ROMIDEPSIN- romidepsin
Teva Parenteral Medicines, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ROMIDEPSIN safely and effectively. See full prescribing information for ROMIDEPSIN.
Romidepsin for injection, for intravenous use Initial U.S. Approval: 2009 RECENT MAJOR CHANGES
INDICATIONS AND USAGERomidepsin is a histone deacetylase (HDAC) inhibitor indicated for the treatment of cutaneous T-cell lymphoma (CTCL) in adult patients who have received at least one prior systemic therapy (1). DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSFor injection: 10 mg of romidepsin as a lyophilized powder in single-dose vial for reconstitution (3). CONTRAINDICATIONSNone (4). WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reactions (≥30%), excluding laboratory abnormalities, are nausea, fatigue, infections, vomiting, anorexia, electrocardiogram ST-T-wave changes, dysgeusia, constipation and pruritis. Grade 3‐4 laboratory abnormalities (≥10%) include lymphopenia, neutropenia, anemia and thrombocytopenia (6). To report SUSPECTED ADVERSE REACTIONS, contact Teva Pharmaceuticals USA, Inc. at 1-888-838-2872 or the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2021 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Romidepsin is indicated for the treatment of cutaneous T-cell lymphoma (CTCL) in adult patients who have received at least one prior systemic therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended dosage of romidepsin is 14 mg/m2 administered intravenously over a 4-hour period on days 1, 8, and 15 of a 28-day cycle. Cycles should be repeated every 28 days provided that the patient continues to benefit from and tolerates the drug.
2.2 Dosage Modification
Nonhematologic toxicities except alopecia
- •
- Grade 2 or 3 toxicity: Treatment with romidepsin should be delayed until toxicity returns to Grade 0-1 or baseline, then therapy may be restarted at 14 mg/m2. If Grade 3 toxicity recurs, treatment with romidepsin should be delayed until toxicity returns to Grade 0-1 or baseline and the dose should be permanently reduced to 10 mg/m2.
- •
- Grade 4 toxicity: Treatment with romidepsin should be delayed until toxicity returns to Grade 0-1 or baseline, then the dose should be permanently reduced to 10 mg/m2.
- •
- Romidepsin should be discontinued if Grade 3 or 4 toxicities recur after dose reduction.
Hematologic toxicities
- •
- Grade 3 or 4 neutropenia or thrombocytopenia: Treatment with romidepsin should be delayed until the specific cytopenia returns to ANC greater than or equal to 1.5×109/L and platelet count greater than or equal to 75×109/L or baseline, then therapy may be restarted at 14 mg/m2.
- •
- Grade 4 febrile (greater than or equal to 38.5ºC) neutropenia or thrombocytopenia that requires platelet transfusion: Treatment with romidepsin should be delayed until the specific cytopenia returns to less than or equal to Grade 1 or baseline, and then the dose should be permanently reduced to 10 mg/m2.
2.3 Dosage in Patients with Hepatic Impairment
For patients with moderate or severe hepatic impairment, reduce the starting dose of romidepsin as shown in Table 1 and monitor for toxicities more frequently. Dosage adjustment is not required for patients with mild hepatic impairment.
| ULN=Upper limit of normal. | ||
|
Hepatic Impairment |
Bilirubin Levels |
Romidepsin Dose |
|
Moderate |
greater than 1.5 × ULN to less than or equal to 3 × ULN |
7 mg/m2 |
|
Severe |
greater than 3 × ULN |
5 mg/m2 |
2.4 Instructions for Preparation and Intravenous Administration
Romidepsin is a hazardous drug. Use appropriate handling procedures.1
Romidepsin must be reconstituted with the supplied diluent and further diluted with 0.9% Sodium Chloride Injection, USP, before intravenous infusion.
Romidepsin and diluent vials contain an overfill to ensure the recommended volume can be withdrawn at a concentration of 5 mg/mL.
- •
- Each 10 mg single-dose vial of romidepsin must be reconstituted with 2.2 mL of the supplied diluent.
- •
- With a suitable syringe, aseptically withdraw 2.2 mL from the supplied diluent vial, and slowly inject it into the romidepsin for injection vial. Swirl the contents of the vial until there are no visible particles in the resulting solution. The reconstituted solution will contain romidepsin 5 mg/mL. The reconstituted romidepsin vial will contain 2 mL of deliverable volume of drug product. The reconstituted romidepsin solution is chemically stable for up to 8 hours at room temperature.
