BRISDELLE- paroxetine capsule
Noven Therapeutics, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BRISDELLE safely and effectively. See full prescribing information for BRISDELLE.
BRISDELLE ® (paroxetine) capsules, for oral use Initial U.S. Approval: 1992 WARNING: SUICIDAL THOUGHTS AND BEHAVIORSSee full prescribing information for complete boxed warning.RECENT MAJOR CHANGESWarnings and Precautions, Serotonin Synrome and amphetamines ( 5.2) 03/2017 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATIONThe recommended dosage of BRISDELLE is 7.5 mg once daily, at bedtime ( 2.1) DOSAGE FORMS AND STRENGTHSCapsules: 7.5 mg ( 3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reactions (≥ 2%) reported in clinical trials were: headache, fatigue, and nausea/vomiting ( 6.1) To report SUSPECTED ADVERSE REACTIONS, contact Noven Therapeutics, LLC at 1-800-455-8070 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONSParoxetine is a strong CYP2D6 inhibitor. Co-administration of BRISDELLE can alter concentrations of other drugs that are metabolized by CYP2D6. Consider potential drug interactions prior to and during therapy ( 5.3, 7.1, 7.3). See Full Prescribing Information for a list of clinically significant drug interactions ( 7.1, 7.2, 7.3) See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 12/2014 |
FULL PRESCRIBING INFORMATION
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants, including selective serotonin reuptake inhibitors (SSRIs), have been shown to increase the risk of suicidal thoughts and behavior in pediatric and young adult patients when used to treat major depressive disorder and other psychiatric disorders. Because BRISDELLE is an SSRI, monitor patients closely for worsening and for emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber [see Warnings and Precautions ( 5.1)] .
1 INDICATIONS AND USAGE
BRISDELLE is indicated for the treatment of moderate to severe vasomotor symptoms (VMS) associated with menopause.
Limitation of Use:
BRISDELLE is not indicated for the treatment of any psychiatric condition. BRISDELLE contains a lower dose of paroxetine than that used to treat depression, obsessive compulsive disorder, panic disorder, generalized anxiety disorder, social anxiety disorder, and post-traumatic stress disorder. The safety and efficacy of this lower dose of paroxetine in BRISDELLE have not been established for any psychiatric condition. Patients who require paroxetine for treatment of a psychiatric condition should discontinue BRISDELLE and initiate a paroxetine-containing medication that is indicated for such use.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended dosage of BRISDELLE for the treatment of moderate to severe VMS is 7.5 mg once daily, at bedtime, with or without food.
2.2 Use of BRISDELLE Before or After a Monoamine Oxidase Inhibitor (MAOI)
Wait at least 14 days after discontinuation of an MAOI before initiating therapy with BRISDELLE. Conversely, allow at least 14 days after stopping BRISDELLE before starting an MAOI [see Contraindications ( 4.1), Warnings and Precautions ( 5.2) and Drug Interactions ( 7.3)] .
3 DOSAGE FORMS AND STRENGTHS
BRISDELLE is available as 7.5 mg pink capsules printed with black edible ink with “NOVEN” and “7.5 mg” on the capsule. Each capsule contains 9.69 mg paroxetine mesylate equivalent to 7.5 mg paroxetine base.
4 CONTRAINDICATIONS
4.1 Monoamine Oxidase Inhibitors
Concomitant use of an MAOI with BRISDELLE or within 14 days of stopping treatment with BRISDELLE is contraindicated because of an increased risk of serotonin syndrome. The use of BRISDELLE within 14 days of stopping an MAOI is also contraindicated [see Dosage and Administration ( 2.2), Warnings and Precautions ( 5.2) and Drug Interactions ( 7.3)] .
Starting BRISDELLE in a patient who is being treated with linezolid or intravenous methylene blue, both of which inhibit monoamine oxidase, is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration ( 2.2), Warnings and Precautions ( 5.2) and Drug Interactions ( 7.3)] .
4.2 Thioridazine
Concomitant use of BRISDELLE with thioridazine is contraindicated, because thioridazine prolongs the QT interval, and paroxetine can increase thioridazine levels [see Drug Interactions ( 7.1)] .
4.3 Pimozide
Concomitant use of BRISDELLE with pimozide is contraindicated because pimozide prolongs the QT interval, and paroxetine increases pimozide levels [see Drug Interactions ( 7.1)] .
4.4 Hypersensitivity to any Ingredient in BRISDELLE
BRISDELLE is contraindicated in patients with a history of hypersensitivity to paroxetine or any of the other ingredients in BRISDELLE.
4.5 Pregnancy
Menopausal VMS does not occur during pregnancy and BRISDELLE may cause fetal harm [see Use in Specific Populations ( 8.1)] .
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors
BRISDELLE is not approved for any psychiatric condition.
Antidepressants, including those that contain an SSRI, increase the risk of suicidal thinking and behavior (suicidality) in pediatric and young adult patients when used to treat major depressive disorder (MDD) and other psychiatric disorders. There is limited information regarding suicidality in women who use BRISDELLE for treatment of VMS. The BRISDELLE trials excluded women with a presence or history of previous psychiatric disorders.
Consider discontinuing BRISDELLE in patients with worsening depression or those who experience emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient’s presenting symptoms.
All patients being treated with BRISDELLE should be observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of treatment.
Anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania have been reported in patients being treated with antidepressants for MDD as well as for other psychiatric and nonpsychiatric indications. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Families and caregivers of patients being treated with BRISDELLE should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to healthcare providers.
5.2 Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome has been reported with SSRIs, including paroxetine, alone but particularly with concomitant use of serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John’s Wort), and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat depression and others such as linezolid and intravenous methylene blue).
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Monitor patients for the emergence of serotonin syndrome.
The concomitant use of BRISDELLE with MAOIs is contraindicated. Do not start BRISDELLE in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. All reports with methylene blue that provided information on the route of administration involved intravenous administration in the dose range of 1 mg/kg to 8 mg/kg. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection) or at lower doses. There may be circumstances when it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking BRISDELLE. BRISDELLE should be discontinued before initiating treatment with the MAOI [see Contraindications ( 4.1) and Dosage and Administration ( 2.2)] .
If concomitant use of BRISDELLE with other serotonergic drugs (e.g., triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John’s Wort) is clinically warranted, consider the increased risk of serotonin syndrome and carefully observe the patient, particularly during treatment initiation [see Contraindications ( 4.1) Drug Interactions ( 7.3)] .
Discontinue BRISDELLE and any concomitant serotonergic agents immediately if the above events occur and initiate supportive symptomatic treatment.
5.3 Potential Impact on Tamoxifen Efficacy
It is uncertain whether the co-administration of paroxetine and tamoxifen has a significant adverse effect on the efficacy of tamoxifen. Some studies have shown that the efficacy of tamoxifen, as measured by the risk of breast cancer relapse/mortality, may be reduced when co-prescribed with paroxetine as a result of paroxetine’s irreversible inhibition of CYP2D6 [see Drug Interactions ( 7.1)] . However, other studies have failed to demonstrate such a risk. When tamoxifen is used for the treatment or prevention of breast cancer, weigh the likely benefit of BRISDELLE for treating VMS vs. the risk of possible decreased tamoxifen effectiveness, and consider avoiding the concomitant use of BRISDELLE for VMS treatment.
