Label: FUROSEMIDE tablet

- NDC Code(s): 72789-030-30, 72789-030-60
- Packager: PD-Rx Pharmaceuticals, Inc.
- This is a repackaged label.
- Source NDC Code(s): 43547-401
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 13, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
WARNING
Furosemide is a potent diuretic which, if given in excessive amounts, can lead to a profound diuresis with water and electrolyte depletion. Therefore, careful medical supervision is required and dose and dose schedule must be adjusted to the individual patient's needs (see DOSAGE AND ADMINISTRATION).
-
DESCRIPTION
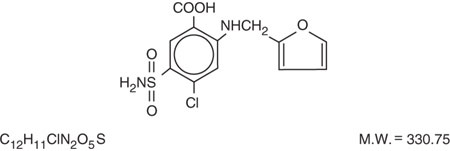
Furosemide is a diuretic which is an anthranilic acid derivative. Furosemide Tablets for oral administration contain furosemide as the active ingredient and the following inactive ingredients: corn starch NF, lactose monohydrate NF, magnesium stearate NF, pregelatinized starch NF, and talc USP. Chemically, it is 4-chloro-N-furfuryl-5-sulfamoylanthranilic acid. Furosemide is available as white-off white tablets for oral administration in dosage strengths of 20, 40 and 80 mg. Furosemide is a white to off-white odorless crystalline powder. It is practically insoluble in water, sparingly soluble in alcohol, freely soluble in dilute alkali solutions and insoluble in dilute acids.
The CAS Registry Number is 54-31-9.
The structural formula is as follows:
Tested by USP Dissolution Test 1.
-
CLINICAL PHARMACOLOGY
Investigations into the mode of action of furosemide have utilized micropuncture studies in rats, stop flow experiments in dogs and various clearance studies in both humans and experimental animals. It has been demonstrated that furosemide inhibits primarily the absorption of sodium and chloride not only in the proximal and distal tubules but also in the loop of Henle. The high degree of efficacy is largely due to the unique site of action. The action on the distal tubule is independent of any inhibitory effect on carbonic anhydrase and aldosterone.
Recent evidence suggests that furosemide glucuronide is the only or at least the major biotransformation product of furosemide in man. Furosemide is extensively bound to plasma proteins, mainly to albumin. Plasma concentrations ranging from 1 to 400 mcg/mL are 91 to 99% bound in healthy individuals. The unbound fraction averages 2.3 to 4.1% at therapeutic concentrations.
The onset of diuresis following oral administration is within 1 hour. The peak effect occurs within the first or second hour. The duration of diuretic effect is 6 to 8 hours.
In fasted normal men, the mean bioavailability of furosemide from furosemide tablets and furosemide oral solution is 64% and 60%, respectively, of that from an intravenous injection of the drug. Although furosemide is more rapidly absorbed from the oral solution (50 minutes) than from the tablet (87 minutes), peak plasma levels and area under the plasma concentration-time curves do not differ significantly. Peak plasma concentrations increase with increasing dose but times-to-peak do not differ among doses. The terminal half-life of furosemide is approximately 2 hours.
Significantly more furosemide is excreted in urine following the IV injection than after the tablet or oral solution. There are no significant differences between the two oral formulations in the amount of unchanged drug excreted in urine.
Geriatric Population
Furosemide binding to albumin may be reduced in elderly patients. Furosemide is predominantly excreted unchanged in the urine. The renal clearance of furosemide after intravenous administration in older healthy male subjects (60 to 70 years of age) is statistically significantly smaller than in younger healthy male subjects (20 to 35 years of age). The initial diuretic effect of furosemide in older subjects is decreased relative to younger subjects (see PRECAUTIONS: Geriatric Use).
-
INDICATIONS AND USAGE
Edema
Furosemide tablets are indicated in adults and pediatric patients for the treatment of edema associated with congestive heart failure, cirrhosis of the liver, and renal disease, including the nephrotic syndrome. Furosemide tablets are particularly useful when an agent with greater diuretic potential is desired.
- CONTRAINDICATIONS
-
WARNINGS
In patients with hepatic cirrhosis and ascites, furosemide therapy is best initiated in the hospital. In hepatic coma and in states of electrolyte depletion, therapy should not be instituted until the basic condition is improved. Sudden alterations of fluid and electrolyte balance in patients with cirrhosis may precipitate hepatic coma; therefore, strict observation is necessary during the period of diuresis. Supplemental potassium chloride and, if required, an aldosterone antagonist are helpful in preventing hypokalemia and metabolic alkalosis.
