VITEKTA- elvitegravir tablet, film coated
Gilead Sciences, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use VITEKTA safely and effectively. See full prescribing information for VITEKTA.
VITEKTA ® (elvitegravir) tablets, for oral use Initial U.S. Approval: 2012 INDICATIONS AND USAGEVITEKTA is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor used in combination with an HIV protease inhibitor coadministered with ritonavir and with other antiretroviral drug(s) indicated for the treatment of HIV-1 infection in antiretroviral treatment-experienced adults. ( 1) Limitations of Use:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSTablets: 85 mg and 150 mg ( 3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONSADVERSE REACTIONSThe most common adverse drug reaction to VITEKTA (all grades) is diarrhea. ( 6.1) To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2015 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
VITEKTA in combination with an HIV protease inhibitor coadministered with ritonavir and with other antiretroviral drug(s) is indicated for the treatment of HIV-1 infection in antiretroviral treatment-experienced adults.
Limitations of Use:
- There are no comparative pharmacokinetic or clinical data evaluating VITEKTA with cobicistat as single entities compared to STRIBILD ®.
- VITEKTA coadministered with protease inhibitors and cobicistat is not recommended [see Warnings and Precautions (5.2)] .
- Coadministration of VITEKTA with dosage regimens or HIV-1 protease inhibitors other than those presented in Table 1 is not recommended.
2 DOSAGE AND ADMINISTRATION
VITEKTA must be administered once daily with food in combination with a protease inhibitor coadministered with ritonavir and another antiretroviral drug. The protease inhibitor and ritonavir dosing regimens presented in Table 1 are the recommended regimens for use with VITEKTA. For additional dosing instructions for these protease inhibitors and other concomitant antiretroviral drugs, refer to their respective prescribing information.
| Dosage of VITEKTA | Dosage of Concomitant Protease Inhibitor | Dosage of Concomitant Ritonavir |
|---|---|---|
|
||
| 85 mg orally once daily | Atazanavir 300 mg orally once daily | 100 mg orally once daily |
| Lopinavir 400 mg orally twice daily | 100 mg orally twice daily | |
| 150 mg orally once daily | Darunavir 600 mg orally twice daily | 100 mg orally twice daily |
| Fosamprenavir 700 mg orally twice daily | 100 mg orally twice daily | |
| Tipranavir 500 mg orally twice daily | 200 mg orally twice daily | |
Treatment history and, when available, resistance testing should guide the use of VITEKTA-containing regimens.
3 DOSAGE FORMS AND STRENGTHS
- Tablets: 85 mg green, pentagon-shaped, film-coated, debossed with "GSI" on one side and "85" on the other side.
- Tablets: 150 mg green, triangle-shaped, film-coated, debossed with "GSI" on one side and "150" on the other side.
4 CONTRAINDICATIONS
There are no contraindications to VITEKTA. Due to the need to use VITEKTA with a protease inhibitor coadministered with ritonavir, prescribers should consult the complete prescribing information of the coadministered protease inhibitor and ritonavir for a description of contraindications.
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of VITEKTA and other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Drug Interactions (7)]:
- Loss of therapeutic effect of VITEKTA and possible development of resistance
- Possible clinically significant adverse reactions from greater exposures of concomitant drugs or elvitegravir.
See Table 4 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions (7)] . Consider the potential for drug interactions prior to and during VITEKTA therapy; review concomitant medications during VITEKTA therapy; and monitor for the adverse reactions associated with the concomitant drugs.
5.2 Use with Other Antiretroviral Agents
Use of VITEKTA in combination with the fixed dose combination STRIBILD is not recommended, because elvitegravir is a component of STRIBILD.
VITEKTA is indicated for use in combination with a protease inhibitor coadministered with ritonavir and with other antiretroviral drug(s). VITEKTA in combination with a protease inhibitor and cobicistat is not recommended because dosing recommendations for such combinations have not been established and may result in suboptimal plasma concentrations of VITEKTA and/or the protease inhibitor, leading to loss of therapeutic effect and development of resistance.
5.3 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety assessment of VITEKTA is primarily based on data from a controlled clinical trial, Study 145, in which 712 HIV-1 infected, antiretroviral treatment-experienced adults received VITEKTA (N=354) or raltegravir (N=358), each administered with a background regimen consisting of a fully active protease inhibitor coadministered with ritonavir and with other antiretroviral drug(s) for at least 96 weeks.
The proportion of subjects who discontinued study treatment due to adverse events, regardless of severity, was 3% in the VITEKTA group and 4% in the raltegravir group. The most common adverse reaction (all Grades, incidence greater than or equal to 5%) in subjects receiving VITEKTA in Study 145 was diarrhea. See also Table 2 for the frequency of adverse reactions occurring in at least 2% of subjects in any treatment group in Study 145.
| VITEKTA
N=354 | Raltegravir
N=358 |
|
|---|---|---|
|
||
| Diarrhea | 7% | 5% |
| Nausea | 4% | 3% |
| Headache | 3% | 3% |
Less Common Adverse Reactions Observed in Treatment-Experienced Studies: The following adverse reactions occurred in <2% of subjects receiving VITEKTA combined with a protease inhibitor and ritonavir. These reactions have been included because of their seriousness, increased frequency on VITEKTA compared with raltegravir, or investigator's assessment of potential causal relationship.