5.4 Abnormal Bleeding
SSRIs, including BRISDELLE, may increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Bleeding events related to SSRIs have ranged from ecchymosis, hematoma, epistaxis, and petechiae to life-threatening hemorrhages. Caution patients about the risk of bleeding associated with the concomitant use of BRISDELLE and NSAIDs, aspirin, or other drugs that affect coagulation [see Drug Interactions ( 7.1)] .
5.5 Angle-Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressants and BRISDELLE may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
5.6 Hyponatremia
Hyponatremia may occur as a result of treatment with SSRIs, including BRISDELLE. Elderly patients may be at greater risk. In many cases, the hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported in patients using SSRIs. Also, patients taking diuretics or who are volume-depleted can be at greater risk. Consider discontinuation of BRISDELLE in patients with symptomatic hyponatremia and institute appropriate medical intervention.
Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which can lead to falls. Signs and symptoms associated with more severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death.
5.7 Bone Fracture
Epidemiological studies on bone fracture risk following exposure to SSRIs have reported an association between SSRI treatment and fractures. It is unknown to what extent fracture risk is directly attributable to SSRI treatment. If a BRISDELLE-treated patient presents with unexplained bone pain, point tenderness, swelling, or bruising, consider the possibility of a fragility fracture.
5.8 Screening Patients for Bipolar Disorder and Monitoring for Mania/Hypomania
BRISDELLE is only indicated for the treatment of moderate to severe VMS and is not approved for use in treating either depression or bipolar depression. However, prior to initiating treatment with BRISDELLE, all patients should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It is generally believed (though not established in controlled trials) that use of an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder.
5.9 Seizures
In premarketing testing of paroxetine, seizures occurred in 0.1% of paroxetine-treated patients. Use BRISDELLE cautiously in patients with a history of seizures or with conditions that potentially lower the seizure threshold. Evaluate and consider discontinuing use in any patient who develops seizures.
5.10 Akathisia
The use of paroxetine or other SSRIs has been associated with the development of akathisia, which is characterized by an inner sense of restlessness and psychomotor agitation such as an inability to sit or stand still usually associated with subjective distress. This is most likely to occur within the first few weeks of treatment. Discontinue treatment with BRISDELLLE if akathisia occurs.
5.11 Potential for Cognitive and Motor Impairment
BRISDELLE has the potential to impair judgment, thinking, or motor skills. Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that the drug treatment does not affect them adversely.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in labeling:
- Suicidality [see Warnings and Precautions ( 5.1)]
- Serotonin syndrome [see Warnings and Precautions ( 5.2)]
- Abnormal bleeding [see Warnings and Precautions ( 5.4)]
- Angle-Closure Glaucoma [see Warnings and Precautions ( 5.5)]
- Hyponatremia [see Warnings and Precautions ( 5.6)]
- Bone Fracture [see Warnings and Precautions ( 5.7)]
- Mania/Hypomania [see Warnings and Precautions ( 5.8)]
- Seizure [see Warnings and Precautions ( 5.9)]
- Akathisia [see Warnings and Precautions ( 5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot directly be compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to BRISDELLE in the one 8-week Phase 2 randomized, placebo-controlled trial and the two Phase 3 randomized, placebo-controlled, 12-week and 24-week trials for the treatment of moderate to severe VMS [see Clinical Studies ( 14)] . In these trials, a total of 635 women were exposed to BRISDELLE 7.5 mg administered orally once daily and 641 women received placebo. The majority of BRISDELLE-treated patients were Caucasian (68%) and African American (30%), with a mean age of 55 years (range 40 to 73 years). Women with a history of suicidal ideation or suicidal behavior were excluded from these studies.
Adverse Reactions Leading to Study Discontinuation: A total of 4.7% of women taking BRISDELLE discontinued from the clinical trials due to an adverse reaction, compared to 3.7% of women on placebo; the most frequent adverse reactions leading to discontinuation among paroxetine-treated women were: abdominal pain (0.3%), attention disturbances (0.3%), headache (0.3%), and suicidal ideation (0.3%).
Common Adverse Reactions: Overall, based on investigators’ determinations about what events were likely to be drug-related, about 20% of women treated with BRISDELLE reported at least 1 adverse reaction in the three controlled studies. The most common adverse reactions (≥ 2% and more common among BRISDELLE-treated women) reported in these studies were headache, fatigue/malaise/lethargy, and nausea/vomiting. Of these commonly reported adverse reactions, nausea occurred primarily within the first 4 weeks of treatment and fatigue occurred primarily within the first week of treatment, and decreased in frequency with continued therapy.
The adverse reactions that occurred in at least 2% of patients in the BRISDELLE group and at a higher incidence than placebo are shown in Table 1 for the pooled Phase 2 and Phase 3 trials.
| Frequency n (%) | ||
| BRISDELLE (n = 635) | Placebo (n = 641) | |
| Nervous system disorders | ||
| Headache | 40 (6.3) | 31 (4.8) |
| General disorders and administration site conditions | ||
| Fatigue, malaise, lethargy | 31 (4.9) | 18 (2.8) |
| Gastrointestinal disorders | ||
| Nausea, vomiting | 27 (4.3) | 15 (2.3) |
Certain symptoms were seen more frequently in women at the time of discontinuation of BRISDELLE compared to women discontinuing placebo, and have also been reported upon discontinuation of other formulations of paroxetine, particularly when abrupt. These include increased dreaming/nightmares, muscle cramps/spasms/twitching, headache, nervousness/anxiety, fatigue/tiredness, restless feeling in legs, and trouble sleeping/insomnia. While these events are generally self-limiting, there have been reports of serious discontinuation symptoms with other formulations of paroxetine.
Serious Adverse Reactions: In the pooled Phase 2 and Phase 3 trials, three BRISDELLE-treated patients reported a serious adverse reaction of suicidal ideation and one BRISDELLE-treated patient reported a serious adverse reaction of suicide attempt. There were no serious adverse reactions of suicidal ideation or suicide attempt reported among the placebo-treated patients.
6.2 Postmarketing Experience
The following adverse reactions have been identified from clinical studies of paroxetine and during post-approval use of other formulations of paroxetine. Because some of these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: Idiopathic thrombocytopenic purpura, Events related to impaired hematopoiesis (including aplastic anemia, pancytopenia, bone marrow aplasia, agranulocytosis).
Cardiac Disorders: Atrial fibrillation, Pulmonary edema, Ventricular fibrillation, Ventricular tachycardia (including torsades de pointes).
Gastrointestinal Disorders: Pancreatitis, Pancreatitis hemorrhagic, Vomiting.
General Disorders and Administration Site Conditions: Death, Drug withdrawal syndrome, Malaise.
Hepatobiliary Disorders: Drug-induced liver injury, Hepatic failure, Jaundice.
Immune System Disorders: Anaphylactoid reaction, Angioedema, Toxic epidermal necrolysis.
Investigations: Elevated liver tests (the most severe cases were deaths due to liver necrosis, and grossly elevated transaminases associated with severe liver dysfunction).
Metabolism and Nutrition Disorders: Diabetes mellitus inadequate control, Type 2 diabetes mellitus.