- •
- Extract the appropriate amount of romidepsin from the vials to deliver the desired dose, using proper aseptic technique. Before intravenous infusion, further dilute romidepsin in 500 mL 0.9% Sodium Chloride Injection, USP.
- •
- Infuse over 4 hours.
The diluted solution is compatible with polyvinyl chloride (PVC), ethylene vinyl acetate (EVA), polyethylene (PE) infusion bags as well as glass bottles, and is chemically stable for up to 24 hours when stored at room temperature. However, it should be administered as soon after dilution as possible.
Parenteral drug products should be inspected visually for particulate matter and discoloration before administration, whenever solution and container permit.
3 DOSAGE FORMS AND STRENGTHS
For Injection: 10 mg of romidepsin as a lyophilized white powder in a single-dose vial for reconstitution and further dilution.
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Treatment with romidepsin can cause thrombocytopenia, leukopenia (neutropenia and lymphopenia), and anemia. Monitor blood counts regularly during treatment with romidepsin and modify the dose as necessary [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.2 Infections
Fatal and serious infections have been reported in clinical trials of romidepsin, including pneumonia, sepsis, and viral reactivation, including reactivation of Epstein Barr and hepatitis B viruses. These infections can occur during and following treatment. The risk of life-threatening infections may be greater in patients with a history of prior treatment with monoclonal antibodies directed against lymphocyte antigens and in patients with disease involvement of the bone marrow [see Adverse Reactions (6.1)].
Reactivation of hepatitis B virus infection was reported in 1% of patients in clinical trials. In patients with evidence of prior hepatitis B infection, consider monitoring for reactivation, and consider antiviral prophylaxis.
Reactivation of Epstein Barr viral infection leading to liver failure has occurred in recipients of romidepsin including after ganciclovir prophylaxis.
5.3 Electrocardiographic Changes
Several treatment-emergent morphological changes in ECGs (including T-wave and ST-segment changes) have been reported in clinical studies. The clinical significance of these changes is unknown [see Adverse Reactions (6.1)].
In patients with congenital long QT syndrome, patients with a history of significant cardiovascular disease, and patients taking anti-arrhythmic medicines or medicinal products that lead to significant QT prolongation, consider cardiovascular monitoring of ECGs at baseline and periodically during treatment.
Confirm that potassium and magnesium levels are within normal range before administration of romidepsin [see Adverse Reactions (6.1)].
5.4 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS) has been reported to occur in recipients of romidepsin, including in 1% of patients with tumor stage CTCL. Patients with advanced stage disease and/or high tumor burden are at greater risk, should be closely monitored, and managed as appropriate.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, romidepsin can cause fetal harm when administered to a pregnant woman. In an animal reproductive study, romidepsin was embryocidal and caused adverse developmental outcomes at exposures below those in patients at the recommended dose of 14 mg/m2. Advise females of reproductive potential to use effective contraception during treatment and for 1 month after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 1 month after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following adverse reactions are described in more detail in other sections of the prescribing information.
- •
- Myelosuppression [see Warnings and Precautions (5.1)]
- •
- Infections [see Warnings and Precautions (5.2)]
- •
- Electrocardiographic Changes [see Warnings and Precautions (5.3)]
- •
- Tumor Lysis Syndrome [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS reflect exposure to romidepsin in four clinical trials involving 363 patients with T-cell lymphoma, including 185 patients with CTCL. Romidepsin was administered as a single agent at a dosage of 14 mg/m2 on days 1, 8, and 15 of a 28-day cycle. Among 363 patients who received romidepsin, 21% were exposed for 6 months or longer and 13% were exposed for greater than one year.
Cutaneous T-Cell Lymphoma
The safety of romidepsin was evaluated in 185 patients with CTCL in 2 single arm clinical studies in which patients received a dosage of 14 mg/m2 on days 1, 8, and 15 of a 28-day cycle. Treatment continued as long as the patient benefitted from and tolerated the drug. The mean duration of treatment in these studies was 5.6 months (range: <1 to 83.4 months).