- Gastrointestinal Disorders: abdominal pain, dyspepsia, vomiting
- General Disorders and Administration Site Conditions: fatigue
- Psychiatric Disorders: depression, insomnia, suicidal ideation and suicide attempt (<1%, most in subjects with a pre-existing history of depression or psychiatric illness)
- Skin and Subcutaneous Tissue Disorders: rash
Laboratory Abnormalities: The frequency of laboratory abnormalities (Grades 3–4), occurring in at least 2% of subjects in either treatment group in Study 145, is presented in Table 3.
| Laboratory Parameter Abnormality | VITEKTA
N=354 | Raltegravir
N=358 |
|---|---|---|
|
||
| Total Bilirubin (>2.5 × ULN) | 6% | 9% |
| Hematuria (> 75 RBC/HPF) | 6% | 7% |
| Serum Amylase * (> 2.0 × ULN) | 6% | 6% |
| Creatine Kinase (≥ 10.0 × ULN) | 6% | 4% |
| Total Cholesterol (> 300 mg/dL) | 5% | 5% |
| Total Triglycerides (>750 mg/dL) | 5% | 4% |
| Hyperglycemia (> 250 mg/dL) | 5% | 3% |
| Urine Glucose (4 +) | 4% | 3% |
| GGT (> 5.0 × ULN) | 3% | 7% |
| Neutrophils (< 750/mm 3) | 3% | 3% |
| ALT (> 5.0 × ULN) | 2% | 5% |
| AST (> 5.0 × ULN) | 2% | 6% |
7 DRUG INTERACTIONS
See also Dosage and Administration (2), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3).
7.1 Effect of Concomitant Drugs on the Pharmacokinetics of Elvitegravir
Elvitegravir is metabolized by CYP3A. Drugs that induce CYP3A activity are expected to increase the clearance of elvitegravir, as well as ritonavir. This may result in decreased plasma concentrations of elvitegravir and/or a concomitantly administered protease inhibitor and lead to loss of therapeutic effect and to possible resistance.
7.2 Established and Other Potentially Significant Interactions
Table 4 provides dosing recommendations as a result of potentially clinically significant drug interactions with VITEKTA. These recommendations are based on either drug-drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of therapeutic effect.
For additional drug-drug interactions related to protease inhibitors coadministered with ritonavir, consult the prescribing information of the coadministered protease inhibitor and ritonavir.
The table is not all-inclusive [see Clinical Pharmacology (12.3), Tables 6– 7] .
| Concomitant Drug Class: Drug Name | Effect on Concentration † | Clinical Comment |
|---|---|---|
| Antiretroviral Agents: Protease Inhibitors (PIs) ‡ | ||
| Atazanavir § | ↔ atazanavir
↑ elvitegravir | There are no data available to make dosing recommendations for coadministration with doses of atazanavir/ritonavir other than 300/100 mg once daily. Please refer to Section 2 for dosage adjustments. |
| Lopinavir/ritonavir § | ↔ lopinavir
↑ elvitegravir | There are no data available to make dosing recommendations for coadministration with doses of lopinavir/ritonavir other than 400/100 mg twice daily. Please refer to Section 2 for dosage adjustments. |
| Other Protease Inhibitors (with or without ritonavir) | Effect is unknown | There are no data available to make dosing recommendations for coadministration with protease inhibitors other than atazanavir, lopinavir/ritonavir, darunavir, fosamprenavir, and tipranavir. |
| Antiretroviral Agents: Nucleoside Reverse Transcriptase Inhibitors (NRTIs) | ||
| Didanosine § | ↔ didanosine
↔ elvitegravir | As didanosine is administered on an empty stomach, administer didanosine at least 1 hour before or 2 hours after VITEKTA (which is administered with food). |
| Antiretroviral Agents: Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | ||
| Efavirenz | ↓ elvitegravir | Coadministration of VITEKTA with efavirenz is not recommended. |
| Nevirapine | ↓ elvitegravir | Coadministration of VITEKTA with nevirapine is not recommended. |
| Other Agents: | ||
| Acid Reducing Agents:
antacids § | ↓ elvitegravir | Separate VITEKTA and antacid administration by at least 2 hours. |
| Anticonvulsants:
carbamazepine oxcarbazepine phenobarbital phenytoin | ↓ elvitegravir | Coadministration of VITEKTA with phenobarbital, phenytoin, carbamazepine, or oxcarbazepine is not recommended. |
| Antifungals:
ketoconazole § | ↑ elvitegravir
↑ ketoconazole | No dose adjustment of VITEKTA is required when coadministered with ketoconazole.
When ketoconazole is used concomitantly with VITEKTA in combination with protease inhibitors/ritonavir; the maximum daily dose of ketoconazole should not exceed 200 mg per day. Consult the prescribing information of coadministered protease inhibitors for any additional dosing recommendation for ketoconazole. |
| Antimycobacterials:
rifampin rifapentine | ↓ elvitegravir | Coadministration of VITEKTA with rifampin or rifapentine is not recommended. |
| rifabutin § | ↑ rifabutin
↑ 25- O-desacetylrifabutin ↓ elvitegravir | When rifabutin is used concomitantly with VITEKTA in combination with a protease inhibitor/ritonavir, dose reduction of rifabutin by at least 75% of the usual dose of 300 mg/day (eg, 150 mg every other day or 3 times per week) is recommended. Increased monitoring for rifabutin-associated adverse events is warranted. Consult the prescribing information of coadministered protease inhibitors for any additional dosing recommendation for rifabutin.
No dose adjustment of VITEKTA is required when coadministered with the reduced dose of rifabutin. |
| Systemic Corticosteroids:
dexamethasone | ↓ elvitegravir | Alternative corticosteroids should be considered. |
| Endothelin Receptor Antagonists:
bosentan | ↑ bosentan
↓ elvitegravir | Coadministration of bosentan in patients on VITEKTA:
In patients who have been receiving VITEKTA for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Coadministration of VITEKTA in patients on bosentan: Discontinue use of bosentan at least 36 hours prior to initiation of VITEKTA. After at least 10 days following the initiation of VITEKTA, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability. |
| HCV Protease Inhibitors:
boceprevir | ↓ boceprevir
↑ or ↓ HIV protease inhibitors | Coadministration of VITEKTA with boceprevir is not recommended. |
| Herbal Products:
St. John's wort (Hypericum perforatum) | ↓ elvitegravir | Coadministration of VITEKTA with St. John's wort is not recommended. |
| Hormonal Contraceptives:
norgestimate/ethinyl estradiol | ↑ norgestimate
↓ ethinyl estradiol ↔ elvitegravir | Alternative methods of non-hormonal contraception are recommended. |
| Narcotic Analgesics:
buprenorphine/ naloxone * methadone | ↑ buprenorphine
↑ norbuprenorphine ↓ naloxone ↓ methadone | No dose adjustment of buprenorphine/naloxone is required upon coadministration with VITEKTA. Patients should be closely monitored for sedation and cognitive effects.
