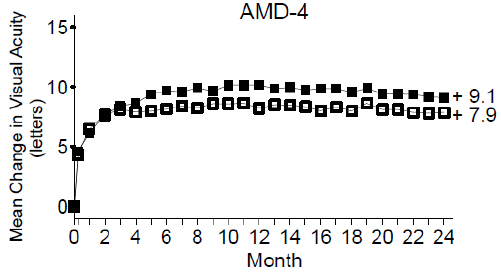

Ranibizumab 0.5 mg Monthly (n=275) 
Ranibizumab 0.5 mg Less Frequent than Monthly (n=275) Figure 4
Distribution of Injections from Month 3 to Month 24 in the Less Frequent Dosing Arm in Study AMD-4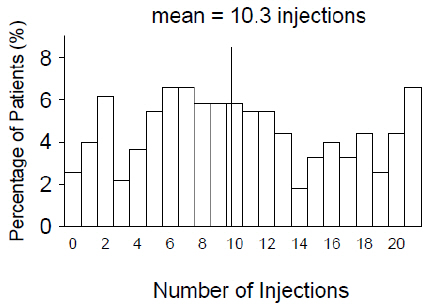
14.2 Macular Edema Following Retinal Vein Occlusion (RVO)
The safety and efficacy of ranibizumab were assessed in two randomized, double-masked, 1-year studies in patients with macular edema following RVO. Sham controlled data are available through Month 6. Patient age ranged from 20 to 91 years, with a mean age of 67 years. A total of 789 patients (ranibizumab 0.3 mg, 266 patients; ranibizumab 0.5 mg, 261 patients; sham, 262 patients) were enrolled, with 739 (94%) patients completing through Month 6. All patients completing Month 6 were eligible to receive ranibizumab injections guided by pre-specified re-treatment criteria until the end of the studies at Month 12.
In Study RVO-1, patients with macular edema following branch or hemi-RVO, received monthly ranibizumab 0.3 mg or 0.5 mg intravitreal injections or monthly sham injections for 6 months. All patients were eligible for macular focal/grid laser treatment beginning at Month 3 of the 6-month treatment period. Macular focal/grid laser treatment was given to 26 of 131 (20%) patients treated with 0.5 mg ranibizumab and 71 of 132 (54%) patients treated with sham.
In Study RVO-2, patients with macular edema following central RVO received monthly ranibizumab 0.3 mg or 0.5 mg intravitreal injections or monthly sham injections for 6 months.
At Month 6, after monthly treatment with 0.5 mg ranibizumab, the following clinical results were observed:
Table 5 Visual Acuity Outcomes at Month 6 in Study RVO-1 and Study RVO-2 Outcome Measure Study * Sham Ranibizumab
0.5 mgEstimated Difference (95% CI) † Gain of ≥15 letters in visual acuity (%) RVO-1 29% 61% 31%
(20%, 43%)Gain of ≥15 letters in visual acuity (%) RVO-2 17% 48% 30%
(20%, 41%)Figure 5
Mean Change in Visual Acuity from Baseline to Month 6 in Study RVO-1 and Study RVO-2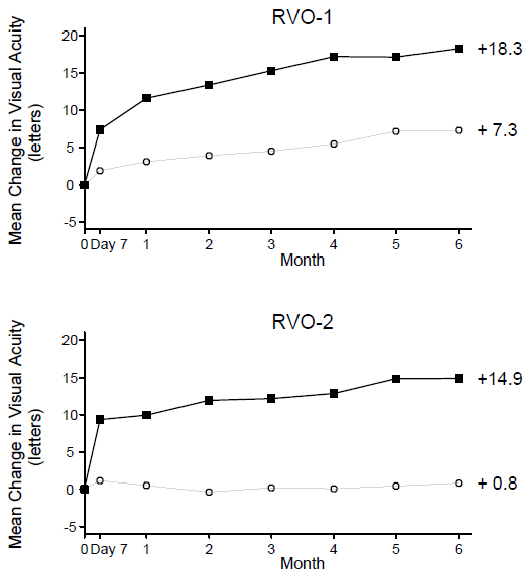
RVO-1: RVO-2: 
Ranibizumab 0.5 mg (n=131) 
Ranibizumab 0.5 mg (n=130) 
Sham (n=132) 
Sham (n=130) p < 0.01 for all time points 14.3 Myopic Choroidal Neovascularization (mCNV)
The efficacy and safety data of ranibizumab were assessed in a randomized, double-masked, active-controlled 3- month study in patients with mCNV. Patients age ranged from 18 to 87 years, with a mean age of 55 years. A total of 276 patients (222 patients in the ranibizumab treated Groups I and II; 55 patients in the active control PDT group) were enrolled. Patients randomized to the ranibizumab groups received injections guided by pre- specified re-treatment criteria. The retreatment criteria in Group I were vision stability guided, with the Best Corrected Visual Acuity (BCVA) at the current visit being assessed for changes compared with the two preceding monthly BCVA values. The retreatment criteria in Group II were disease activity guided, based on BCVA decrease from the previous visit that was attributable to intra- or sub-retinal fluid or active leakage secondary to mCNV as assessed by OCT and/or FA compared to the previous monthly visit.