Common Adverse Reactions
Table 2 summarizes the most frequent adverse reactions (>20%) regardless of causality using the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE, Version 3.0). Due to methodological differences between the studies, the AE data are presented separately for Study 1 and Study 2. Adverse reactions are ranked by their incidence in Study 1. Laboratory abnormalities commonly reported (>20%) as adverse reactions are included in Table 2.
|
Adverse Reactions n (%) |
Study 1
|
Study 2
|
||
|
All grades |
Grade 3 or 4 |
All grades |
Grade 3 or 4 |
|
|
Any adverse reactions |
99 (97) |
36 (35) |
83 (100) |
68 (82) |
|
Nausea |
57 (56) |
3 (3) |
71 (86) |
5 (6) |
|
Asthenia/Fatigue |
54 (53) |
8 (8) |
64 (77) |
12 (14) |
|
Infections |
47 (46) |
11 (11) |
45 (54) |
27 (33) |
|
Vomiting |
35 (34) |
1 (<1) |
43 (52) |
8 (10) |
|
Anorexia |
23 (23) |
1 (<1) |
45 (54) |
3 (4) |
|
Hypomagnesemia |
22 (22) |
1 (<1) |
23 (28) |
0 |
|
Diarrhea |
20 (20) |
1 (<1) |
22 (27) |
1 (1) |
|
Pyrexia |
20 (20) |
4 (4) |
19 (23) |
1 (1) |
|
Anemia |
19 (19) |
3 (3) |
60 (72) |
13 (16) |
|
Thrombocytopenia |
17 (17) |
0 |
54 (65) |
12 (14) |
|
Dysgeusia |
15 (15) |
0 |
33 (40) |
0 |
|
Constipation |
12 (12) |
2 (2) |
32 (39) |
1 (1) |
|
Neutropenia |
11 (11) |
4 (4) |
47 (57) |
22 (27) |
|
Hypotension |
7 (7) |
3 (3) |
19 (23) |
3 (4) |
|
Pruritus |
7 (7) |
0 |
26 (31) |
5 (6) |
|
Hypokalemia |
6 (6) |
0 |
17 (20) |
2 (2) |
|
Dermatitis/Exfoliative dermatitis |
4 (4) |
1 (<1) |
22 (27) |
7 (8) |
|
Hypocalcemia |
4 (4) |
0 |
43 (52) |
5 (6) |
|
Leukopenia |
4 (4) |
0 |
38 (46) |
18 (22) |
|
Lymphopenia |
4 (4) |
0 |
47 (57) |
31 (37) |
|
Alanine aminotransferase increased |
3 (3) |
0 |
18 (22) |
2 (2) |
|
Aspartate aminotransferase increased |
3 (3) |
0 |
23 (28) |
3 (4) |
|
Hypoalbuminemia |
3 (3) |
1 (<1) |
40 (48) |
3 (4) |
|
Electrocardiogram ST-T wave changes |
2 (2) |
0 |
52 (63) |
0 |
|
Hyperglycemia |
2 (2) |
2 (2) |
42 (51) |
1 (1) |
|
Hyponatremia |
1 (<1) |
1 (<1) |
17 (20) |
2 (2) |
|
Hypermagnesemia |
0 |
0 |
22 (27) |
7 (8) |
|
Hypophosphatemia |
0 |
0 |
22 (27) |
8 (10) |
|
Hyperuricemia |
0 |
0 |
27 (33) |
7 (8) |
Serious Adverse Reactions
Infections were the most common type of SAE reported in both studies with 8 patients (8%) in Study 1 and 26 patients (31%) in Study 2 experiencing a serious infection. Serious adverse reactions reported in >2% of patients in Study 1 were sepsis and pyrexia (3%). In Study 2, serious adverse reactions in >2% of patients were fatigue (7%), supraventricular arrhythmia, central line infection, neutropenia (6%), hypotension, hyperuricemia, edema (5%), ventricular arrhythmia, thrombocytopenia, nausea, leukopenia, dehydration, pyrexia, aspartate aminotransferase increased, sepsis, catheter related infection, hypophosphatemia and dyspnea (4%).
There were eight deaths not due to disease progression. In Study 1, there were two deaths: one due to cardiopulmonary failure and one due to acute renal failure. There were six deaths in Study 2: four due to infection and one each due to myocardial ischemia and acute respiratory distress syndrome.