Nervous System Disorders: Neuroleptic malignant syndrome, Paresthesia, Somnolence, Tremor.
Psychiatric Disorders: Aggression, Agitation, Anxiety, Confusional state, Depression, Disorientation, Homicidal ideation, Insomnia, Restlessness.
Respiratory, Thoracic and Mediastinal Disorders: Pulmonary hypertension.
Skin and Subcutaneous Tissue Disorders: Hyperhidrosis, Stevens-Johnson syndrome.
7 DRUG INTERACTIONS
No drug-drug interaction studies have been conducted with BRISDELLE.
7.1 Potential for BRISDELLE to Affect Other Drugs
Paroxetine is a strong CYP2D6 inhibitor. Clinical drug interaction studies have been performed with substrates of CYP2D6 and show that paroxetine can inhibit the metabolism of drugs metabolized by CYP2D6 [see Clinical Pharmacology ( 12.3)] . Table 2 contains examples of drugs with a metabolism that may be affected by co-administration with BRISDELLE.
| Concomitant
Drug Name | Effect of Paroxetine on
Other Drugs | Clinical Recommendations |
| Thioridazine
| Increased plasma concentrations of thioridazine
Potential QTc prolongation | Concomitant use of thioridazine and BRISDELLE is
contraindicated. |
| Pimozide
| Increased plasma concentrations
of pimozide. Potential QTc prolongation | Concomitant use of pimozide and BRISDELLE is
contraindicated. |
| Tamoxifen
| Reduced plasma concentrations
of active tamoxifen metabolite | Consider avoiding concomitant use of tamoxifen
and BRISDELLE. |
|
Tricyclic Antidepressant (TCA) (e.g., Desipramine) | Increased plasma concentrations
and elimination half-life | Plasma TCA concentrations may need to be
monitored and the dose of TCA may need to be reduced if a TCA is co-administered with BRISDELLE. Monitor tolerability. |
| Risperidone
| Increased plasma concentrations
of risperidone | A lower dosage of risperidone may be necessary
(see the Full Prescribing Information for risperidone). Monitor tolerability. |
| Atomoxetine
| Increased exposure of
atomoxetine | A lower dosage of atomoxetine may be necessary
(see Full Prescribing Information for atomoxetine). Monitor tolerability. |
| Drugs Highly Bound
to Plasma Protein (e.g., Warfarin) | Increased free plasma
concentrations | The dosage of warfarin may need to be reduced.
Monitor tolerability and the International Normalized Ratio. |
| Digoxin
| Decreased plasma concentrations
of digoxin | Dosage of digoxin may need to be increased.
Monitor digoxin concentrations and clinical effect. |
| Theophylline
| Increased plasma concentrations
of theophylline | Dosage of theophylline may need to be decreased.
Monitor theophylline concentrations and tolerability. |
Use caution if co-administering BRISDELLE with other drugs that are metabolized by CYP2D6, including nortriptyline, amitriptyline, imipramine, desipramine, fluoxetine, phenothiazines, risperidone, and Type 1C antiarrhythmics (e.g., propafenone, flecainide, and encainide).
7.2 Potential for Other Drugs to Affect BRISDELLE
The metabolism and pharmacokinetics of paroxetine may be affected by the induction and inhibition of drug metabolizing enzymes such as CYP2D6. Table 3 contains a list of drugs that may affect the pharmacokinetics of BRISDELLE when administered concomitantly [see Clinical Pharmacology ( 12.3)] .
| Concomitant
Drug Name | Effect of Concomitant Drug
on Paroxetine | Clinical Recommendations
|
| Phenobarbital
| Decreased paroxetine exposure
| |
|
Phenytoin | Decreased paroxetine exposure
| |
|
Fosamprenavir/ Ritonavir | Decreased plasma concentration
of paroxetine | No dose adjustment for BRISDELLE.
Monitor clinical effect of BRISDELLE. |
| Cimetidine | Increased plasma concentration
of paroxetine |
Use caution if co-administering BRISDELLE with other drugs that inhibit CYP2D6 (e.g., quinidine).
7.3 Other Potentially Significant Drug Interactions
Monoamine Oxidase Inhibitors (MAOIs)
Serious adverse reactions such as serotonin syndrome have been reported in patients receiving a concomitant SSRI and MAOI, in patients started on an SSRI who recently received an MAOI and in patients started on an MAOI who recently received an SSRI. Therefore, concomitant use of MAOIs with BRISDELLE or use of BRISDELLE and an MAOI within 14 days of each other is contraindicated
[see Dosage and Administration (
2.2), Contraindications (
4.1) and Warnings and Precautions (
5.2)]
.
Serotonergic Drugs
If concomitant use of BRISDELLE with other serotonergic drugs (e.g., triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John’s Wort) is clinically warranted, consider the increased risk of serotonin syndrome and carefully observe the patient, particularly during treatment initiation
[see Warnings and Precautions (
5.2)]
.
An interaction between paroxetine and tryptophan may occur when they are co-administered. Adverse experiences, consisting primarily of headache, nausea, sweating, and dizziness, have been reported when tryptophan was administered to patients taking paroxetine. Consequently, concomitant use of BRISDELLE with tryptophan is not recommended.
If concomitant use of BRISDELLE with a serotonergic drug is warranted, carefully observe the patient, particularly during treatment initiation. There have been postmarketing reports of serotonin syndrome with the use of an SSRI and a triptan.
BRISDELLE contains paroxetine, which is also the active ingredient in other drugs. The concomitant use of BRISDELLE with other paroxetine products is not recommended [see Indications and Usage ( 1)] .
Drugs that Interfere with Hemostasis (e.g., NSAIDs, Aspirin, and Warfarin)
Altered anticoagulant effects, including increased bleeding, have been reported when SSRIs are co-administered with NSAIDs, aspirin, and warfarin or other drugs that affect coagulation. There may be a pharmacodynamic interaction between paroxetine and warfarin that causes an increased bleeding diathesis despite unaltered prothrombin time. Carefully monitor patients receiving warfarin therapy when BRISDELLE is initiated or discontinued
[see Warnings and Precautions (
5.4)]
.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category X
Risk Summary
BRISDELLE is contraindicated in pregnant women because menopausal VMS does not occur during pregnancy and paroxetine can cause fetal harm. Epidemiological studies have shown that infants exposed to paroxetine in the first trimester of pregnancy may have an increased risk of cardiovascular malformations. Cardiac malformations are a common congenital abnormality. These data would suggest that the risk of a cardiac abnormality following paroxetine exposure in the first trimester may increase the risk from 1% to 2%. Exposure to SSRIs in late pregnancy may lead to an increased risk for neonatal complications requiring prolonged hospitalization, respiratory support, and tube feeding, and/or persistent pulmonary hypertension of the newborn (PPHN). No teratogenicity was seen in reproductive development studies conducted in rats and rabbits. However, an increase in rat pup deaths was seen during the first 4 days of lactation when dosing occurred during the last trimester of gestation and continued throughout lactation, at a dose approximately equal to the maximum recommended human dose (MRHD) for VMS (7.5 mg) on an mg/m 2 basis. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
Human Data
First-Trimester Pregnancy Exposure
- Epidemiologic studies which include data from the Swedish National Registry, a retrospective cohort study using United Healthcare data and a meta-analysis of studies (1992-2008) have shown a less than 2-fold increased risk of cardiac malformations, primarily ventricular septal and atrial septal defects, with first-trimester paroxetine exposure. Two case-control studies using separate databases with > 9000 birth defect cases and > 4000 controls showed 7 and 6 paroxetine-exposed infants respectively, with right ventricular outflow tract obstructions, a 2- to 3-fold increased risk. An increase in overall congenital malformations with first-trimester paroxetine use was not observed in all studies.