If increasing azotemia and oliguria occur during treatment of severe progressive renal disease, furosemide should be discontinued.
Cases of tinnitus and reversible or irreversible hearing impairment and deafness have been reported. Reports usually indicate that furosemide ototoxicity is associated with rapid injection, severe renal impairment, the use of higher than recommended doses, hypoproteinemia or concomitant therapy with aminoglycoside antibiotics, ethacrynic acid, or other ototoxic drugs. If the physician elects to use high dose parenteral therapy, controlled intravenous infusion is advisable (for adults, an infusion rate not exceeding 4 mg furosemide per minute has been used) (see PRECAUTIONS: Drug Interactions).
-
PRECAUTIONS
General
Excessive diuresis may cause dehydration and blood volume reduction with circulatory collapse and possibly vascular thrombosis and embolism, particularly in elderly patients. As with any effective diuretic, electrolyte depletion may occur during furosemide therapy, especially in patients receiving higher doses and a restricted salt intake. Hypokalemia may develop with furosemide, especially with brisk diuresis, inadequate oral electrolyte intake, when cirrhosis is present, or during concomitant use of corticosteroids, ACTH, licorice in large amounts, or prolonged use of laxatives. Digitalis therapy may exaggerate metabolic effects of hypokalemia, especially myocardial effects.
All patients receiving furosemide therapy should be observed for these signs or symptoms of fluid or electrolyte imbalance (hyponatremia, hypochloremic alkalosis, hypokalemia, hypomagnesemia or hypocalcemia): dryness of mouth, thirst, weakness, lethargy, drowsiness, restlessness, muscle pains or cramps, muscular fatigue, hypotension, oliguria, tachycardia, arrhythmia, or gastrointestinal disturbances such as nausea and vomiting. Increases in blood glucose and alterations in glucose tolerance tests (with abnormalities of the fasting and 2-hour postprandial sugar) have been observed, and rarely, precipitation of diabetes mellitus has been reported.
In patients with severe symptoms of urinary retention (because of bladder emptying disorders, prostatic hyperplasia, urethral narrowing), the administration of furosemide can cause acute urinary retention related to increased production and retention of urine. Thus, these patients require careful monitoring, especially during the initial stages of treatment.
In patients at high risk for radiocontrast nephropathy, furosemide can lead to a higher incidence of deterioration in renal function after receiving radiocontrast compared to high-risk patients who received only intravenous hydration prior to receiving radiocontrast.
In patients with hypoproteinemia (e.g., associated with nephrotic syndrome), the effect of furosemide may be weakened and its ototoxicity potentiated.
Asymptomatic hyperuricemia can occur and gout may rarely be precipitated.
Patients allergic to sulfonamides may also be allergic to furosemide. The possibility exists of exacerbation or activation of systemic lupus erythematosus.
As with many other drugs, patients should be observed regularly for the possible occurrence of blood dyscrasias, liver or kidney damage, or other idiosyncratic reactions.
Information for Patients
Patients receiving furosemide should be advised that they may experience symptoms from excessive fluid and/or electrolyte losses. The postural hypotension that sometimes occurs can usually be managed by getting up slowly. Potassium supplements and/or dietary measures may be needed to control or avoid hypokalemia.
Patients with diabetes mellitus should be told that furosemide may increase blood glucose levels and thereby affect urine glucose tests. The skin of some patients may be more sensitive to the effects of sunlight while taking furosemide.
Hypertensive patients should avoid medications that may increase blood pressure, including over-the-counter products for appetite suppression and cold symptoms.
Laboratory Tests
Serum electrolytes (particularly potassium), CO 2, creatinine and BUN should be determined frequently during the first few months of furosemide therapy and periodically thereafter. Serum and urine electrolyte determinations are particularly important when the patient is vomiting profusely or receiving parenteral fluids. Abnormalities should be corrected or the drug temporarily withdrawn. Other medications may also influence serum electrolytes.
Reversible elevations of BUN may occur and are associated with dehydration, which should be avoided, particularly in patients with renal insufficiency.
Urine and blood glucose should be checked periodically in diabetics receiving furosemide, even in those suspected of latent diabetes.
Furosemide may lower serum levels of calcium (rarely cases of tetany have been reported) and magnesium. Accordingly, serum levels of these electrolytes should be determined periodically.
In premature infants furosemide may precipitate nephrocalcinosis/nephrolithiasis, therefore renal function must be monitored and renal ultrasonography performed (see PRECAUTIONS: Pediatric Use).