Other Clinical Trials Experience
The following common adverse reactions have been reported following administration of romidepsin as a single agent in 178 patients with peripheral T-cell lymphoma, for which romidepsin is not indicated or recommended. The most common adverse reactions (≥30%) included nausea (63%), fatigue (61%), thrombocytopenia (49%), vomiting (39%), neutropenia (39%), pyrexia (38%), diarrhea (36%) and anemia (35%). Other common (≥10%) clinically significant adverse reactions included dysgeusia (22%), headache (20%), cough (19%), dyspnea (15%), abdominal pain (13%) and stomatitis (10%). Grade 3 and higher adverse reactions in ≥10% were hematologic toxicities (including thrombocytopenia, neutropenia, leukopenia and anemia) and fatigue.
7 DRUG INTERACTIONS
7.1 Warfarin or Coumarin Derivatives
Prolongation of PT and elevation of INR were observed in a patient receiving romidepsin concomitantly with warfarin. Monitor PT and INR more frequently in patients concurrently receiving romidepsin and warfarin [see Clinical Pharmacology (12.3)].
7.2 Drugs That Inhibit CYP3A4 Enzymes
Strong CYP3A4 inhibitors increase concentrations of romidepsin [see Clinical Pharmacology (12.3)]. Monitor for toxicity related to increased romidepsin exposure and follow the dose modifications for toxicity [see Dosage and Administration (2.2)] when romidepsin is initially co-administered with strong CYP3A4 inhibitors.
7.3 Drugs That Induce CYP3A4 Enzymes
Rifampin (a potent CYP3A4 inducer) increased the concentrations of romidepsin [see Clinical Pharmacology (12.3)]. Avoid co-administration of romidepsin with rifampin. The use of other potent CYP3A4 inducers should be avoided when possible.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings from animal studies, romidepsin can cause embryo-fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
There are no available data on romidepsin use in pregnant women to inform a drug associated risk of major birth defects and miscarriage. In an animal reproductive study, romidepsin was embryocidal and caused adverse developmental outcomes including embryo-fetal toxicity and malformations at exposures below those in patients at the recommended dose (see Data). Advise pregnant women of the potential risk to a fetus and to avoid becoming pregnant while receiving romidepsin and for at least 1 month after the last dose.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Romidepsin was administered intravenously to pregnant rats during the period of organogenesis at doses of 0.1, 0.2, or 0.5 mg/kg/day. Substantial resorption or postimplantation loss was observed at the high-dose of 0.5 mg/kg/day, a maternally toxic dose. Adverse embryo-fetal effects were noted at romidepsin doses of ≥0.1 mg/kg/day, with systemic exposures (AUC) ≥0.2% of the human exposure at the recommended dose of 14 mg/m2/week. Drug-related fetal effects consisted of reduced fetal body weights, folded retina, rotated limbs, and incomplete sternal ossification.
8.2 Lactation
Risk Summary
There are no data on the presence of romidepsin or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in the breastfed child, advise lactating women not to breastfeed during treatment with romidepsin and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Romidepsin can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Perform pregnancy testing in females of reproductive potential within 7 days prior to initiating therapy with romidepsin.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with romidepsin and for 1 month after the last dose. Romidepsin may reduce the effectiveness of estrogen-containing contraceptives. Therefore, alternative methods of non-estrogen containing contraception (e.g., condoms, intrauterine devices) should be used in patients receiving romidepsin.
Infertility
Based on findings in animals, romidepsin has the potential to affect male and female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of romidepsin in pediatric patients have not been established.
8.5 Geriatric Use
Of the 186 patients with CTCL who received romidepsin in clinical studies, 51 (28%) were 65 years of age and older, while 16 (9%) were 75 years of age. No overall differences in safety or effectiveness were observed between patients 65 years or age and over and younger patients; however, greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
In a hepatic impairment study, romidepsin was evaluated in 19 patients with advanced cancer and mild (8), moderate (5), or severe (6) hepatic impairment. There were 4 deaths during the first cycle of treatment: 1 patient with mild hepatic impairment, 1 patient with moderate hepatic impairment, and 2 patients with severe hepatic impairment. No dose adjustments are recommended for patients with mild hepatic impairment. Reduce the romidepsin starting dose for patients with moderate and severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. Monitor patients with hepatic impairment more frequently for toxicity, especially during the first cycle of therapy.
10 OVERDOSAGE
No specific information is available on the treatment of overdosage of romidepsin.
Toxicities in a single-dose study in rats or dogs, at intravenous romidepsin doses up to 2.2-fold the recommended human dose based on the body surface area, included irregular respiration, irregular heartbeat, staggering gait, tremor, and tonic convulsions.