Third-Trimester Pregnancy Exposure
- Neonates exposed to SSRIs late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying. These features are consistent with either a direct toxic effect of SSRIs or, possibly, a drug discontinuation syndrome. It should be noted that in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions ( 5.2)] .
- Infants exposed to SSRIs in late pregnancy may have an increased risk for persistent pulmonary hypertension of the newborn (PPHN). PPHN occurs in 1 – 2 per 1000 live births in the general population and is associated with substantial neonatal morbidity and mortality. In a retrospective case-control study of 377 women whose infants were born with PPHN and 836 women whose infants were born healthy, the risk for developing PPHN was approximately 6-fold higher for infants exposed to SSRIs after the 20th week of gestation compared to infants who had not been exposed to antidepressants during pregnancy. There is currently no corroborative evidence regarding the risk for PPHN following exposure to SSRIs in pregnancy; this is the first study that has investigated the potential risk. The study did not include enough cases with exposure to individual SSRIs to determine if all SSRIs posed similar levels of PPHN risk.
Animal Data
Reproduction studies were performed at doses up to 50 mg/kg/day in rats and 6 mg/kg/day in rabbits administered during organogenesis. These doses are approximately 65 (rat) and 16 (rabbit) times the maximum recommended human dose (MRHD) for VMS on an mg/m 2 basis. There were no teratogenic effects. However, in rats, there was an increase in pup deaths during the first 4 days of lactation when dosing occurred during the last trimester of gestation and continued throughout lactation. This effect occurred at a dose of 1 mg/kg/day or approximately equal to the MRHD for VMS on an mg/m 2 basis. The no-effect dose for rat pup mortality was not determined. The cause of these deaths is unknown.
8.3 Nursing Mothers
Paroxetine is excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from BRISDELLE, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established; BRISDELLE is not indicated in the pediatric population.
8.5 Geriatric Use
Clinical studies of BRISDELLE did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Elderly patients may have elevated paroxetine plasma concentrations compared to younger patients. However, no BRISDELLE dose adjustment is considered necessary in elderly patients [see Clinical Pharmacology ( 12.3)] .
SSRIs have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse event [see Warnings and Precautions ( 5.6)] .
8.6 Renal Impairment
No BRISDELLE dose adjustment is considered necessary in patients with renal impairment [see Clinical Pharmacology ( 12.3)] .
8.7 Hepatic Impairment
No BRISDELLE dose adjustment is considered necessary in patients with liver impairment [see Clinical Pharmacology ( 12.3)] .
10 OVERDOSAGE
10.1 Human Experience with Overdosage
There is limited clinical experience with BRISDELLE overdosage in humans, as there were no overdoses reported in the clinical studies.
Spontaneous cases of deliberate or accidental overdosage during paroxetine treatment have been reported; some of these cases were fatal and some of the fatalities appeared to involve paroxetine alone. Of nonfatal cases with known outcome, most recovered without sequelae. The largest known ingestion involved 2000 mg of paroxetine (267 times the maximum recommended daily dose) in a patient who recovered.
Commonly reported adverse reactions associated with paroxetine overdosage include somnolence, coma, nausea, tremor, tachycardia, confusion, vomiting, and dizziness. Other notable signs and symptoms observed with overdoses involving paroxetine (alone or with other substances) include mydriasis, convulsions (including status epilepticus), ventricular dysrhythmias (including torsades de pointes), hypertension, aggressive reactions, syncope, hypotension, stupor, bradycardia, dystonia, rhabdomyolysis, symptoms of hepatic dysfunction (including hepatic failure, hepatic necrosis, jaundice, hepatitis, and hepatic steatosis), serotonin syndrome, manic reactions, myoclonus, acute renal failure, and urinary retention.
10.2 Management of Overdosage
Treatment should consist of those general measures employed in the management of overdosage with any SSRI. Consult with a certified poison control center for up-to-date guidance and advice on treatment of overdosage.
Ensure an adequate airway, oxygenation, and ventilation. Monitor cardiac rhythm and vital signs. General supportive and symptomatic measures are also recommended. Induction of emesis is not recommended. In managing overdosage, consider the possibility of multiple drug involvement.
11 DESCRIPTION
BRISDELLE (paroxetine) is an orally administered selective serotonin reuptake inhibitor (SSRI) for the treatment of moderate to severe VMS associated with menopause. It is identified chemically as (-)- trans -4R- (4’-fluorophenyl) - 3S - [(3’, 4’-methylenedioxyphenoxy) methyl] piperidine mesylate and has the empirical formula of C 19H 20FNO 3•CH 3SO 3H. The molecular weight is 425.5 (329.4 as free base). The structural formula is:
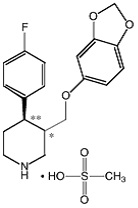
The mesylate salt of paroxetine is an odorless, off-white powder, having a melting point range of 147° to 150°C and a solubility of more than 1 g/mL in water.
Each pink capsule contains 9.69 mg paroxetine mesylate equivalent to 7.5 mg paroxetine base.
Inactive ingredients consist of: dibasic calcium phosphate, sodium starch glycolate, magnesium stearate, gelatin, titanium dioxide, FD&C Yellow #6, FD&C Red #3, FD&C Red #40, shellac, and black iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nonclinical studies have shown that paroxetine is an SSRI. BRISDELLE is not an estrogen, and its mechanism of action for the treatment of VMS is unknown.
12.2 Pharmacodynamics
Studies at clinically relevant doses in humans have demonstrated that paroxetine blocks the uptake of serotonin into human platelets. In vitro studies in animals also suggest that paroxetine is a selective inhibitor of neuronal serotonin reuptake and has weak effects on norepinephrine and dopamine neuronal reuptake. In vitro radioligand binding studies indicate that paroxetine has little affinity for muscarinic alpha 1-, alpha 2-, beta-adrenergic-, dopamine (D 2)-, 5-HT 1-, 5-HT 2-, and histamine (H 1)-receptors.
12.3 Pharmacokinetics
Absorption, Distribution, Metabolism and Excretion
-
Absorption
- Paroxetine is completely absorbed after oral dosing of the mesylate salt. In a study in which healthy postmenopausal women (n=24) received BRISDELLE 7.5 mg capsules as a daily dose for 14 days, steady-state paroxetine concentrations were achieved by approximately 12 days of dosing for most subjects, although it may take substantially longer in an occasional patient. Peak concentrations were reached at a median of 6 hours (3 to 8 hours range). Steady-state mean values of C
max, C
min, and AUC
0-last were 13.10 ng/mL (CV 91%), 7.17 ng/mL (CV 99%), and 237 hr*ng/mL (CV 94%), respectively.