Visual gains for the two ranibizumab 0.5 mg treatment arms were superior to the active control arm. The mean change in BCVA from baseline at Month 3 was: +12.1 letters for Group I, +12.5 letters for Group II and +1.4 letters for the PDT group. (Figure 6; Table 6). Efficacy was comparable between Group I and Group II.
Table 6 Mean Change in Visual Acuity and Proportion of Patients who Gained ≥15 letters from Baseline at Month 3 Study Arms Mean change in BCVA from baseline (Letters) Proportion of patients who gained ≥15 letters from baseline Mean (SD) Estimated Difference (95% CI)* Percent Estimated Difference (95% CI)* - *
- Adjusted estimates based on stratified models; p < 0.01
Group I 12.1 (10.2) 10.9
(7.6, 14.3)37.1 22.6
(9.5, 35.7)Group II 12.5 (8.8) 11.4
(8.3, 14.5)40.5 26.0
(13.1, 38.9)Control (PDT) 1.4 (12.2) 14.5 Figure 6
Mean Change in Visual Acuity from Baseline to Month 3 in mCNV Study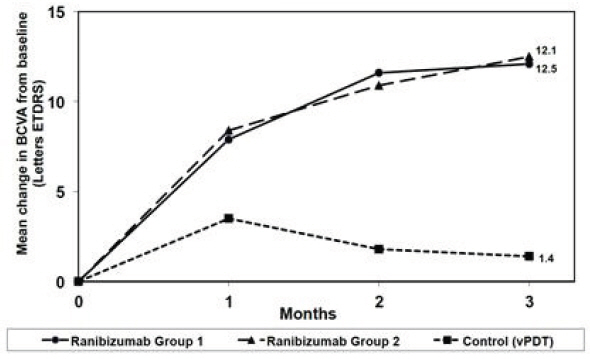
The proportion of patients who gained ≥15 letters (ETDRS) by Month 3 was 37.1% and 40.5% for ranibizumab Groups I and II, respectively and 14.5% for the PDT group. The mean number of injections between baseline and Month 3 was 2.5 and 1.8 for Groups I and II, respectively. 41% of patients received 1, 2 or 3 injections between baseline and Month 3 with no injections afterwards.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each BYOOVIZ 0.5 mg carton (NDC 64406-019-01) contains a single-dose, 2-mL glass vial designed to deliver 0.05 mL of 10 mg/mL ranibizumab-nuna solution that is clear to slightly opalescent and colorless to pale yellow.
EACH CARTON IS FOR SINGLE-EYE USE ONLY.
BYOOVIZ should be refrigerated at 2°C to 8°C (36°F to 46°F). DO NOT FREEZE. Do not use beyond the date stamped on the label. Protect BYOOVIZ vials from light and store in the original carton until time of use.
Prior to use, the unopened vial can be stored at temperatures up to 86°F (30°C) for up to 72 hours.
-
17 PATIENT COUNSELING INFORMATION
Advise patients that in the days following BYOOVIZ administration, patients are at risk of developing endophthalmitis. If the eye becomes red, sensitive to light, painful, or develops a change in vision, advise the patient to seek immediate care from an ophthalmologist [see Warnings and Precautions (5.1)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 0.5 mg Vial Carton
NDC 64406-019-01
Rx only
Byooviz™ 0.5mg wAMD RVO mCNO
(ranibizumab-nuna)
Injection
0.5 mg single-dose vial.
Discard unused portion.
IMPORTANT:
A 5-micron sterile filter needle (19-gauge x 1-1/2 inch) is required for preparation, but not included.
See enclosed package insert.
For intravitreal injection.
INDICATED FOR:
Neovascular (wet) age-related macular degeneration (AMD)
Macular edema following retinal vein occlusion (RVO)
Myopic choroidal neovascularization (mCNV)
KEEP REFRIGERATED.
DO NOT FREEZE.
PROTECT VIAL FROM LIGHT.
SAMSUNG BIOEPIS, Biogen®
-
INGREDIENTS AND APPEARANCE
BYOOVIZ
ranibizumab-nuna injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64406-019 Route of Administration INTRAVITREAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ranibizumab (UNII: ZL1R02VT79) (ranibizumab - UNII:ZL1R02VT79) ranibizumab 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) HISTIDINE (UNII: 4QD397987E) TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLYSORBATE 20 (UNII: 7T1F30V5YH) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64406-019-01 1 in 1 CARTON 06/01/2022 1 NDC:64406-019-07 0.05 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:64406-019-08 1 in 1 CARTON 10/03/2022 2 NDC:64406-019-09 0.05 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761202 06/01/2022 Labeler - BIOGEN INC. (121376230) Registrant - Samsung Bioepis Co., Ltd. (557822226)