PHENYTOIN SODIUM- phenytoin sodium injection, solution
X-GEN Pharmaceuticals, Inc.
----------
PHENYTOIN SODIUM INJECTION, USP
BOXED WARNING
WARNING: CARDIOVASCULAR RISK ASSOCIATED WITH RAPID INFUSION The rate of intravenous Phenytoin administration should not exceed 50 mg per minute in adults and 1-3 mg/kg/min (or 50 mg per minute, whichever is slower) in pediatric patients because of the risk of severe hypotension and cardiac arrhythmias. Careful cardiac monitoring is needed during and after administering intravenous Phenytoin. Although the risk of cardiovascular toxicity increases with infusion rates above the recommended infusion rate, these events have also been reported at or below the recommended infusion rate. Reduction in rate of administration or discontinuation of dosing may be needed (see WARNINGS and DOSAGE AND ADMINISTRATION).
DESCRIPTION
Phenytoin Sodium Injection, USP is a ready-mixed solution of phenytoin sodium in a vehicle containing 40% propylene glycol and 10% alcohol in water for injection, adjusted to pH 12 with sodium hydroxide.
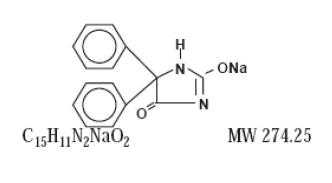
Phentoin sodium is related to the barbiturates in chemical structure, but has a five-membered ring. The chemical name is sodium 5,5-diphenyl-2, 4-imidazolidinedione represented by the following structural formula:
CLINICAL PHARMACOLOGY
Mechanism of Action
Phenytoin is an anticonvulsant which may be useful in the treatment of generalized tonic-clonic status epilepticus. The primary site of action appears to be the motor cortex where spread of seizure activity is inhibited. Possibly by promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of posttetanic potentiation at synapses. Loss of posttetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas. Phenytoin reduces the maximal activity of brain stem centers responsible for the tonic phase of generalized tonic-clonic seizures.
Pharmacokinetics and Drug Metabolism
The plasma half-life in man after intravenous administration ranges from 10 to 15 hours. Optimum control without clinical signs of toxicity occurs most often with serum levels between 10 and 20 mcg/mL.
Phenytoin is metabolized by the cytochrome P450 enzymes CYP2C9 and CYP2C19.
A fall in plasma levels may occur when patients are changed from oral to intramuscular administration. The drop is caused by slower absorption, as compared to oral administration, due to the poor water solubility of phenytoin. Intravenous administration is the preferred route for producing rapid therapeutic serum levels.
When intramuscular administration may be required, a sufficient dose must be administered intramuscularly to maintain the plasma level within the therapeutic range. Where oral dosage is resumed following intramuscular usage, the oral dose should be properly adjusted to compensate for the slow, continuing IM absorption to avoid toxic symptoms.
Patients stabilized on a daily oral regimen of phenytoin experience a drop in peak blood levels to 50‑60 percent of stable levels if crossed over to an equal dose administered intramuscularly. However, the intramuscular depot of poorly soluble material is eventually absorbed, as determined by urinary excretion of 5-(p-hydroxyphenyl)‑5‑ phenylhydantoin (HPPH), the principal metabolite, as well as the total amount of drug eventually appearing in the blood. As phenytoin is highly protein bound, free phenytoin levels may be altered in patients whose protein binding characteristics differ from normal.
A short-term (one week) study indicates that patients do not experience the expected drop in blood levels when crossed over to the intramuscular route if the phenytoin IM dose is increased by 50 percent over the previously established oral dose. To avoid drug cumulation due to absorption from the muscle depots, it is recommended that for the first week back on oral phenytoin, the dose be reduced to half of the original oral dose (one-third of the IM dose). Experience for periods greater than one week is lacking and blood level monitoring is recommended.
Special Populations
Patients with Renal or Hepatic Disease: Due to an increased fraction of unbound phenytoin in patients with renal or hepatic disease, or in those with hypoalbuminemia, the interpretation of total phenytoin plasma concentrations should be made with caution (see DOSAGE AND ADMINISTRATION). Unbound phenytoin concentrations may be more useful in these patient populations.