- Steady-state AUC
0-24 values were about 3 times those of AUC
0-inf following a single dose, indicating non-linear pharmacokinetics. Steady-state C
max values were approximately 5 times greater than those attained after a single dose and steady-state exposure based on AUC
0-24 was about 10 times greater than AUC
0-24 after a single dose.
- The nonlinear kinetics and excess accumulation are due to the fact that CYP2D6, an enzyme that is in part responsible for paroxetine metabolism, is readily saturable.
- The effects of food on the bioavailability of paroxetine were studied with paroxetine tablets at higher strength. AUC was only slightly increased (6%) when drug was administered with food but the C
max was 29% greater, while the time to reach peak plasma concentration decreased from 6.4 hours post-dosing to 4.9 hours. BRISDELLE can be taken with or without food.
-
Distribution
- Paroxetine distributes throughout the body, including the central nervous system, with only 1% remaining in the plasma.
- Approximately 95% and 93% of paroxetine is bound to plasma protein at 100 ng/mL and 400 ng/mL, respectively. Under clinical conditions, paroxetine concentrations would normally be less than 100 ng/mL. Paroxetine does not alter the
in vitro protein binding of phenytoin or warfarin.
-
Metabolism
- Paroxetine is extensively metabolized after oral administration. The principal metabolites are polar and conjugated products of oxidation and methylation, which are readily cleared. Conjugates with glucuronic acid and sulfate predominate, and major metabolites have been isolated and identified. Data indicate that the metabolites have no more than 1/50 the potency of the parent compound at inhibiting serotonin uptake. The metabolism of paroxetine is accomplished in part by cytochrome CYP2D6. Saturation of this enzyme at clinical doses appears to account for the nonlinearity of paroxetine kinetics with increasing dose and increasing duration of treatment. The role of this enzyme in paroxetine metabolism also suggests potential drug-drug interactions
[see Drug Interactions (
7)]
. At steady state, when the CYP2D6 pathway is essentially saturated, paroxetine clearance is governed by alternative P450 isozymes, which, unlike CYP2D6, show no evidence of saturation.
-
Excretion
- Approximately 64% of a 30 mg oral solution of paroxetine was excreted in the urine with 2% as the parent compound and 62% as metabolites over a 10-day post-dosing period. About 36% of the dose was excreted in the feces (probably via the bile), mostly as metabolites and less than 1% as the parent compound over the 10-day post-dosing period.
Specific Populations
-
Renal and Liver Impairment
- Increased plasma concentrations of paroxetine occur in subjects with renal and hepatic impairment. The mean plasma concentration in patients with creatinine clearance below 30 mL/min was approximately 4 times greater than seen in normal volunteers. Patients with creatinine clearance of 30 to 60 mL/min and patients with hepatic impairment had about a 2-fold increase in plasma concentrations (AUC, C
max). No BRISDELLE dose adjustment is considered necessary in patients with renal or hepatic impairment.
-
Elderly Patients
- In a multiple-dose study in the elderly at daily paroxetine doses of 20, 30, and 40 mg, C
min concentrations were about 70% to 80% greater than the respective C
min concentrations in nonelderly subjects. No BRISDELLE dose adjustment is considered necessary in elderly patients.
Drug Interaction Studies
-
Potential Effect of BRISDELLE on Other Drugs
-
Drugs Metabolized by CYP3A4
- An
in vivo drug interaction study involving the co-administration under steady-state conditions of paroxetine and terfenadine, a substrate for cytochrome CYP3A4, revealed no effect of paroxetine on terfenadine pharmacokinetics.
In vitro studies have shown ketoconazole, a potent inhibitor of CYP3A4 activity, to be at least 100 times more potent than paroxetine as an inhibitor of the metabolism of several substrates for CYP3A4, including astemizole, triazolam, and cyclosporine. Based on the assumption that the relationship between paroxetine’s
in vitro Ki and its lack of effect on terfenadine’s
in vivo clearance predicts its effect on other CYP3A4 substrates, paroxetine’s extent of inhibition of CYP3A4 activity is not likely to be of clinical significance.
-
Drugs Metabolized by CYP2D6
- Many drugs are metabolized by the cytochrome P450 isozyme CYP2D6. Like other agents that are metabolized by CYP2D6, paroxetine may significantly inhibit the activity of this isozyme. In most patients (> 90%), this CYP2D6 isozyme is saturated early during paroxetine dosing.
- Specific studies investigating the effect of paroxetine on drugs metabolized by CYP2D6 are listed below:
-
Pimozide: Higher doses of paroxetine have been shown to elevate plasma levels of pimozide. In a controlled study of healthy volunteers, after paroxetine was titrated to 60 mg daily, co-administration of a single dose of 2 mg pimozide was associated with mean increases in pimozide AUC of 151% and C
max of 62%, compared to pimozide administered alone
[see Drug Interactions (
7.1)]
.
-
Tamoxifen: It is uncertain whether the co-administration of paroxetine and tamoxifen has a significant adverse effect on the efficacy of tamoxifen. Some studies have shown that the efficacy of tamoxifen, as measured by the risk of breast cancer relapse/mortality, may be reduced when co-prescribed with paroxetine as a result of paroxetine’s irreversible inhibition of CYP2D6. However, other studies have failed to demonstrate such a risk
[see Warnings and Precautions (
5.3) and Drug Interactions (
7.1)]
.
-
Desipramine: In one study, daily dosing of paroxetine (20 mg once daily) under steady-state conditions increased single dose desipramine (100 mg) C
max, AUC, and T
1/2 by an average of approximately 2-, 5-, and 3-fold, respectively
[see Drug Interactions (
7.1)]
.
-
Risperidone: Daily dosing of paroxetine 20 mg in patients stabilized on risperidone (4 to 8 mg/day), a CYP2D6 substrate, increased mean plasma concentrations of risperidone approximately 4-fold, decreased 9-hydroxyrisperidone concentrations approximately 10%, and increased concentrations of the active moiety (the sum of risperidone plus 9-hydroxyrisperidone) approximately 1.4-fold
[see Drug Interactions (
7.1)]
.
-
Atomoxetine: The effect of paroxetine on the pharmacokinetics of atomoxetine has been evaluated when both drugs were at steady state. In healthy volunteers who were extensive metabolizers of CYP2D6, paroxetine 20 mg daily was given in combination with 20 mg atomoxetine every 12 hours. This resulted in increases in steady-state atomoxetine AUC values that were 6- to 8-fold greater and in atomoxetine C
max values that were 3- to 4-fold greater than when atomoxetine was given alone
[see Drug Interactions (
7.1)]
.
-
Digoxin: Mean digoxin AUC at steady state decreased by 15% in the presence of paroxetine
[see Drug Interactions (
7.1)]
.
-
Beta Blockers: In a study in which propranolol (80 mg twice daily) was dosed orally for 18 days, the steady-state plasma concentrations of propranolol were unaltered during co-administration with paroxetine (30 mg once daily) for the final 10 days. The effects of propranolol on paroxetine have not been evaluated.
-
-
Potential Effect of Other Drugs on BRISDELLE
-
Concomitant use of paroxetine with other drugs that alter CYP enzymes activities including CYP2D6 may affect the plasma concentrations of paroxetine. Specific studies investigating the effect of other drugs on paroxetine are listed below:
-
Cimetidine: Cimetidine inhibits many cytochrome P450 enzymes. In a study in which paroxetine (30 mg once daily) was dosed orally for 4 weeks, steady-state plasma concentrations of paroxetine were increased by approximately 50% during co-administration with oral cimetidine (300 mg three times daily) for the final week
[see Drug Interactions (
7.2)]
.