In the event of an overdose, it is reasonable to employ the usual supportive measures, e.g., clinical monitoring and supportive therapy, if required. There is no known antidote for romidepsin and it is not known if romidepsin is dialyzable.
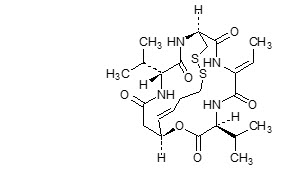
11 DESCRIPTION
Romidepsin, a histone deacetylase (HDAC) inhibitor, is a bicyclic depsipeptide. At room temperature, romidepsin is a white powder and is described chemically as (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-bis(1-methylethyl)-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone. The empirical formula is C24H36N4O6S2.
The molecular weight is 540.71 and the structural formula is:
Romidepsin for injection is intended for intravenous infusion only after reconstitution with the supplied diluent and after further dilution with 0.9% Sodium Chloride, USP.
Romidepsin is supplied as a kit containing 2 vials.
Romidepsin for injection is a sterile lyophilized white powder and is supplied in a 10 mg single-dose vial containing 11 mg romidepsin, 22 mg povidone, USP, and hydrochloric acid, NF, as a pH adjuster.
Diluent for romidepsin is a sterile clear solution and is supplied in a single-dose vial containing 2.4 mL (2.2 mL deliverable volume). Diluent for romidepsin contains 80% (v/v) propylene glycol, USP and 20% (v/v) dehydrated alcohol, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Romidepsin is a histone deacetylase (HDAC) inhibitor. HDACs catalyze the removal of acetyl groups from acetylated lysine residues in histones, resulting in the modulation of gene expression. HDACs also deacetylate non-histone proteins, such as transcription factors. In vitro, romidepsin causes the accumulation of acetylated histones, and induces cell cycle arrest and apoptosis of some cancer cell lines with IC50 values in the nanomolar range. The mechanism of the antineoplastic effect of romidepsin observed in nonclinical and clinical studies has not been fully characterized.
12.2 Pharmacodynamics
Cardiac Electrophysiology
At doses of 14 mg/m2 as a 4-hour intravenous infusion, and at doses of 8 (0.57 times the recommended dose), 10 (0.71 times the recommended dose) or 12 (0.86 times the recommended dose) mg/m2 as a 1-hour infusion, no large changes in the mean QTc interval (>20 milliseconds) from baseline based on Fridericia correction method were detected. Small increase in mean QT interval (< 10 milliseconds) and mean QT interval increase between 10 to 20 milliseconds cannot be excluded.
Romidepsin was associated with a delayed concentration-dependent increase in heart rate in patients with advanced cancer with a maximum mean increase in heart rate of 20 beats per minute occurring at the 6-hour time point after start of romidepsin infusion for patients receiving 14 mg/m2 as a 4-hour infusion.
12.3 Pharmacokinetics
In patients with T-cell lymphomas who received 14 mg/m2 of romidepsin intravenously over a 4-hour period on days 1, 8, and 15 of a 28-day cycle, geometric mean values of the maximum plasma concentration (Cmax) and the area under the plasma concentration versus time curve (AUC0-∞) were 377 ng/mL and 1549 ng*hr/mL, respectively. Romidepsin exhibited linear pharmacokinetics across doses ranging from 1.0 (0.07 times the recommended dose) to 24.9 (1.76 times the recommended dose) mg/m2 when administered intravenously over 4 hours in patients with advanced cancers.
Distribution
Romidepsin is highly protein bound in plasma (92% to 94%) over the concentration range of 50 ng/mL to 1000 ng/mL with α1-acid-glycoprotein (AAG) being the principal binding protein. Romidepsin is a substrate of the efflux transporter P-glycoprotein (P-gp, ABCB1).
In vitro, romidepsin accumulates into human hepatocytes via an unknown active uptake process. Romidepsin is not a substrate of the following uptake transporters: BCRP, BSEP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3, or OCT2. In addition, romidepsin is not an inhibitor of BCRP, MRP2, MDR1 or OAT3. Although romidepsin did not inhibit OAT1, OCT2, and OATP1B3 at concentrations seen clinically (1 μmol/L), modest inhibition was observed at 10 μmol/L. Romidepsin was found to be an inhibitor of BSEP and OATP1B1.