Dosage of methadone may need to be increased when coadministered with VITEKTA |
7.3 Drugs without Clinically Significant Interactions with Elvitegravir
Based on drug interaction studies conducted with elvitegravir, no clinically significant drug interactions have been either observed or expected when elvitegravir is combined with the following drugs: abacavir, darunavir, emtricitabine, etravirine, fosamprenavir, maraviroc, stavudine, tipranavir, tenofovir disoproxil fumarate, zidovudine; H 2-receptor antagonists such as famotidine; proton-pump inhibitors such as omeprazole; and the HMG-CoA reductase inhibitors atorvastatin, pravastatin, and rosuvastatin.
When any of the above drugs are used concomitantly with VITEKTA in combination with a protease inhibitor coadministered with ritonavir, consult the prescribing information of the protease inhibitor for dosing recommendation for these drugs.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category B
There are no adequate and well-controlled studies of VITEKTA in pregnant women. Because animal reproduction studies are not always predictive of human response, VITEKTA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Antiretroviral Pregnancy Registry
To monitor fetal outcomes of pregnant women exposed to VITEKTA, an Antiretroviral Pregnancy Registry has been established. Healthcare providers are encouraged to register patients by calling 1-800-258-4263.
Animal Data
Elvitegravir studies in animals have shown no evidence of teratogenicity or an effect on reproductive function. In offspring from rat and rabbit dams treated with VITEKTA during pregnancy, there were no toxicologically significant effects on developmental endpoints. The exposures (AUC) at the embryo-fetal No Observed Adverse Effects Levels (NOAELs) in rats and rabbits were respectively 23 and 0.2 times higher than the exposure in humans at the recommended daily dose of 150 mg.
8.3 Nursing Mothers
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breastfeed their infants, to avoid risking postnatal transmission of HIV. Studies in rats have demonstrated that elvitegravir is secreted in milk. It is not known whether elvitegravir is excreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breastfeed if they are receiving VITEKTA.
8.4 Pediatric Use
Safety and efficacy in pediatric patients have not been established.
Adolescents (12 through 17 Years Old)
Study 152 was an open-label, multicenter trial of VITEKTA in HIV-1 infected, antiretroviral treatment-experienced adolescent subjects 12 through 17 years of age. The trial included a 10-day pharmacokinetic evaluation phase of VITEKTA followed by an optional extended treatment phase. Dosage regimens were similar to those evaluated in adults, either VITEKTA 150 mg plus darunavir/ritonavir, fosamprenavir/ritonavir, or tipranavir/ritonavir (n=11) or VITEKTA 85 mg plus lopinavir/ritonavir or atazanavir/ritonavir (n=14).
Twenty-five subjects were enrolled and 23 completed the pharmacokinetic phase [see Clinical Pharmacology (12.3)] . Nine subjects with baseline HIV-1 RNA greater than 1,000 copies/mL who completed the 10-day pharmacokinetic evaluation phase enrolled in the optional 48 week treatment phase. All nine completed treatment through 48 weeks; 2/9 subjects (22%) achieved HIV-1 RNA less than 50 copies/mL at Week 48, and 4/9 (44%) achieved HIV-1 RNA less than 400 copies/mL. During the treatment phase of the trial, 8/9 subjects (89%) were found to have undetectable elvitegravir levels during treatment, suggesting that adherence to the regimen was poor and may have contributed to the low response rate in this trial. Although adolescents achieved acceptable VITEKTA plasma levels in the pharmacokinetic phase, the 48-week treatment phase data were insufficient to establish safety and effectiveness in this age group.
8.5 Geriatric Use
Clinical trials of VITEKTA did not include sufficient numbers of subjects aged 65 and older, to determine whether they respond differently from younger subjects. In general, dose selection for elderly patients should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)] .
8.6 Renal Impairment
No clinically relevant differences in elvitegravir pharmacokinetics were observed between subjects with severe renal impairment and healthy subjects. No dose adjustment of VITEKTA is required for patients with renal impairment [see Clinical Pharmacology (12.3)] .
8.7 Hepatic Impairment
No clinically relevant differences in elvitegravir pharmacokinetics were observed between subjects with moderate hepatic impairment (Child-Pugh Class B) and healthy subjects. No dose adjustment of VITEKTA is required in patients with mild (Child-Pugh Class A) or moderate hepatic impairment. VITEKTA has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, VITEKTA is not recommended for use in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
If overdose occurs the patient must be monitored for evidence of toxicity. Treatment of overdose with VITEKTA consists of general supportive measures including monitoring of vital signs, as well as observation of the clinical status of the patient.
Limited clinical experience is available at doses higher than the therapeutic dose of elvitegravir. The effects of higher doses are not known. As elvitegravir is highly bound to plasma proteins, it is unlikely that it will be significantly removed by hemodialysis or peritoneal dialysis.
11 DESCRIPTION
VITEKTA (elvitegravir) is a human immunodeficiency virus-1 (HIV-1) integrase strand transfer inhibitor.
The chemical name of elvitegravir is 6-(3-Chloro-2-fluorobenzyl)-1-[(2 S)-1-hydroxy-3-methylbutan-2-yl]-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. It has a molecular formula of C 23H 23ClFNO 5 and a molecular weight of 447.9. It has the following structural formula:
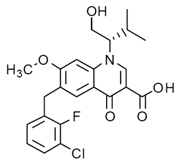
Elvitegravir is a white-to-pale yellow solid with a solubility of less than 0.3 mcg/mL in water at 20 °C.