-
Phenobarbital: Phenobarbital induces many cytochrome P450 enzymes. When a single oral 30 mg dose of paroxetine was administered at phenobarbital steady state (100 mg once daily for 14 days), paroxetine AUC and T
1/2 were reduced (by an average of 25% and 38%, respectively) compared to paroxetine administered alone. The effect of paroxetine on phenobarbital pharmacokinetics was not studied. Because paroxetine exhibits nonlinear pharmacokinetics, the results of this study may not address the case where the 2 drugs are both being chronically dosed
[see Drug Interactions (
7.2)]
.
-
Phenytoin: When a single oral 30 mg dose of paroxetine was administered at phenytoin steady state (300 mg once daily for 14 days), paroxetine AUC and T
1/2 were reduced (by an average of 50% and 35%, respectively) compared to paroxetine administered alone. In a separate study, when a single oral 300 mg dose of phenytoin was administered at paroxetine steady state (30 mg once daily for 14 days), phenytoin AUC was slightly reduced (12% on average) compared to phenytoin administered alone. Because both drugs exhibit nonlinear pharmacokinetics, the above studies may not address the case where the 2 drugs are both being chronically dosed
[see Drug Interactions (
7.2)]
.
-
Digoxin: A clinical drug interaction study showed that concurrent use of digoxin did not affect paroxetine exposure.
- Diazepam: A clinical drug interaction study showed that concurrent use of diazepam did not affect paroxetine exposure.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies were conducted in rodents given paroxetine in the diet at 1, 5, and 25 mg/kg/day (mice) and 1, 5, and 20 mg/kg/day (rats). The doses used in these carcinogenicity studies were approximately 16 (mouse) and 26 (rat) times the MHRD for VMS. There was a significantly greater number of male rats in the high-dose group with reticulum cell sarcomas (1/100, 0/50, 0/50, and 4/50 for control, low-, middle-, and high-dose groups, respectively) and a significantly increased linear trend across groups for the occurrence of lymphoreticular tumors in male rats. Female rats were not affected. Although there was a dose-related increase in the number of tumors in mice, there was no drug-related increase in the number of mice with tumors. The relevance of these findings to humans is unknown.
Mutagenesis
Paroxetine produced no genotoxic effect in a battery of 5 in vitro and 2 in vivo assays that included the following: bacterial mutation assay, mouse lymphoma mutation assay, unscheduled DNA synthesis assay, and tests for cytogenetic aberrations in vivo in mouse bone marrow and in vitro in human lymphocytes and in a dominant lethal test in rats.
Impairment of Fertility
A reduced pregnancy rate was found in reproduction studies in rats at a paroxetine dose of 15 mg/kg/day, which is 19 times the MRHD for VMS on an mg/m 2 basis. Irreversible lesions occurred in the reproductive tract of male rats after dosing in toxicity studies for 2 to 52 weeks. These lesions consisted of vacuolation of epididymal tubular epithelium at 50 mg/kg/day and atrophic changes in the seminiferous tubules of the testes with arrested spermatogenesis at 25 mg/kg/day (65 times and 32 times the MHRD for VMS on an mg/m 2 basis, respectively).
14 CLINICAL STUDIES
The efficacy of BRISDELLE as a treatment for moderate to severe VMS associated with menopause was established in two Phase 3 studies (at a dose of 7.5 mg once daily at bedtime) in 1174 postmenopausal women with a minimum of 7-8 moderate to severe vasomotor symptoms per day at baseline (≥ 50 per week) for 30 days prior to receiving study drug.
Study 1 was a 12-week, randomized, double-blind, placebo-controlled clinical trial with a total of 606 postmenopausal women (average age 55 years, 65% Caucasian and 33% African American, 18% surgically menopausal and 82% naturally menopausal).
Study 2 was a 24-week, randomized, double-blind, placebo-controlled clinical trial with a total of 568 postmenopausal women (average age 54 years, 76% Caucasian and 22% African American, 20% surgically menopausal and 81% naturally menopausal).
The co-primary efficacy endpoints for both studies were the reduction from baseline in VMS frequency and severity at Weeks 4 and 12. Data from Study 1 showed a statistically significant reduction from baseline in the frequency of moderate to severe vasomotor symptoms at Week 4 and Week 12 and a statistically significant reduction in the severity of moderate to severe VMS at Week 4 for BRISDELLE compared to placebo ( Table 4). Data from Study 2 showed a statistically significant reduction from baseline in the frequency and severity of moderate to severe vasomotor symptoms at Week 4 and Week 12 for BRISDELLE compared to placebo ( Table 5).
| MITT population: all consented and randomized subjects with valid baseline daily hot flash diary data who had taken at least 1 dose of study medication and had at least 1 day of on-treatment daily hot flash diary data.
* Treatment Difference: the difference between the median changes from baseline. #P-value is obtained from rank-ANCOVA model. |
|||||
| Frequency
| Severity
|
||||
| BRISDELLE
| Placebo
| BRISDELLE
| Placebo
|
||
| Baseline | |||||
| n | 301 | 305 | 301 | 305 | |
| Median | 10.4 | 10.4 | 2.5 | 2.5 | |
| Change from baseline at Week 4 | |||||
| n | 289 | 293 | 281 | 289 | |
| Median | -4.3 | -3.1 | -0.05 | 0.00 | |
| Treatment Difference* | -1.2 | -0.05 | |||
| P-value # | <0.01 | <0.01 | |||
| Change from baseline at Week 12 | |||||
| n | 264 | 274 | 236 | 253 | |
| Median | -5.9 | -5.0 | -0.06 | -0.02 | |
| Treatment Difference* | -0.9 | -0.04 | |||
|
P-value
#
| <0.01 | 0.17 | |||
| MITT population: all consented and randomized subjects with valid baseline daily hot flash diary data who had taken at least 1 dose of study medication and had at least 1 day of on-treatment daily hot flash diary data.
* Treatment Difference: the difference between the median changes from baseline. #P-value is obtained from rank-ANCOVA model. |
|||||
| Frequency
| Severity
|
||||
| BRISDELLE
| Placebo
| BRISDELLE
| Placebo
|
||
| Baseline | |||||
| n | 284 | 284 | 284 | 284 | |
| Median | 9.9 | 9.6 | 2.5 | 2.5 | |
| Change from baseline at Week 4 | |||||
| n | 276 | 274 | 268 | 271 | |
| Median | -3.8 | -2.5 | -0.04 | -0.01 | |
| Treatment Difference* | -1.3 | -0.03 | |||
| P-value # | <0.01 | 0.04 | |||
| Change from baseline at Week 12 | |||||
| n | 257 | 244 | 245 | 236 | |
| Median | -5.6 | -3.9 | -0.05 | 0.00 | |
| Treatment Difference* | -1.7 | -0.05 | |||
|
P-value
#
| <0.01 | <0.01 | |||
Persistence of benefit at 24 weeks in Study 2 was evaluated with a responder analysis where responders were defined as those patients who achieved ≥ 50% reduction from baseline in the frequency of moderate to severe VMS at Week 24. The proportion of patients achieving a ≥ 50% reduction in the frequency of moderate to severe VMS from baseline to Week 24 was 48% in the BRISDELLE group and 36% in the placebo group at Week 24.