Metabolism
Romidepsin undergoes extensive metabolism in vitro primarily by CYP3A4 with minor contribution from CYP3A5, CYP1A1, CYP2B6, and CYP2C19. At therapeutic concentrations, romidepsin did not competitively inhibit CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 in vitro.
At therapeutic concentrations, romidepsin did not cause notable induction of CYP1A2, CYP2B6 and CYP3A4 in vitro. Therefore, pharmacokinetic drug-drug interactions are unlikely to occur due to CYP450 induction or inhibition by romidepsin when co-administered with CYP450 substrates.
Excretion
Following 4-hour intravenous administration of romidepsin at 14 mg/m2 on days 1, 8, and 15 of a 28-day cycle in patients with T-cell lymphomas, the terminal half-life (t1/2) was approximately 3 hours. No accumulation of plasma concentration of romidepsin was observed after repeated dosing.
Drug Interactions
Ketoconazole: Following co-administration of 8 mg/m2 romidepsin (4-hour infusion) with ketoconazole, the overall romidepsin exposure was increased by approximately 25% and 10% for AUC0-∞ and Cmax, respectively, compared to romidepsin alone, and the difference in AUC0-∞ between the 2 treatments was statistically significant.
Rifampin: Following co-administration of 14 mg/m2 romidepsin (4-hour infusion) with rifampin, the overall romidepsin exposure was increased by approximately 80% and 60% for AUC0-∞ and Cmax, respectively, compared to romidepsin alone, and the difference between the 2 treatments was statistically significant. Co-administration of rifampin decreased the romidepsin clearance and volume of distribution by 44% and 52%, respectively. The increase in exposure seen after co-administration with rifampin is likely due to rifampin's inhibition of an undetermined hepatic uptake process that is predominant for the disposition of romidepsin.
Drugs that inhibit P-glycoprotein: Drugs that inhibit p-glycoprotein may increase the concentration of romidepsin.
Specific Populations
Effect of Age, Gender, Race or Renal Impairment
The pharmacokinetics of romidepsin was not influenced by age (27 to 83 yrs), gender, race (white vs. black) or mild (estimated creatinine clearance 50 - 80 mL/min), moderate (estimated creatinine clearance 30-50 mL/min), or severe (estimated creatinine clearance <30 mL/min) renal impairment. The effect of end-stage renal disease (estimated creatine clearance less than 15 mL/min) on romidepsin pharmacokinetics has not been studied.
Hepatic Impairment
Romidepsin clearance decreased with increased severity of hepatic impairment. In patients with cancer, the geometric mean Cmax values after administration of 14, 7, and 5 mg/m2 romidepsin in patients with mild (B1: bilirubin ≤ULN and AST >ULN; B2: bilirubin >ULN but ≤1.5 × ULN and any AST), moderate (bilirubin >1.5 × ULN to ≤3 × ULN and any AST), and severe (bilirubin >3 × ULN and any AST) hepatic impairment were approximately 111%, 96%, and 86% of the corresponding value after administration of 14 mg/m2 romidepsin in patients with normal (bilirubin ≤upper limit of normal (ULN) and aspartate aminotransferase (AST) ≤ULN) hepatic function, respectively. The geometric mean AUCinf values in patients with mild, moderate, and severe hepatic impairment were approximately 144%, 114%, and 116% of the corresponding value in patients with normal hepatic function, respectively. Among these 4 cohorts, moderate interpatient variability was noted for the exposure parameters Cmax and AUCinf, as the coefficient of variation (CV) ranged from 30% to 54%.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with romidepsin. Romidepsin was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test) or the mouse lymphoma assay. Romidepsin was not clastogenic in an in vivo rat bone marrow micronucleus assay when tested to the maximum tolerated dose (MTD) of 1 mg/kg in males and 3 mg/kg in females (6 and 18 mg/m2 in males and females, respectively). These doses were up to 1.3-fold the recommended human dose, based on body surface area.