Drug Interactions
Furosemide may increase the ototoxic potential of aminoglycoside antibiotics, especially in the presence of impaired renal function. Except in life-threatening situations, avoid this combination.
Furosemide should not be used concomitantly with ethacrynic acid because of the possibility of ototoxicity. Patients receiving high doses of salicylates concomitantly with furosemide, as in rheumatic disease, may experience salicylate toxicity at lower doses because of competitive renal excretory sites.
There is a risk of ototoxic effects if cisplatin and furosemide are given concomitantly. In addition, nephrotoxicity of nephrotoxic drugs such as cisplatin may be enhanced if furosemide is not given in lower doses and with positive fluid balance when used to achieve forced diuresis during cisplatin treatment.
Furosemide has a tendency to antagonize the skeletal muscle-relaxing effect of tubocurarine and may potentiate the action of succinylcholine.
Lithium generally should not be given with diuretics because they reduce lithium's renal clearance and add a high risk of lithium toxicity.
Furosemide combined with angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers may lead to severe hypotension and deterioration in renal function, including renal failure. An interruption or reduction in the dosage of furosemide, angiotensin-converting enzyme inhibitors, or angiotensin receptor blockers may be necessary.
Potentiation occurs with ganglionic or peripheral adrenergic blocking drugs.
Furosemide may decrease arterial responsiveness to norepinephrine. However, norepinephrine may still be used effectively.
Simultaneous administration of sucralfate and furosemide tablets may reduce the natriuretic and antihypertensive effects of furosemide. Patients receiving both drugs should be observed closely to determine if the desired diuretic and/or antihypertensive effect of furosemide is achieved. The intake of furosemide and sucralfate should be separated by at least two hours.
In isolated cases, intravenous administration of furosemide within 24 hours of taking chloral hydrate may lead to flushing, sweating attacks, restlessness, nausea, increase in blood pressure, and tachycardia. Use of furosemide concomitantly with chloral hydrate is therefore not recommended.
Phenytoin interferes directly with renal action of furosemide. There is evidence that treatment with phenytoin leads to decreased intestinal absorption of furosemide, and consequently to lower peak serum furosemide concentrations.
Methotrexate and other drugs that, like furosemide, undergo significant renal tubular secretion may reduce the effect of furosemide. Conversely, furosemide may decrease renal elimination of other drugs that undergo tubular secretion. High-dose treatment of both furosemide and these other drugs may result in elevated serum levels of these drugs and may potentiate their toxicity as well as the toxicity of furosemide.
Furosemide can increase the risk of cephalosporin-induced nephrotoxicity even in the setting of minor or transient renal impairment.
Concomitant use of cyclosporine and furosemide is associated with increased risk of gouty arthritis secondary to furosemide-induced hyperurecemia and cyclosporine impairment of renal urate excretion.
High doses (> 80 mg) of furosemide may inhibit the binding of thyroid hormones to carrier proteins and result in transient increase in free thyroid hormones, followed by an overall decrease in total thyroid hormone levels.
One study in six subjects demonstrated that the combination of furosemide and acetylsalicylic acid temporarily reduced creatinine clearance in patients with chronic renal insufficiency. There are case reports of patients who developed increased BUN, serum creatinine and serum potassium levels, and weight gain when furosemide was used in conjunction with NSAIDs.
Literature reports indicate that co-administration of indomethacin may reduce the natriuretic and antihypertensive effects of furosemide in some patients by inhibiting prostaglandin synthesis. Indomethacin may also affect plasma renin levels, aldosterone excretion, and renin profile evaluation. Patients receiving both indomethacin and furosemide should be observed closely to determine if the desired diuretic and/or antihypertensive effect of furosemide is achieved.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Furosemide was tested for carcinogenicity by oral administration in one strain of mice and one strain of rats. A small but significantly increased incidence of mammary gland carcinomas occurred in female mice at a dose 17.5 times the maximum human dose of 600 mg. There were marginal increases in uncommon tumors in male rats at a dose of 15 mg/kg (slightly greater than the maximum human dose) but not at 30 mg/kg.
Furosemide was devoid of mutagenic activity in various strains of Salmonella typhimurium when tested in the presence or absence of an in vitro metabolic activation system, and questionably positive for gene mutation in mouse lymphoma cells in the presence of rat liver S9 at the highest dose tested. Furosemide did not induce sister chromatid exchange in human cells in vitro, but other studies on chromosomal aberrations in human cells in vitro gave conflicting results. In Chinese hamster cells it induced chromosomal damage but was questionably positive for sister chromatid exchange. Studies on the induction by furosemide of chromosomal aberrations in mice were inconclusive. The urine of rats treated with this drug did not induce gene conversion in Saccharomyces cerevisiae.