VITEKTA tablets are for oral administration and contain 85 mg or 150 mg of elvitegravir. The 85 mg and the 150 mg tablets each include the following inactive ingredients: microcrystalline cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate. The tablets are film-coated with a coating material containing polyvinyl alcohol, polyethylene glycol, titanium dioxide, talc, indigo carmine, FD&C Blue #2 aluminum lake, and iron oxide yellow.
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of multiple doses of elvitegravir 125 mg (1.5 times the lowest recommended dosage) and 250 mg (1.7 times the maximum recommended dosage) (coadministered with 100 mg ritonavir) on QT interval was evaluated in a randomized, placebo- and active-controlled (moxifloxacin 400 mg) parallel group thorough QT study in 126 healthy subjects. No clinically meaningful changes in QTc interval were observed with either 125 mg dose or the 250 mg dose. The dose of 250 mg elvitegravir (with 100 mg ritonavir) is expected to cover the high exposure clinical scenario.
12.3 Pharmacokinetics
Absorption
Following oral administration of VITEKTA and ritonavir with food, in HIV-1 infected subjects, peak elvitegravir plasma concentrations were observed approximately 4 hours post-dose. The steady-state mean elvitegravir pharmacokinetic parameters are presented in Table 5. Elvitegravir plasma exposures increased in a less than dose proportional manner, likely due to solubility-limited absorption.
| Parameter Mean ± SD | Elvitegravir 85 mg | Elvitegravir 150 mg |
|---|---|---|
| SD = Standard Deviation | ||
|
||
| C max (mcg/mL) | 1.2 ± 0.36 | 1.5 ± 0.37 |
| AUC tau (mcg∙hr/mL) | 18 ± 7.1 | 18 ± 6.5 |
| C trough (mcg/mL) | 0.42 ± 0.24 | 0.35 ± 0.20 |
| Inhibitory Quotient * | ~9.5 | ~7.9 |
VITEKTA must be taken with food.
Distribution
Elvitegravir is 98–99% bound to human plasma proteins and the binding is independent of drug concentration over the range of 1 ng/mL to 1.6 µg/mL. The mean plasma-to-blood drug concentration ratio is 1.37.
Metabolism and Elimination
Elvitegravir undergoes primarily oxidative metabolism via CYP3A, and is secondarily glucuronidated via UGT1A1/3 enzymes. Following oral administration of [ 14C]elvitegravir/ritonavir, elvitegravir was the predominant species in plasma, representing ~94% of the circulating radioactivity. Aromatic and aliphatic hydroxylation or glucuronidation metabolites were present in very low levels, displayed considerably lower anti-HIV activity and did not contribute to the overall antiviral activity of elvitegravir.
Following oral administration of [ 14C]elvitegravir/ritonavir, 94.8% of the dose was recovered in feces, consistent with the hepatobiliary excretion of elvitegravir; 6.7% of the administered dose was recovered in urine as metabolites. The median terminal plasma half-life of elvitegravir following administration of VITEKTA and ritonavir was approximately 8.7 hours.
Specific Populations
Race
Population pharmacokinetics analysis of elvitegravir in HIV-1 infected subjects indicated that race had no clinically relevant effect on the exposure of elvitegravir/ritonavir.
Gender
No clinically relevant pharmacokinetic differences have been observed between men and women for elvitegravir/ritonavir.
Pediatric Patients
The pharmacokinetics of elvitegravir in pediatric patients less than 12 years of age have not been established [see Use in Specific Populations (8.4)].
Exposures of elvitegravir in adolescents were comparable to those in adults. The steady-state mean elvitegravir C max, AUC tau, and C trough (mean ± SD) following multiple doses of boosted VITEKTA in HIV-1 infected pediatric subjects 12 to less than 18 years were 2.1 ± 0.96 mcg/mL, 25 ± 11 mcg∙hr/mL, and 0.63 ± 0.43 mcg/mL, respectively, for the 85 mg dose, and 2.1 ± 0.74 mcg/mL, 21 ± 7.6 mcg∙hr/mL, and 0.32 ± 0.24 mcg/mL, respectively, for the 150 mg dose of elvitegravir, with inhibitory quotients of ~14 and ~7.1 (ratio of C trough: protein binding-adjusted EC 95 for wild-type HIV-1 virus for the 85 mg and 150 mg doses, respectively).
Geriatric Patients
Pharmacokinetics of elvitegravir have not been fully evaluated in the elderly (65 years of age and older) [see Use in Specific Populations (8.5)].
Patients with Renal Impairment
No clinically relevant differences in elvitegravir pharmacokinetics were observed between subjects with severe renal impairment (estimated creatinine clearance below 30 mL/min) and healthy subjects in a clinical trial [see Use in Specific Populations (8.6)] .
Patients with Hepatic Impairment
Elvitegravir is primarily metabolized and eliminated by the liver. No clinically relevant differences in elvitegravir pharmacokinetics were observed between subjects with moderate hepatic impairment (Child-Pugh Class B) and healthy subjects in a clinical trial. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of elvitegravir has not been studied [see Use in Specific Populations (8.7)] .
Drug Interaction Studies
The drug interaction studies described were conducted with VITEKTA coadministered with ritonavir.
Elvitegravir is primarily metabolized by cytochrome CYP3A. Coadministration of VITEKTA with drugs that induce CYP3A may result in decreased plasma concentrations of elvitegravir.