16 HOW SUPPLIED/STORAGE AND HANDLING
BRISDELLE is available as 7.5 mg pink capsules printed with black edible ink with “NOVEN” and “7.5 mg” on each capsule.
NDC 68968-9075-3, blister packs of 30
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling ( Medication Guide).
Instruct patients to read the Medication Guide before starting therapy with BRISDELLE and to reread it each time the prescription is renewed.
- Advise patients, their families, and their caregivers to look for the emergence of suicidality, especially early during treatment
[see Boxed Warning and Warnings and Precautions (
5.1)]
.
- Instruct patients not to take BRISDELLE with an MAOI or within 14 days of stopping an MAOI and allow 14 days after stopping BRISDELLE before starting an MAOI
[see Dosage and Administration (
2.2) and Contraindications (
4.1)]
.
- Advise patients not to take BRISDELLE with thioridazine or pimozide
[see Contraindications (
4.2 and
4.3)]
.
- Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of BRISDELLE with triptans, tricyclic antidepressants, linezolid, tramadol, amphetamines, St. John’s Wort, lithium, tryptophan supplements, other serotonergic agents, or antipsychotic drugs
[see Warnings and Precautions (
5.2) and Drug Interactions (
7.3)]
.
- Caution patients that efficacy of tamoxifen may be reduced when administered concomitantly and counsel them about the likely benefit of paroxetine for treating VMS vs. the risk of possible decreased tamoxifen effectiveness
[see Warnings and Precautions (
5.3)]
.
- Caution patients about the concomitant use of BRISDELLE and NSAIDs, aspirin, warfarin, and other anticoagulants because combined use of drugs that interfere with serotonin reuptake has been associated with an increased risk of bleeding
[see Warnings and Precautions (
5.4)]
.
- Advise patients that taking BRISDELLE can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle-closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle-closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle-closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible
[See Warnings and Precautions (
5.5)]
.
- Caution patients about the risk of hyponatremia, particularly elderly patients and those who are taking diuretics or are volume-depleted
[see Warnings and Precautions (
5.6)]
.
- Inform patients that there is the possibility for an increased risk of fracture
[see Warnings and Precautions (
5.7)]
.
- Advise patients, their families, and their caregivers to observe for signs of activation of mania/hypomania
[see Warnings and Precautions (
5.8)]
.
- Advise patients to notify their physician if they become pregnant during therapy
[see Contraindications (
4.5) and Use in Specific Populations (
8.1)]
.Caution patients about operating hazardous machinery, including motor vehicles, until they are reasonably certain that paroxetine therapy does not affect their ability to engage in such activities
[see Warnings and Precautions (
5.11)]
.
- Advise patients to inform their healthcare provider if they are taking, or plan to take, any prescription or over-the-counter drugs, including herbal supplements, because there is a potential for interaction with paroxetine
[see Drug Interactions (
7.3)]
.
- Advise patients that paroxetine, the active ingredient in BRISDELLE, is also the active ingredient in certain other drugs and these medications should not be taken concomitantly
[see Indications and Usage (
1) and Drug Interactions (
7.3)]
.
Distributed by: Sebela Pharmaceuticals Inc., 645 Hembree Parkway, Suite I, Roswell, GA 30076
www.sebelapharma.com
Toll Free 1-844-732-3521
BRISDELLE is a registered trademark of Sebela International Limited.
© 2017 Sebela Pharmaceuticals Inc. All rights reserved.
MEDICATION GUIDE
BRISDELLE ® (bris-del)
(Paroxetine)
Capsules
Read the Medication Guide that comes with BRISDELLE before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or treatment. Talk with your healthcare provider if there is something you do not understand or want to learn more about. BRISDELLE contains a lower dose of paroxetine, a medicine also used to treat a number of psychiatric disorders. The lower dose of paroxetine in BRISDELLE has not been studied in any psychiatric conditions and BRISDELLE is not approved for any psychiatric uses.
What is the most important information I should know about BRISDELLE?
BRISDELLE may cause serious side effects.
Call your healthcare provider right away if you have any of the following symptoms, or go to the nearest emergency room:
1. Suicidal thoughts or actions:
- BRISDELLE, and related antidepressant medicines, may increase suicidal thoughts or actions within the first few months of treatment.
- Depression or other serious mental illnesses are the most important causes of suicidal thoughts or actions.
- Watch for these changes and call your healthcare provider right away if you notice:
- New or sudden changes in mood, behavior, actions, thoughts, or feelings, especially if severe.
- Pay particular attention to such changes when BRISDELLE is started.
- Keep all follow-up visits with your healthcare provider and call between visits if you are worried about symptoms.
-
Call your healthcare provider right away or go to the nearest emergency room if you have any of the following symptoms, especially if they are new, worse, or worry you:
- attempts to commit suicide
- acting on dangerous impulses
- acting aggressive or violent
- thoughts about suicide or dying
- new or worse depression
- new or worse anxiety or panic attacks
- feeling agitated, restless, angry or irritable
- trouble sleeping
- an increase in activity or talking more than what is normal for you
- other unusual changes in behavior or mood.
2. Serotonin Syndrome. This condition can be life-threatening and may include:
- agitation (nervousness), hallucinations, coma or other changes in mental status
- coordination problems or muscle twitching (small movements of the muscles that you cannot control)
- racing heartbeat, high or low blood pressure
- sweating or fever
- nausea, vomiting, or diarrhea
- muscle rigidity
- dizziness
- flushing
- tremors
- seizures
3. Reduced effectiveness of tamoxifen. Tamoxifen (a medicine used to treat breast cancer) may not work as well if it is taken while you take BRISDELLE. If you are taking tamoxifen, tell your healthcare provider before starting BRISDELLE.
4. Abnormal bleeding. BRISDELLE may increase your risk of bleeding or bruising, especially if you take the blood thinner warfarin or non-steroidal anti-inflammatory drugs (NSAIDs) like ibuprofen, naproxen, or aspirin.
5. Visual problems.
• Eye pain
• Changes in vision • Swelling or redness in or around the eye
Only some people are at risk for these problems. You may want to undergo an eye examination to see if you are at risk and receive preventative treatment if you are.
6. Low salt (sodium) levels in the blood. Elderly people may be at greater risk for this condition. Symptoms may include:
- headache
- weakness or feeling unsteady
- confusion, problems concentrating or thinking or memory problems.
7. Bone Fractures. Women who take BRISDELLE may have a higher risk of bone fractures. Contact your healthcare provider if you have pain in a bone.
8. Manic episodes:
- greatly increased energy
- severe trouble sleeping
- racing thoughts
- reckless behavior
- unusually grand ideas
- excessive happiness or irritability
- talking more or faster than usual.
9. Seizures or convulsions.
10. Restlessness. Women who take BRISDELLE may feel an inner restlessness, agitation (nervousness), or be unable to sit still or stand still especially when they start taking BRISDELLE. Call your healthcare provider if this happens to you.