Age: Phenytoin clearance tends to decrease with increasing age (20% less in patients over 70 years of age relative to that in patients 20-30 years of age). Phenytoin dosing requirements are highly variable and must be individualized (see DOSAGE AND ADMINISTRATION).
Gender and Race: Gender and race have no significant impact on phenytoin pharmacokinetics.
Pediatrics: A loading dose of 15-20 mg/kg of Phenytoin Sodium Injection intravenously will usually produce plasma concentrations of phenytoin within the generally accepted therapeutic range (10-20 mcg/mL). The drug should be injected slowly intravenously at a rate not exceeding 1-3 mg/kg/min or 50 mg per minute, whichever is slower.
INDICATIONS AND USAGE
Parenteral Phenytoin Sodium Injection is indicated for the control of generalized tonic-clonic status epilepticus, and prevention and treatment
of seizures occurring during neurosurgery. Parenteral phenytoin should be used only when oral phenytoin administration is not possible.
CONTRAINDICATIONS
Phenytoin is contraindicated in patients with a history of hypersensitivity to phenytoin, its inactive ingredients, or other hydantoins.
Because of its effect on ventricular automaticity, phenytoin is contraindicated in sinus bradycardia, sino-atrial block, second and third degree A-V block, and patients with Adams-Stokes syndrome.
Coadministration of phenytoin is contraindicated with delavirdine due to potential for loss of virologic response and possible resistance to delavirdine or to the class of non-nucleoside reverse transcriptase inhibitors.
WARNINGS
Cardiovascular Risk Associated with Rapid Infusion
Because of the increased risk of adverse cardiovascular reactions associated with rapid administration, intravenous administration should not exceed 50 mg per minute in adults. In pediatric patients, the drug should be administered at a rate not exceeding 1-3 mg/kg/min or 50 mg per minute, whichever is slower.
As non-emergency therapy, Phenytoin Sodium Injection should be administered more slowly as either a loading dose or by intermittent infusion. Because of the risks of cardiac and local toxicity associated with intravenous phenytoin, oral phenytoin should be used whenever possible.
Because adverse cardiovascular reactions have occurred during and after infusions, careful cardiac monitoring is needed during and after the administration of intravenous Phenytoin Sodium Injection. Reduction in rate of administration or discontinuation of dosing may be needed.
Adverse cardiovascular reactions include severe hypotension and cardiac arrhythmias. Cardiac arrhythmias have included bradycardia, heart block, ventricular tachycardia, and ventricular fibrillation which have resulted in asystole, cardiac arrest, and death. Severe complications are most commonly encountered in critically ill patients, elderly patients, and patients with hypotension and severe myocardial insufficiency. However, cardiac events have also been reported in adults and children without underlying cardiac disease or comorbidities and at recommended doses and infusion rates.
Withdrawal Precipitated Seizure, Status Epilepticus
Antiepileptic drugs should not be abruptly discontinued because of the possibility of increased seizure frequency, including status epilepticus. When, in the judgement of the clinician, the need for dosage reduction, discontinuation, or substitution of alternative antiepileptic medication arises, this should be done gradually. However, in the event of an allergic or hypersensitivity reaction, rapid substitution of alternative therapy may be necessary. In this case, alternative therapy should be an antiepileptic drug not belonging to the hydantoin chemical class.
Serious Dermatologic Reactions
Serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN) and Stevens-Johnson syndrome (SJS), have been reported with phenytoin treatment. The onset of symptoms is usually within 28 days, but can occur later. Phenytoin should be discontinued at the first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest SJS/TEN, use of this drug should not be resumed and alternative therapy should be considered. If a rash occurs, the patient should be evaluated for signs and symptoms of Drug Reaction with Eosinophilia and Systemic Symptoms (see DRESS/Multiorgan hypersensitivity below).
Studies in patients of Chinese ancestry have found a strong association between the risk of developing SJS/TEN and the presence of HLA-B*1502, an inherited allelic variant of the HLA B gene, in patients using carbamazepine. Limited evidence suggests that HLA B*1502 may be a risk factor for the development of SJS/TEN in patients of Asian ancestry taking other antiepileptic drugs associated with SJS/TEN, including phenytoin. Consideration should be given to avoiding phenytoin as an alternative for carbamazepine in patients positive for HLA-B*1502.