In drug interaction studies conducted with elvitegravir coadministered with ritonavir or cobicistat, there was no clinically significant interaction observed between elvitegravir and abacavir, emtricitabine, etravirine, famotidine, omeprazole, stavudine, tenofovir disoproxil fumarate, or zidovudine. The effects of coadministered drugs on the exposure of elvitegravir/ritonavir are shown in Table 6. The effects of elvitegravir and ritonavir on the exposure of coadministered drugs are shown in Table 7.
| Coadministered Drug | Dose of Coadministered Drug (mg) | Elvitegravir Dose (mg) | Ritonavir Dose (mg) | N | Mean Ratio of Elvitegravir/Ritonavir Pharmacokinetic Parameters † (90% CI); No Effect = 1.00 | ||
|---|---|---|---|---|---|---|---|
| C max | AUC | C min | |||||
|
|||||||
| Antacids | 20 mL single dose given 4 hours before elvitegravir | 50 once daily | 100 once daily | 8 | 0.95
(0.84, 1.07) | 0.96
(0.88, 1.04) | 1.04
(0.93, 1.17) |
| 20 mL single dose given 4 hours after elvitegravir | 10 | 0.98
(0.88, 1.10) | 0.98
(0.91, 1.06) | 1.00
(0.90, 1.11) |
|||
| 20 mL single dose given 2 hours before elvitegravir | 11 | 0.82
(0.74, 0.91) | 0.85
(0.79, 0.91) | 0.90
(0.82, 0.99) |
|||
| 20 mL single dose given 2 hours after elvitegravir | 10 | 0.79
(0.71, 0.88) | 0.80
(0.75, 0.86) | 0.80
(0.73, 0.89) |
|||
| Atazanavir | 300 once daily | 200 once daily | 100 once daily | 33 | 1.85
(1.69, 2.03) | 2.00
(1.85, 2.16) | 2.88
(2.53, 3.27) |
| 300 once daily | 85 once daily | 100 once daily | 20 | 0.91
(0.81, 1.02) ‡ | 1.07
(0.95, 1.21) ‡ | 1.38
(1.18, 1.61) ‡ |
|
| Carbamazepine | 200 twice daily | 150 daily | 150 once daily § | 12 | 0.55
(0.49,0.61) | 0.31
(0.28,0.33) | 0.03
(0.02,0.04) |
| Darunavir | 600 twice daily | 125 once daily | 100 twice daily | 21 | 1.13
(1.03, 1.24) | 1.10
(0.99, 1.22) | 1.18
(1.06, 1.31) |
| Didanosine | 400 single dose | 200 once daily | 100 once daily | 32 | 0.95
(0.90, 1.01) | 0.97
(0.92, 1.01) | 1.06
(1.01, 1.12) |
| Ketoconazole | 200 twice daily | 150 once daily | 100 once daily | 18 | 1.17
(1.04, 1.33) | 1.48
(1.36, 1.62) | 1.67
(1.48, 1.88) |
| Lopinavir/ritonavir | 400 twice daily | 125 once daily | 100 twice daily | 14 | 1.52
(1.29, 1.79) | 1.75
(1.50, 2.05) | 2.38
(1.81, 3.13) |
| Maraviroc | 150 twice daily | 150 once daily | 100 once daily | 17 | 1.01
(0.89, 1.15) | 1.07
(0.96, 1.18) | 1.09
(0.95, 1.26) |
| Rifabutin | 150 once every other day | 300 once daily | 100 once daily | 19 | 0.92
(0.84, 1.00) | 0.96
(0.90, 1.02) | 0.94
(0.82, 1.09) |
| Rosuvastatin | 10 single dose | 150 single dose | NA §,¶ | 10 | 0.94
(0.83, 1.07) # | 1.02
(0.91, 1.14) # | 0.98
(0.83, 1.16) # |
| Tipranavir | 500 twice daily | 200 once daily | 200 twice daily | 26 | 1.06
(0.89, 1.26) # | 0.92
(0.79, 1.08) # | 0.90
(0.70, 1.17) # |
| Coadministered Drug | Dose of Coadministered Drug (mg) | Elvitegravir Dose (mg) | Ritonavir Dose (mg) | N | Mean Ratio of Coadministered Drug Pharmacokinetic Parameters † (90% CI); No Effect = 1.00 | ||
|---|---|---|---|---|---|---|---|
| C max | AUC | C min | |||||
|
|||||||
| Atazanavir | 300 once daily | 200 once daily | 100 once daily | 33 | 0.84
(0.78, 0.91) | 0.79
(0.74, 0.85) | 0.65
(0.59, 0.73) |
| 300 once daily | 85 once daily | 100 once daily | 20 | 0.97
(0.87, 1.08) | 0.89
(0.80, 0.99) | 0.83
(0.72, 0.95) |
|
| Buprenorphine | 16 – 24 once daily | 150 once daily | 150 once daily ¶ | 17 | 1.12
(0.98, 1.27) | 1.35
(1.18, 1.55) | 1.66
(1.43, 1.93) |
| Norburprenorphine | 1.24
(1.03, 1.49) | 1.42
(1.22, 1.67) | 1.57
(1.31, 1.88) |
||||
| Carbamazepine | 200 twice daily | 150 once daily | 150 once daily ¶ | 12 | 1.40
(1.32,1.49) | 1.43
(1.36,1.52) | 1.51
(1.41,1.62) |
| Carbamazepine-10,11-epoxide | 0.73
(0.70,0.78) | 0.65
(0.63,0.66) | 0.59
(0.57,0.61) |
||||
| Darunavir | 600 twice daily | 125 once daily | 100 twice daily | 22 | 0.89
(0.85, 0.94) | 0.89
(0.82, 0.96) | 0.83
(0.74, 0.93) |
| Didanosine | 400 single dose | 200 once daily | 100 once daily | 32 | 0.84
(0.67, 1.05) | 0.86
(0.75, 0.99) | NC ‡ |
| Lopinavir/ritonavir | 400 twice daily | 125 once daily | 100 twice daily | 13 | 0.99
(0.88, 1.12) | 0.97
(0.85, 1.09) | 0.92
(0.79, 1.08) |
| Maraviroc | 150 twice daily | 150 once daily | 100 once daily | 11 | 2.15
(1.71, 2.69) | 2.86
(2.33, 3.51) | 4.23
(3.47, 5.16) |
| Naloxone | 4 – 6 once daily | 150 once daily | 150 once daily ¶ | 17 | 0.72
(0.61, 0.85) | 0.72
(0.59, 0. 87) | NC |
| Rifabutin | 150 once every other day | 300 once daily | 100 once daily | 18 | 0.92
(0.83, 1.02) § | 0.94
(0.86, 1.03) § | 1.16
(1.02, 1.31) § |
| 25- O-desacetyl-rifabutin | 5.40
(4.66, 6.25) § | 9.51
(8.09, 11.18) § | 19.36
(15.85, 23.65) § |
||||
| Rosuvastatin | 10 single dose | 150 single dose | NA ¶,‡ | 10 | 1.89
(1.48, 2.42) | 1.38
(1.14, 1.67) | 1.43
(1.08, 1.89) |
| Tipranavir | 500 twice daily | 200 once daily | 200 twice daily | 26 | 0.92
(0.84, 1.00) | 0.89
(0.80, 0.99) | 0.89
(0.77, 1.02) |
12.4 Microbiology
Mechanism of Action
Elvitegravir is an HIV-1 integrase strand transfer inhibitor (INSTI). Integrase is an HIV-1 encoded enzyme that is required for viral replication. Inhibition of integrase prevents the integration of HIV-1 DNA into host genomic DNA, blocking the formation of the HIV-1 provirus and propagation of the viral infection. Elvitegravir does not inhibit human topoisomerases I or II.