Based on nonclinical findings, male and female fertility may be compromised by treatment with romidepsin. In a 26-week toxicology study, romidepsin administration resulted in testicular degeneration in rats at 0.33 mg/kg/dose (2 mg/m2/dose) following the clinical dosing schedule. This dose resulted in AUC0-∞ values that were approximately 2% the exposure level in patients receiving the recommended dose of 14 mg/m2/dose. A similar effect was seen in mice after 4 weeks of drug administration at higher doses. Seminal vesicle and prostate organ weights were decreased in a separate study in rats after 4 weeks of daily drug administration at 0.1 mg/kg/day (0.6 mg/m2/day), approximately 30% the estimated human daily dose based on body surface area. Romidepsin showed high affinity for binding to estrogen receptors in pharmacology studies. In a 26-week toxicology study in rats, atrophy was seen in the ovary, uterus, vagina and mammary gland of females administered doses as low as 0.1 mg/kg/dose (0.6 mg/m2/dose) following the clinical dosing schedule. This dose resulted in AUC0-∞ values that were 0.3% of those in patients receiving the recommended dose of 14 mg/m2/dose. Maturation arrest of ovarian follicles and decreased weight of ovaries were observed in a separate study in rats after 4 weeks of daily drug administration at 0.1 mg/kg/day (0.6 mg/m2/day). This dose is approximately 30% the estimated human daily dose based on body surface area.
14 CLINICAL STUDIES
Romidepsin was evaluated in 2 multicenter, single-arm clinical studies in patients with CTCL (Study 1 [NCT00106431] and Study 2 [NCT00007345]). Overall, 167 patients with CTCL were treated in the US, Europe, and Australia. Study 1 included 96 patients with confirmed CTCL after failure of at least 1 prior systemic therapy. Study 2 included 71 patients with a primary diagnosis of CTCL who received at least 2 prior skin directed therapies or one or more systemic therapies. Patients were treated with romidepsin at a starting dose of 14 mg/m2 infused over 4 hours on days 1, 8, and 15 every 28 days.
In both studies, patients could be treated until disease progression at the discretion of the investigator and local regulators. Objective disease response was evaluated according to a composite endpoint that included assessments of skin involvement, lymph node and visceral involvement, and abnormal circulating T-cells ("Sézary cells").
The primary efficacy endpoint for both studies was overall objective disease response rate (ORR) based on the investigator assessments, and was defined as the proportion of patients with confirmed complete response (CR) or partial response (PR). CR was defined as no evidence of disease and PR as ≥ 50% improvement in disease. Secondary endpoints in both studies included duration of response and time to response.
Baseline Patient Characteristics
Demographic and disease characteristics of the patients in Study 1 and Study 2 are provided in Table 3.
|
Characteristic |
Study 1
|
Study 2
|
|
Age |
||
|
N |
96 |
71 |
|
Mean (SD) |
57 (12) |
56 (13) |
|
Median (Range) |
57 (21, 89) |
57 (28, 84) |
|
Sex, n (%) |
||
|
Men |
59 (61) |
48 (68) |
|
Women |
37 (39) |
23 (32) |
|
Race, n (%) |
||
|
White |
90 (94) |
55 (77) |
|
Black |
5 (5) |
15 (21) |
|
Other/Not Reported |
1 (1) |
1 (1) |
|
Stage of Disease at Study Entry, n (%) |
||
|
IA |
0 (0) |
1 (1) |
|
IB |
15 (16) |
6 (9) |
|
IIA |
13 (14) |
2 (3) |
|
IIB |
21 (22) |
14 (20) |
|
III |
23 (24) |
9 (13) |
|
IVA |
24 (25) |
27 (38) |
|
IVB |
0 (0) |
12 (17) |
|
Number of Prior Skin-Directed Therapies |
||
|
Median (Range) |
2 (0, 6) |
1 (0, 3) |
|
Number of Prior Systemic Therapies |
||
|
Median (Range) |
2 (1, 8) |
2 (0, 7) |
Clinical Results
Efficacy outcomes for CTCL patients are provided in Table 4. Median time to first response was 2 months (range 1 to 6) in both studies. Median time to CR was 4 months in Study 1 and 6 months in Study 2 (range 2 to 9).
| * Denotes censored value. | |||||
|
Response Rate |
Study 1
|
Study 2
|
|||
|
ORR (CR + PR), n (%) |
33 (34) |
25 (35) |
|||
|
CR, n (%) |
6 (6) |
4 (6) |
|||
|
PR, n (%) |
27 (28) |
21 (30) |
|||
|
Duration of Response (months) |
|||||
|
N |
33 |
25 |
|||
|
Median (range) |
15 (1, 20*) |
11 (1, 66*) |
|||
15 REFERENCES
1. OSHA Hazardous Drugs. OSHA. [Accessed on 09/11/2018, from http://www.osha.gov/SLTC/hazardousdrugs/index.html]
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Romidepsin is supplied as a kit including a sterile, lyophilized powder in a 10 mg single-dose vial containing 11 mg of romidepsin, 22 mg of the bulking agent, povidone, USP, and hydrochloric acid, NF, as a pH adjuster. In addition, each kit includes a single-dose sterile diluent vial containing 2.4 mL (2.2 mL deliverable volume) of 80% propylene glycol, USP, and 20% dehydrated alcohol, USP.