Furosemide produced no impairment of fertility in male or female rats, at 100 mg/kg/day (the maximum effective diuretic dose in the rat and 8 times the maximal human dose of 600 mg/day).
Pregnancy
Furosemide has been shown to cause unexplained maternal deaths and abortions in rabbits at 2, 4 and 8 times the maximal recommended human dose. There are no adequate and well-controlled studies in pregnant women. Furosemide should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Treatment during pregnancy requires monitoring of fetal growth because of the potential for higher birth weights.
The effects of furosemide on embryonic and fetal development and on pregnant dams were studied in mice, rats and rabbits.
Furosemide caused unexplained maternal deaths and abortions in the rabbit at the lowest dose of 25 mg/kg (2 times the maximal recommended human dose of 600 mg/day). In another study, a dose of 50 mg/kg (4 times the maximal recommended human dose of 600 mg/day) also caused maternal deaths and abortions when administered to rabbits between Days 12 and 17 of gestation. In a third study, none of the pregnant rabbits survived a dose of 100 mg/kg. Data from the above studies indicate fetal lethality that can precede maternal deaths.
The results of the mouse study and one of the three rabbit studies also showed an increased incidence and severity of hydronephrosis (distention of the renal pelvis and, in some cases, of the ureters) in fetuses derived from the treated dams as compared with the incidence in fetuses from the control group.
Nursing Mothers
Because it appears in breast milk, caution should be exercised when furosemide is administered to a nursing mother.
Furosemide may inhibit lactation.
Pediatric Use
In premature infants furosemide may precipitate nephrocalcinosis/nephrolithiasis. Nephrocalcinosis/nephrolithiasis has also been observed in children under 4 years of age with no history of prematurity who have been treated chronically with furosemide. Monitor renal function, and renal ultrasonography should be considered, in pediatric patients receiving furosemide.
If furosemide is administered to premature infants during the first weeks of life, it may increase the risk of persistence of patent ductus arteriosus.
Geriatric Use
Controlled clinical studies of furosemide did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for the elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection and it may be useful to monitor renal function (see PRECAUTIONS: General and DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS
Adverse reactions are categorized below by organ system and listed by decreasing severity.
Gastrointestinal System Reactions
- 1. hepatic encephalopathy in patients with hepatocellular insufficiency
- 6. oral and gastric irritation
- 7. cramping
2. pancreatitis
8. diarrhea
- 3. jaundice (intrahepatic cholestatic jaundice)
9. constipation
4. increased liver enzymes
10. nausea
5. anorexia
- 11. vomiting
Systemic Hypersensitivity Reactions
- 1. severe anaphylactic or anaphylactoid reactions (e.g., with shock)
3. interstitial nephritis
- 2. systemic vasculitis
4. necrotizing angiitis
Central Nervous System Reactions
1. tinnitus and hearing loss
5. headache
2. paresthesias
6. blurred vision
3. vertigo
7. xanthopsia
4. dizziness
Hematologic Reactions
- 1. aplastic anemia
- 5. leukopenia
- 2. thrombocytopenia
- 6. anemia
- 3. agranulocytosis
- 7. eosinophilia
- 4. hemolytic anemia
Dermatologic-Hypersensitivity Reactions
- 1. toxic epidermal necrolysis
- 7. bullous pemphigoid
- 2. Stevens-Johnson Syndrome
- 8. purpura
- 3. erythema multiforme
- 9. fever
- 4. drug rash with eosinophilia and systemic symptoms
- 10. rash
- 5. acute generalized exanthematous pustulosis
- 11. pruritus
- 6. exfoliative dermatitis
- 12. urticaria
Cardiovascular Reaction
- Orthostatic hypotension may occur and be aggravated by alcohol, barbiturates or narcotics.
- Increase in cholesterol and triglyceride serum levels
Other Reactions
- 1. hyperglycemia
- 6. restlessness
- 2. glycosuria
- 7. urinary bladder spasm
- 3. hyperuricemia
- 8. thrombophlebitis
- 4. muscle spasm
- 9. fever
- 5. weakness
Whenever adverse reactions are moderate or severe, furosemide dosage should be reduced or therapy withdrawn.
To report SUSPECTED ADVERSE REACTIONS, contact Solco Healthcare US, LLC at 1-866-257-2597 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
The principal signs and symptoms of overdose with furosemide are dehydration, blood volume reduction, hypotension, electrolyte imbalance, hypokalemia and hypochloremic alkalosis, and are extensions of its diuretic action.