Antiviral Activity in Cell Culture
The antiviral activity of elvitegravir against laboratory and clinical isolates of HIV-1 was assessed in T lymphoblastoid cells, monocyte/macrophage cells, and primary peripheral blood lymphocytes. The 50% effective concentration (EC 50) values ranged from 0.02 to 1.7 nM. Elvitegravir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC 50 values ranged from 0.1 to 1.3 nM) and activity against HIV-2 (EC 50 value of 0.53 nM). The antiviral activity of elvitegravir with antiretroviral drugs in two-drug combination studies was not antagonistic when combined with the INSTI raltegravir, NNRTIs (efavirenz, etravirine, or nevirapine), NRTIs (abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, or zidovudine), PIs (amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, or tipranavir), the fusion inhibitor enfuvirtide, or the CCR5 co-receptor antagonist maraviroc.
Elvitegravir did not show inhibition of replication of HBV or HCV in cell culture.
Resistance
In Cell Culture
HIV-1 isolates with reduced susceptibility to elvitegravir were selected in cell culture. Reduced susceptibility to elvitegravir was associated with the primary integrase substitutions T66A/I, E92G/Q, S147G, and Q148R. Additional integrase substitutions observed in cell culture selection included D10E, S17N, H51Y, F121Y, S153F/Y, E157Q, D232N, R263K, and V281M.
Clinical Studies
Pooled resistance analysis was performed on virus samples from subjects receiving elvitegravir-containing regimens in 6 clinical trials of elvitegravir (as single drug in combination with a regimen containing a protease inhibitor/ritonavir or as the fixed dose combination STRIBILD) who were viremic with HIV-1 RNA greater than 400 copies/mL at the time of efficacy evaluation (up to 192 weeks). Development of substitutions T66A/I/K, E92G/Q, T97A, S147G, Q148H/K/R, and N155H in the HIV-1 integrase protein was primarily associated with resistance to elvitegravir. In addition to these primary elvitegravir resistance-associated substitutions, E92A, F121C/Y, P145S, Q146I/L/R, and N155S were also occasionally observed and were shown to confer reduced susceptibility to elvitegravir. Substitutions at positions E92 and N155 were the most frequently observed (39% and 27% of those evaluated subjects, respectively). In virus isolates harboring the observed primary elvitegravir resistance-associated substitutions, additional substitutions in integrase were detected including H51Y, L68I/V, G70R, V72A/N, I73V, Q95K/R, S119R, E138A/K, G140A/C/S, E157Q, K160N, E170A, S230R, and D232N.
Treatment-Experienced HIV-1-Infected Subjects
By Week 96, evidence of emerging primary elvitegravir resistance-associated substitutions T66A/I, E92G/Q, T97A, S147G, Q148R, or N155H was observed in 23 of the 74 subjects with evaluable genotypic data in Study 145. Post-baseline virus isolates harboring primary elvitegravir resistance-associated substitutions had median decreases in susceptibility to elvitegravir of 8-fold (29 isolates, ranging from 2- to greater than 158-fold) and of 5-fold (26 isolates, ranging from 1- to greater than 58-fold) compared to wild-type reference HIV-1 and to their respective baseline isolates, respectively.
Cross-Resistance
Cross-resistance has been observed among INSTIs. Among the 23 subjects who developed genotypic resistance to elvitegravir with evidence of emerging primary elvitegravir resistance-associated substitutions in Study 145, 12/21 (57%) subjects with evaluable drug susceptibility data had HIV-1 with reduced susceptibility to raltegravir (greater than 1.5-fold, above the biological cutoff for raltegravir).
Elvitegravir-resistant viruses showed varying degrees of cross-resistance in cell culture to raltegravir in the INSTI class depending on the type and number of substitutions in HIV-1 integrase. Of the primary elvitegravir resistance-associated substitutions tested (T66A/I/K, E92G/Q, T97A, S147G, Q148H/K/R, and N155H), all but three (T66I, E92G, and S147G) conferred greater than 1.5-fold reduced susceptibility to raltegravir when introduced individually into a wild-type virus by site-directed mutagenesis. Of the primary raltegravir resistance-associated substitutions tested (Y143C/H/R, Q148H/K/R, and N155H), all but one (Y143H) conferred greater than 2.5-fold reductions in susceptibility to elvitegravir (above the biological cutoff for elvitegravir).