The use of HLA-B*1502 genotyping has important limitations and must never substitute for appropriate clinical vigilance and patient management. The role of other possible factors in the development of, and morbidity from, SJS/TEN, such as antiepileptic drug (AED) dose, compliance, concomitant medications, comorbidities, and the level of dermatologic monitoring have not been studied.
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan hypersensitivity, has been reported in patients taking antiepileptic drugs, including phenytoin. Some of these events have been fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, and/or lymphadenopathy, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Phenytoin Sodium Injection should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
Hyersensitivity
Phenytoin and other hydantoins are contraindicated in patients who have experienced phenytoin hypersensitivity (see CONTRAINDICATIONS). Additionally, consider alternatives to structurally similar drugs such as carboxamides (e.g., carbamazepine), barbiturates, succinimides, and oxazolidinediones (e.g., trimethadione) in these same patients. Similarly, if there is a history of hypersensitivity reactions to these structurally similar drugs in the patient or immediate family members, consider alternatives to phenytoin.
Hepatic Injury
Cases of acute hepatotoxicity, including infrequent cases of acute hepatic failure, have been reported with phenytoin. These events may be part of the spectrum of DRESS or may occur in isolation. Other common manifestations include jaundice, hepatomegaly, elevated serum transaminase levels, leukocytosis, and eosinophilia. The clinical course of acute phenytoin hepatotoxicity ranges from prompt recovery to fatal outcomes. In these patients with acute hepatotoxicity, phenytoin should be immediately discontinued and not re-administered.
Hematopoietic System
Hematopoietic complications, some fatal, have occasionally been reported in association with administration of phenytoin. These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression. There have been a number of reports suggesting a relationship between phenytoin and the development of lymphadenopathy (local or generalized) including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s disease. Although a cause and effect relationship has not been established, the occurrence of lymphadenopathy indicates the need to differentiate such a condition from other types of lymph node pathology. Lymph node involvement may occur with or without symptoms and signs resembling DRESS. In all cases of lymphadenopathy, follow-up observation for an extended period is indicated and every effort should be made to achieve seizure control using alternative antiepileptic drugs.
Local Toxicity (including Purple Glove Syndrome)
Soft tissue irritation and inflammation has occurred at the site of injection with and without extravasation of intravenous phenytoin. Edema, discoloration and pain distal to the site of injection (described as “purple glove syndrome”) have also been reported following peripheral intravenous phenytoin injection. Soft tissue irritation may vary from slight tenderness to extensive necrosis, and sloughing. The syndrome may not develop for several days after injection. Although resolution of symptoms may be spontaneous, skin necrosis and limb ischemia have occurred and required such interventions as fasciotomies, skin grafting, and, in rare cases, amputation. Because of the risk of local toxicity, intravenous Phenytoin Sodium Injection should be administered directly into a large peripheral or central vein through a large-gauge catheter. Prior to the administration, the patency of the IV catheter should be tested with a flush of sterile saline. Each injection of parenteral Phenytoin Sodium Injection should then be followed by a flush of sterile saline through the same catheter to avoid local venous irritation due to the alkalinity of the solution. Intramuscular Phenytoin Sodium Injection administration may cause pain, necrosis, and abscess formation at the injection site (see DOSAGE AND ADMINISTRATION).
Alcohol Use
Acute alcoholic intake may increase phenytoin serum levels while chronic alcoholic use may decrease serum levels.
Exacerbation of Porphyria
In view of isolated reports associating phenytoin with exacerbation of porphyria, caution should be exercised in using this medication in patients suffering from this disease.
Usage In pregnancy
Clinical Risks to Mother
An increase in seizure frequency may occur during pregnancy because of altered phenytoin pharmacokinetics. Periodic measurement of plasma phenytoin concentrations may be valuable in the management of pregnant women as a guide to appropriate adjustment of dosage (see PRECAUTIONS, Laboratory Tests). However, postpartum restoration of the original dosage will probably be indicated.