11. Driving. BRISDELLE may affect your ability to make decisions, think clearly, or react quickly. Do not drive, operate heavy machinery, or do other potentially dangerous activities until you know how BRISDELLE affects you.
What is BRISDELLE?
BRISDELLE is a prescription medicine used to reduce moderate to severe hot flashes associated with menopause. BRISDELLE is a selective serotonin reuptake inhibitor (SSRI). It is not a hormone. The way BRISDELLE treats hot flashes associated with menopause is not known. BRISDELLE does not prevent or treat osteoporosis or dryness, itching or burning in and around the vagina.
BRISDELLE is not for psychiatric problems such as depression, obsessive compulsive disorder, panic disorder, generalized anxiety disorder, social anxiety disorder, and post-traumatic stress disorder.
BRISDELLE is not for use in children.
Talk to your healthcare provider if you do not think that your hot flashes are getting better while taking BRISDELLE.
Who should not take BRISDELLE?
Do not take BRISDELLE if you:
-
take a Monoamine Oxidase Inhibitor (MAOI). Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI, including the antibiotic linezolid.
- Do not take an MAOI within 14 days of stopping BRISDELLE unless directed to do so by your healthcare provider.
- Do not start BRISDELLE if you stopped taking an MAOI in the last 14 days unless directed to do so by your healthcare provider.
-
People who take BRISDELLE close in time to an MAOI may have serious or life-threatening side effects. Get medical help right away if you have any of these symptoms:
- high fever
- uncontrolled muscle spasms
- stiff muscles
- rapid changes in heart rate or blood pressure
- confusion
- loss of consciousness (pass out)
- take thioridazine. Do not take thioridazine together with BRISDELLE because this can cause serious heart rhythm problems or sudden death.
- take the antipsychotic medicine pimozide. Do not take pimozide together with BRISDELLE because this can cause serious heart problems.
- are allergic to paroxetine or any of the ingredients in BRISDELLE. See the end of this Medication Guide for a complete list of ingredients in BRISDELLE.
- are pregnant. BRISDELLE is not for pregnant women. Paroxetine, the active ingredient in BRISDELLE, can harm your unborn baby. Risks to your unborn baby include an increased risk of birth defects, particularly heart defects. Your baby may also have certain other serious symptoms shortly after birth.
What should I tell my healthcare provider before taking BRISDELLE?
Before starting BRISDELLE, tell your healthcare provider if you:
- have liver problems
- have kidney problems
- have or had seizures or convulsions
- have bipolar disorder or mania
- have low sodium levels in your blood
- have or had bleeding problems
- have glaucoma (high pressure in the eye)
- have any other medical conditions
- are breastfeeding or plan to breastfeed. BRISDELLE passes into breast milk. Talk to your healthcare provider before taking BRISDELLE if you are breast-feeding.
Tell your healthcare provider about all the medicines that you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. BRISDELLE and some medicines may interact with each other, may not work as well, or may cause serious side effects when taken together.
| If you take BRISDELLE, you should not take any other medicines that contain paroxetine,
including Paxil, Paxil CR and Pexeva. |
Especially tell your healthcare provider if you take:
- triptans used to treat migraine headache
- medicines used to treat mood, anxiety, psychotic or thought disorders, including MAOIs, SSRIs, tricyclics, lithium, buspirone, or antipsychotics
- tramadol, fentanyl or over-the-counter supplements such as tryptophan or St. John’s Wort
- amphetamines
- thioridazine
- pimozide
- tamoxifen
- atomoxetine
- cimetidine
- digoxin
- theophylline
- medicines to treat irregular heart rate (like propafenone, flecainide, and encainide)
- medicines used to treat schizophrenia
- certain medicines used to treat HIV infection
- the blood thinner warfarin
- nonsteroidal anti-inflammatory drugs (NSAIDs) (like ibuprofen, naproxen, or aspirin)
- certain medicines used to treat seizures (like phenobarbital and phenytoin)
- other drugs containing paroxetine, the medicine in BRISDELLE.
Ask your healthcare provider if you are not sure if you are taking any of these medications.
Your healthcare provider or pharmacist can tell you if it is safe to take BRISDELLE with your other medicines. Do not start or stop any medicine while taking BRISDELLE without talking to your healthcare provider first.
How should I take BRISDELLE?
- Take BRISDELLE exactly as your healthcare provider tells you to take it.
- Take BRISDELLE 1 time each day at bedtime.
- BRISDELLE may be taken with or without food.
- If you miss a dose of BRISDELLE, take the missed dose as soon as you remember. If it is almost time for the next dose, skip the missed dose and take your next dose at the regular time. Do not take two doses of BRISDELLE at the same time.
- If you take too much BRISDELLE, call your healthcare provider or poison control center right away, or go to the nearest emergency room right away.
What should I avoid while taking BRISDELLE?
- BRISDELLE can cause sleepiness or may affect your ability to make decisions, think clearly, or react quickly. You should not drive, operate heavy machinery, or do other dangerous activities until you know how BRISDELLE affects you.
What are the possible side effects of BRISDELLE?
BRISDELLE may cause serious side effects, including:
The most common side effects of BRISDELLE include:
- headache
- tiredness
- nausea and vomiting
Tell your healthcare provider if you have any side effect that bothers you or does not go away. These are not all the possible side effects of BRISDELLE. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store BRISDELLE?
- Store BRISDELLE at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep BRISDELLE out of the light.
- Keep BRISDELLE dry.
- Keep BRISDELLE and all medicines out of the reach of children.
General information about the safe and effective use of BRISDELLE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BRISDELLE for a condition for which it was not prescribed. Do not give BRISDELLE to other people, even if they have the same condition. It may harm them.
This Medication Guide summarizes the most important information about BRISDELLE. If you would like more information, talk with your healthcare provider. You may ask your healthcare provider or pharmacist for information about BRISDELLE that is written for healthcare professionals.
For more information about BRISDELLE call 1-800-455-8070 or go to www.BRISDELLE.com.
What are the ingredients in BRISDELLE?
Active ingredient: paroxetine
Inactive ingredients: dibasic calcium phosphate, sodium starch glycolate, magnesium stearate, gelatin, titanium dioxide, FD&C Yellow #6, FD&C Red #3, FD&C Red #40, shellac and black iron oxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Distributed by: Sebela Pharmaceuticals Inc. 645 Hembree Parkway, Suite I
Roswell, GA 30076
BRISDELLE
® is a registered trademark of Sebela International Ltd.
© 2017 Sebela Pharmaceuticals Inc. All rights reserved.
Revised 03/2017

| BRISDELLE
paroxetine capsule |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Noven Therapeutics, LLC (166888268) |
| Registrant - Sebela Pharmaceuticals Inc. (079104574) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sharp Corporation | 002346625 | pack(68968-9075) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Anderson Brecon Packaging, Inc. | 053217022 | pack(68968-9075) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Quality Chemical Laboratories | 071344167 | analysis(68968-9075) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Packaging Coordinators, LLC | 078525133 | pack(68968-9075) , label(68968-9075) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Norwich Pharmaceuticals, Inc. | 132218731 | manufacture(68968-9075) , analysis(68968-9075) , pack(68968-9075) , label(68968-9075) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Excella GmbH | 329809800 | api manufacture(68968-9075) , analysis(68968-9075) | |