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term carcinogenicity studies of elvitegravir were carried out in mice (104 weeks) and in rats (up to 88 weeks in males and 90 weeks in females). No drug-related increases in tumor incidence were found in mice at doses up to 2000 mg per kg per day alone or in combination with 25 mg per kg per day ritonavir at exposures 3- and 14-fold, respectively, the human systemic exposure at the recommended daily dose of 150 mg. No drug-related increases in tumor incidence were found in rats at doses up to 2000 mg per kg per day at exposures 12- to 27-fold, respectively, in male and female, the human systemic exposure.
Mutagenesis
Elvitegravir was not genotoxic in the reverse mutation bacterial test (Ames test) and the rat micronucleus assay. In an in vitro chromosomal aberration test, elvitegravir was negative with metabolic activation; however, an equivocal response was observed without activation.
Fertility
Elvitegravir did not affect fertility in male and female rats at approximately 16- and 30-fold higher exposures (AUC), respectively, than in humans at the therapeutic 150 mg daily dose.
Fertility was normal in the offspring of rats exposed daily from before birth ( in utero) through sexual maturity at daily exposures (AUC) of approximately 18-fold higher than human exposures at the recommended 150 mg daily dose.
14 CLINICAL STUDIES
14.1 Treatment-Experienced Adults with HIV-1 Infection
The efficacy of VITEKTA in treatment-experienced adult patients with HIV-1 infection is based on the analyses through 96 weeks from one randomized, double-blind, active-controlled trial, Study 145, in treatment-experienced, HIV-1 infected subjects (N=702). In Study 145, subjects were randomized in a 1:1 ratio to receive either VITEKTA (150 mg or 85 mg) once daily or raltegravir 400 mg twice daily, each administered with a background regimen (BR) containing a fully active protease inhibitor coadministered with ritonavir and a second antiretroviral drug. The BR was selected by the investigator based on genotypic/phenotypic resistance testing and prior antiretroviral treatment history.
The mean age of subjects was 45 years (range 19–78); 82% were male, 62% were White, and 34% were Black. The mean baseline plasma HIV-1 RNA was 4.3 log 10 copies/mL (range 1.7–6.6) and 26% of subjects had baseline viral loads greater than 100,000 copies/mL. The mean duration of prior HIV-1 treatment was 9.4 years. The mean baseline CD4+ cell count was 262 cells/mm 3 (range 1–1497), 45% had CD4+ cell counts ≤ 200 cells/mm 3, and 85% had a baseline genotypic sensitivity score ≥ 2.
Virologic outcomes were similar across the treatment arms through 96 weeks as presented in Table 8. The mean increase from baseline in CD4+ cell count at Week 96 was 205 cells/mm 3 in VITEKTA-treated subjects and 198 cells/mm 3 in raltegravir-treated subjects.
| VITEKTA + Protease Inhibitor/Ritonavir + Another Antiretroviral Drug
(N=351) | Raltegravir + Protease Inhibitor/Ritonavir + Another Antiretroviral Drug
(N=351) |
|
|---|---|---|
|
||
| HIV-1 RNA <50 copies/mL † | 52% | 53% |
| HIV-1 RNA ≥50 copies/mL ‡ | 36% | 31% |
| No Virologic Data at Week 96 | 12% | 16% |
| Discontinued Study Drug Due to AE or Death § | 3% | 7% |
| Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA <50 copies/mL ¶ | 8% | 9% |
| Missing Data During Window but on Study Drug | 1% | 1% |
16 HOW SUPPLIED/STORAGE AND HANDLING
VITEKTA tablets are available in bottles containing 30 tablets with a child-resistant closure as follows:
- 85 mg tablets are green, pentagon-shaped, film-coated, debossed with "GSI" on one side and "85" on the other side: NDC 61958-1301-1
- 150 mg tablets are green, triangle-shaped, film-coated, debossed with "GSI" on one side and "150" on the other side: NDC 61958-1302-1
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
A statement to patients and healthcare providers is included on the product's bottle label: ALERT: Find out about medicines that should NOT be taken with VITEKTA. A Patient Package Insert for VITEKTA is available for patient information.
Information for Patients
Advise patients of the following:
- Inform patients that they should remain under the care of a healthcare provider when using VITEKTA.
- Inform patients that VITEKTA is not a cure for HIV infection. Patients must stay on continuous HIV therapy to control HIV infection and decrease HIV-related illnesses. Inform patients that sustained decreases in plasma HIV RNA have been associated with a reduced risk of progression to AIDS and death.
- Advise patients to continue to practice safer sex and to use latex or polyurethane condoms to lower the chance of sexual contact with any body fluids such as semen, vaginal secretions or blood. Advise patients never to re-use or share needles.
- Inform patients that it is important to take VITEKTA on a regular dosing schedule with food and to avoid missing doses.
- Inform patients that VITEKTA must be taken with an HIV protease inhibitor combined with ritonavir in order to achieve adequate drug levels.
- VITEKTA may interact with many drugs; therefore, inform patients of the potential serious drug interactions with VITEKTA, and that some drugs should not be taken with VITEKTA. Advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John's wort [see Warnings and Precautions (5.1)].
- In some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection [see Warnings and Precautions (5.3)].
© 2015 Gilead Sciences, Inc. All rights reserved.
GILEAD, GSI, STRIBILD, TYBOST, and VITEKTA are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks are the property of their respective owners.
Patient Information
VITEKTA
® (vye-TEK-tuh)
(elvitegravir)
tablets
Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with VITEKTA. For more information, see the section " What should I tell my healthcare provider before taking VITEKTA?"