Risks to the Fetus
If this drug is used during pregnancy, or if the patient becomes pregnant while taking the drug, the patient should be apprised of the potential harm to the fetus.
Prenatal exposure to phenytoin may increase the risks for congenital malformations and other adverse development outcomes.
Increased frequencies of major malformations (such as profacial clefts and cardiac defects), minor anomalies (dysmorphic facial features, nail and digit hypoplasia), growth abnormalities (including microcephaly), and mental deficiency have been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy. There have also been several reported cases of malignancies, including neuroblastoma, in children whose mothers received phenytoin during pregnancy. The overall incidence of malformations for children of epileptic women treated with antiepileptic drugs (phenytoin and/or others) during pregnancy is about 10%, or two to three fold that in the general population. However, the relative contribution of antiepileptic drugs and other factors associated with epilepsy to this increased risk are uncertain and in most cases it has not been possible to attribute specific developmental abnormalities to particular antiepileptic drugs. Patients should consult with their physicians to weigh the risks and benefits of phenytoin during pregnancy.
Postpartum Period
A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.
Preclinical
Increased resorption and malformation rates have been reported following administration of phenytoin doses of 75 mg/kg or higher(approximately 120% of the maximum human loading dose or higher on a mg/m2 basis) to pregnant rabbits.
PRECAUTIONS
General
The liver is the site of biotransformation. Patients with impaired liver function, elderly patients, or those who are gravely ill may show early toxicity.
A small percentage of individuals who have been treated with phenytoin have been shown to metabolize the drug slowly. Slow metabolism may be due to limited enzyme availability and lack of induction; it appears to be genetically determined.
Hyperglycemia, resulting from the drug"s inhibitory effect on insulin release, has been reported. Phenytoin may also raise the serum glucose level in diabetic patients.
Phenytoin is not indicated for seizures due to hypoglycemic or other metabolic causes. Appropriate diagnostic procedures should be performed as indicated.
Phenytoin is not effective for absence seizures. If tonic-clonic and absence seizures are present, combined drug therapy is needed.
Serum levels of phenytoin sustained above the optimal range may produce confusional states referred to as "delirium", "psychosis", or "encephalopathy", or rarely irreversible cerebellar dysfunction. Accordingly, at the first sign of acute toxicity, plasma levels are recommended. Dose reduction of phenytoin therapy is indicated if plasma levels are excessive; if symptoms persist, termination is recommended. (See Warnings)
Laboratory Tests
Phenytoin serum level determinations may be necessary to achieve optimal dosage adjustments. Phenytoin doses are usually selected to attain therapeutic plasma total phenytoin concentrations of 10 to 20 μg/mL (unbound phenytoin concentrations of 1 to 2 μg/mL).
Drug Interactions
Phenytoin is extensively bound to serum plasma proteins and is prone to competitive displacement. Phenytoin is metabolized by hepatic cytochrome P450 enzymes CYP2C9 and CYP2C19 and is particularly susceptible to inhibitory drug interactions because it is subject to saturable metabolism. Inhibition of metabolism may produce significant increases in circulating phenytoin concentrations and enhance the risk of drug toxicity. Phenytoin is a potent inducer of hepatic drug-metabolizing enzymes. Serum level determinations for phenytoin are especially helpful when possible drug interactions are suspected. The most commonly occurring drug interactions are listed below:
Note: The list is not intended to be inclusive or comprehensive. Individual drug package inserts should be consulted.
Drugs that affect phenytoin concentrations:
• Drugs that may increase phenytoin serum levels, include: acute alcohol intake, amiodarone, anti-epileptic agents (ethosuximide, felbamate, oxcarbazepine, methsuximide, topiramate), azoles (fluconazole, ketoconazole, itraconazole, voriconazole), capecitabine, chloramphenicol, chlordiazepoxide, cimetidine, diazepam, disulfiram, estrogens, fluorouracil, fluoxetine, fluvastatin, fluvoxamine, H2-antagonists (e.g cimetidine), halothane, isoniazid, methylphenidate, omeprazole, phenothiazines, salicylates, sertraline, succinimides, (e.g., sulfamethizole, sulfaphenazole, sulfadiazine, sulfamethoxazole-trimethoprim), sulfonamides, ticlopidine, tolbutamide, trazodone, and warfarin.