NDC 0703-3125-08: Romidepsin KIT containing 1 vial of romidepsin and 1 vial of diluent for romidepsin per carton.
Storage and Handling
Romidepsin for injection is supplied as a kit containing 2 vials in a single carton. The carton must be stored at 20° to 25°C, excursions permitted between 15° to 30°C. (See USP Controlled Room Temperature.)
Romidepsin is a hazardous drug. Follow applicable special handling and disposal procedures.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Low Blood Counts
- Advise patients that treatment with romidepsin can cause low blood counts and that frequent monitoring of hematologic parameters is required. Patients should be instructed to report fever or other signs of infection, significant fatigue, shortness of breath, or bleeding [see Warnings and Precautions (5.1)].
Infections
- Advise patients that infections may occur during treatment with romidepsin. Advise patients to report fever, cough, shortness of breath with or without chest pain, burning on urination, flu-like symptoms, muscle aches, or worsening skin problems. Advise patients to report any previous history of hepatitis B before starting romidepsin [see Warnings and Precautions (5.2)].
Tumor Lysis Syndrome
- Advise patients of the risk of tumor lysis syndrome (especially those with advanced stage disease and/or high tumor burden) to maintain high fluid intake for at least 72 hours after each dose [see Warnings and Precautions (5.4)].
Nausea and Vomiting
- Advise patients that nausea and vomiting are common following treatment with romidepsin. Prophylactic antiemetics are recommended for all patients. Advise patients to report these symptoms so that appropriate treatment can be instituted [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
- Advise patients that romidepsin can cause fetal harm when administered during pregnancy [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
Contraception
- Advise females of reproductive potential to use effective contraception during treatment with romidepsin and for 1 month after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with romidepsin and for 1 month after the last dose [see Use in Specific Populations (8.3)].
Lactation
- Advise lactating women not to breastfeed during treatment with romidepsin and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
- Advise females and males of reproductive potential that romidepsin may cause infertility [see Nonclinical Toxicology (13.1)].
Distributed by:
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Manufactured by:
Baxter Oncology GmbH
Halle/Westfalen, Germany
ROMTEVPI.010/PPI.010
| This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 07/2021 |
||
|
PATIENT INFORMATION
|
||
|
What is romidepsin? Romidepsin is a prescription medicine used to treat people with a type of cancer called cutaneous T-cell lymphoma (CTCL) after at least one other type of medicine by mouth or injection has been tried. It is not known if romidepsin is safe and effective in children under 18 years of age. |
||
|
Before receiving romidepsin, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how romidepsin works, or romidepsin may affect how other medicines work. Especially tell your healthcare provider if you take or use:
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine. |
||
|
How will I receive romidepsin?
|
||
|
What are the possible side effects of romidepsin? Romidepsin may cause serious side effects, including:
|
||
|
|
|
The most common side effects of romidepsin include:
These are not all the possible side effects of romidepsin. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
General information about the safe and effective use of romidepsin Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. This Patient Information leaflet summarizes the most important information about romidepsin. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about romidepsin that is written for health professionals. |
||
|
What are the ingredients in romidepsin? Active ingredient: romidepsin Inactive ingredients: povidone, hydrochloric acid as a pH adjuster. The diluent contains 80% propylene glycol and 20% dehydrated alcohol. Distributed by: Manufactured by: ROMTEVPPI.010 7/21 |
||
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 0703-3125-08
Rx only
Romidepsin
for Injection Kit
10 mg/vial
Reconstitution and dilution required.
Each kit contains:
- One 10 mg single-dose vial of romidepsin
- One single-dose vial with 2.2 mL of Diluent
For Intravenous Use Only
Single-Dose Vial
Discard Unused Portion
TEVA
| ROMIDEPSIN
romidepsin kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Teva Parenteral Medicines, Inc. (794362533) |