Read this Patient Information before you start taking VITEKTA and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
Also read the Patient Information for ritonavir (NORVIR ®) and the other antiretroviral Human Immunodeficiency Virus-1 (HIV-1) medicines prescribed by your healthcare provider when taking VITEKTA.
What is VITEKTA?
VITEKTA is a type of prescription HIV-1 medicine that is used to treat HIV-1 in adults who are already taking or have taken HIV-1 medicines before. HIV-1 is the virus that causes Acquired Immunodeficiency Syndrome (AIDS).
VITEKTA must be taken with an HIV protease inhibitor medicine along with ritonavir (NORVIR ®). You must also take other antiretroviral medicines prescribed by your healthcare provider.
It is not known if VITEKTA is safe and effective in children.
When used with other HIV-1 medicines to treat HIV-1 infection, VITEKTA may:
- Reduce the amount of HIV-1 in your blood. This is called "viral load".
- Increase the count of CD4+ (T) cells in your blood that help fight off other infections.
Reducing the amount of HIV-1 and increasing the CD4+ (T) cells in your blood may help improve the immune system. This may reduce your risk of death or getting infections that can happen when your immune system is weak (opportunistic infections).
VITEKTA does not cure HIV-1 infections or AIDS. You must stay on continuous HIV-1 therapy to control HIV-1 infection and decrease HIV-related illnesses.
Avoid doing things that can spread HIV-1 infection to others.
- Do not share or re-use needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safer sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Ask your healthcare provider if you have any questions about how to prevent passing HIV-1 to other people.
What should I tell my healthcare provider before taking VITEKTA?
Before you take VITEKTA, tell your healthcare provider if you:
- have liver problems
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if VITEKTA can harm your unborn baby. Tell your healthcare provider if you become pregnant while taking VITEKTA.
Pregnancy Registry. There is a pregnancy registry for women who take antiviral medicines during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk with your healthcare provider about how you can take part in this registry. - are breastfeeding or plan to breastfeed. Do not breastfeed if you take VITEKTA.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- It is not known if VITEKTA can pass to your baby in your breast milk.
- Talk to your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Do not take VITEKTA if you also take:
- cobicistat (TYBOST ®) with a protease inhibitor
- other medicines that contain elvitegravir (STRIBILD ®)
Especially tell your healthcare provider if you take:
- hormone-based contraceptives (birth control pills and patches)
- an antacid medicine. Take antacids at least 2 hours before or after you take VITEKTA.
- medicines used to treat seizures
- any of the following medicines:
- boceprevir (VICTRELIS ®)
- bosentan (TRACLEER ®)
- buprenorphine/naloxone (BUNAVAIL™, SUBOXONE ®, ZUBSOLV ®)
- didanosine (VIDEX ®, VIDEX EC ®)
- medicines that contain dexamethasone
- efavirenz (SUSTIVA ®)
- ketoconazole (EXTINA ®, NIZORAL ®, XOLEGEL ®)
- nevirapine (VIRAMUNE ®)
- rifabutin (MYCOBUTIN ®)
- rifampin (RIFADIN ®, RIFAMATE ®, RIFATER ®, RIMACTANE ®)
- rifapentine (PRIFTIN ®)
- St. John's wort (Hypericum perforatum) or products containing St. John's wort
Ask your healthcare provider or pharmacist if you are not sure if your medicine is one that is listed above. Do not start any new medicines while you are taking VITEKTA without first talking with your healthcare provider or pharmacist.
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take VITEKTA?
- Take VITEKTA exactly as your healthcare provider tells you.
- VITEKTA must be taken with a protease inhibitor medicine which may include one of the following:
- atazanavir (REYATAZ ®) with ritonavir (NORVIR)
- darunavir (PREZISTA ®) with ritonavir (NORVIR)
- fosamprenavir (LEXIVA ®) with ritonavir (NORVIR)
- lopinavir/ritonavir (KALETRA ®)
- tipranavir (APTIVUS ®) with ritonavir (NORVIR)
- Stay under the care of your healthcare provider during treatment with VITEKTA. See your healthcare provider regularly while you take VITEKTA.
- Do not change your dose or stop taking VITEKTA without first talking with your healthcare provider.
- Do not miss a dose of VITEKTA.
- Take VITEKTA 1 time each day.
- Take VITEKTA with food.
- If you take too much VITEKTA, call your healthcare provider or go to the nearest hospital emergency room right away.
- Do not run out of VITEKTA. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to VITEKTA and become harder to treat. When your supply starts to run low, get more from your healthcare provider or pharmacy.
What are the possible side effects of VITEKTA?
VITEKTA may cause serious side effects, including:
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effect of VITEKTA is diarrhea.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of VITEKTA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VITEKTA?
- Store VITEKTA at room temperature below 86 °F (30 °C).
- Keep VITEKTA in its original container.
- Do not use VITEKTA if the seal over the bottle opening is broken or missing.
Keep VITEKTA and all medicines out of the reach of children.
General information about VITEKTA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VITEKTA for a condition for which it was not prescribed. Do not give VITEKTA to other people, even if they have the same symptoms you have. It may harm them.
If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about VITEKTA that is written for health professionals.
For more information, call 1-800-445-3235 or go to www.GILEAD.com.
What are the ingredients in VITEKTA?
Active ingredient: elvitegravir
Inactive ingredients: microcrystalline cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate. The tablets are film-coated and contain polyvinyl alcohol, polyethylene glycol, titanium dioxide, talc, indigo carmine, FD&C Blue #2 aluminum lake, and iron oxide yellow.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured and distributed by:
Gilead Sciences, Inc.
Foster City, CA 94404
© 2015 Gilead Sciences, Inc. All rights reserved.
Revised: July 2015
GILEAD, GSI, STRIBILD, TYBOST, and VITEKTA are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks are the property of their respective owners.
203093-GS-001


| VITEKTA
elvitegravir tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| VITEKTA
elvitegravir tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Gilead Sciences, Inc. (185049848) |