The acute toxicity of furosemide has been determined in mice, rats and dogs. In all three, the oral LD 50 exceeded 1000 mg/kg body weight, while the intravenous LD 50 ranged from 300 to 680 mg/kg. The acute intragastric toxicity in neonatal rats is 7 to 10 times that of adult rats.
The concentration of furosemide in biological fluids associated with toxicity or death is not known.
Treatment of overdosage is supportive and consists of replacement of excessive fluid and electrolyte losses.
Serum electrolytes, carbon dioxide level and blood pressure should be determined frequently. Adequate drainage must be assured in patients with urinary bladder outlet obstruction (such as prostatic hypertrophy).
Hemodialysis does not accelerate furosemide elimination.
-
DOSAGE AND ADMINISTRATION
Edema
Therapy should be individualized according to patient response to gain maximal therapeutic response and to determine the minimal dose needed to maintain that response.
Adults -
The usual initial dose of furosemide tablets is 20 to 80 mg given as a single dose. Ordinarily a prompt diuresis ensues. If needed, the same dose can be administered 6 to 8 hours later or the dose may be increased. The dose may be raised by 20 or 40 mg and given not sooner than 6 to 8 hours after the previous dose until the desired diuretic effect has been obtained. The individually determined single dose should then be given once or twice daily (e.g., at 8 am and 2 pm). The dose of furosemide tablets may be carefully titrated up to 600 mg/day in patients with clinically severe edematous states.
Edema may be most efficiently and safely mobilized by giving furosemide tablets on 2 to 4 consecutive days each week.
When doses exceeding 80 mg/day are given for prolonged periods, careful clinical observation and laboratory monitoring are particularly advisable (see PRECAUTIONS: Laboratory Tests).
Geriatric Patients -
In general, dose selection for the elderly patient should be cautious, usually starting at the low end of the dosing range (see PRECAUTIONS: Geriatric Use).
Pediatric Patients -
The usual initial dose of oral furosemide in pediatric patients is 2 mg/kg body weight, given as a single dose. If the diuretic response is not satisfactory after the initial dose, dosage may be increased by 1 or 2 mg/kg no sooner than 6 to 8 hours after the previous dose. Doses greater than 6 mg/kg body weight are not recommended. For maintenance therapy in pediatric patients, the dose should be adjusted to the minimum effective level.
Hypertension
Therapy should be individualized according to the patient's response to gain maximal therapeutic response and to determine the minimal dose needed to maintain the therapeutic response.
Adults -
The usual initial dose of furosemide tablets for hypertension is 80 mg, usually divided into 40 mg twice a day. Dosage should then be adjusted according to response. If response is not satisfactory, add other antihypertensive agents.
Changes in blood pressure must be carefully monitored when furosemide tablets are used with other antihypertensive drugs, especially during initial therapy. To prevent excessive drop in blood pressure, the dosage of other agents should be reduced by at least 50 percent when furosemide tablets are added to the regimen. As the blood pressure falls under the potentiating effect of furosemide tablets, a further reduction in dosage or even discontinuation of other antihypertensive drugs may be necessary.
Geriatric Patients -
In general, dose selection and dose adjustment for the elderly patient should be cautious, usually starting at the low end of the dosing range (see PRECAUTIONS: Geriatric Use).
-
HOW SUPPLIED
Furosemide Tablets, USP
20 mg: White-off white, oval, debossed "3169" on one side and debossed "V" on the reverse side, available as follows:
- Bottles of 30: NDC 72789-030-30
- Bottles of 60: NDC 72789-030-60
You may report side effects to Solco Healthcare US, LLC at 1-866-257-2597 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
- PRINCIPAL DISPLAY PANEL - 20 mg
-
INGREDIENTS AND APPEARANCE
FUROSEMIDE
furosemide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72789-030(NDC:43547-401) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FUROSEMIDE (UNII: 7LXU5N7ZO5) (FUROSEMIDE - UNII:7LXU5N7ZO5) FUROSEMIDE 20 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color white (off-white) Score no score Shape OVAL Size 8mm Flavor Imprint Code 3169;V Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72789-030-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/05/2019 2 NDC:72789-030-60 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/05/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076796 03/26/2004 Labeler - PD-Rx Pharmaceuticals, Inc. (156893695) Registrant - PD-Rx Pharmaceuticals, Inc. (156893695) Establishment Name Address ID/FEI Business Operations PD-Rx Pharmaceuticals, Inc. 156893695 repack(72789-030)