• Drugs that may decrease phenytoin levels, include: anticancer drugs usually in combination (e.g., bleomycin, carboplatin, cisplatin, doxorubicin, methotrexate), carbamazepine, chronic alcohol abuse, folic acid, fosamprenavir, nelfinavir, reserpine, ritonavir, St. John"s Wort, and vigabatrin.
• Drugs that may either increase or decrease phenytoin serum levels include: phenobarbital, sodium valproate, and valproic acid. Similarly, the effect of phenytoin on phenobarbital, valproic acid and sodium valproate serum levels is unpredictable.
• The addition or withdrawal of the agents in patients on phenytoin therapy may require an adjustment of the phenytoin dose to achieve optimal clinical outcome.
Drugs affected by phenytoin:
• Drugs that should not be coadministered with phenytoin: Delavirdine (see CONTRAINDICATIONS).
• Drugs whose efficacy is impaired by phenytoin include: azoles (fluconazole, ketoconazole, itraconazole, voriconazole, posaconazole), corticosteroids, doxycycline, estrogens, furosemide, irinotecan, oral contraceptives, paclitaxel, paroxetine, quinidine, rifampin, sertraline, tenisposide, theophylline, and Vitamin D.
• Increased and decreased PT/INR responses have been reported when phenytoin is coadministered with warfarin.
• Phenytoin decreases plasma concentrations of certain HIV antivirals ( efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir), anti-epileptic agents (felbamate, topiramate, oxcarbazepine, quetiapine), atorvastatin, cyclosporine, digoxin, fluvastatin, folic acid, mexiletine, nisoldipine, praziquantel, and simvastatin.
• Phenytoin when given with fosamprenavir alone may decrease the concentration of amprenavir, the active metabolite. Phenytoin when given with the combination of fosamprenavir and ritonavir may increase the concentration of amprenavir.
• Resistance to the neuromuscular blocking action of the nondepolarizing neuromuscular blocking agents pancuronium, vecuronium, rocuronium, and cisatracurium has occurred in patients chronically administered phenytoin. Whether or not phenytoin has the same effect on other non-depolarizing agents is unknown. Patients should be monitored closely for more rapid recovery from neuromuscular blockade than expected, and infusion rate requirements may be higher.
• The addition or withdrawal of phenytoin during concomitant therapy with these agents may require adjustment of the dose of these agents to achieve optimal clinical outcome.
Drug-Enteral Feeding/Nutritional Preparations Interaction
Literature reports suggest that patients who have received enteral feeding preparations and/or related nutritional supplements have lower than expected phenytoin plasma levels. It is therefore suggested that phenytoin not be administered concomitantly with an enteral feeding preparation. More frequent serum phenytoin level monitoring may be necessary in these patients.
Drug/Laboratory Test Interactions
Phenytoin may decrease serum concentrations of T4. It may also produce lower than normal values for dexamethasone or metyrapone tests. Phenytoin may also cause increased serum levels of glucose, alkaline phosphatase, and gamma glutamyl transpeptidase (GGT). Care should be taken when using immunoanalytical methods to measure plasma phenytoin concentrations following fosphenytoin administration.
ADVERSE REACTIONS
The most notable signs of toxicity associated with the intravenous use of this drug are cardiovascular collapse and/or central nervous system depression. Hypotension does occur when the drug is administered rapidly by the intravenous route. The rate of administration is very important; it should not exceed 50 mg per minute in adults, and 1-3 mg/ kg/ min (or 50 mg per minute, whichever is slower) in pediatric patients.
Body As a Whole
Allergic reactions in the form of rash and rarely more serious forms (see Skin and Appendages paragraph below) and DRESS (see WARNINGS) have been observed. Anaphylaxis has also been reported. There have also been reports of coarsening of facial features, systemic lupus erythematosus, periarteritis nodosa, and immunoglobulin abnormalities.
Cardiovascular
Severe cardiovascular events and fatalities have been reported with atrial and ventricular conduction depression and ventricular fibrillation. Severe complications are most commonly encountered in elderly or critically ill patients (see WARNINGS).
Nervous System
The most common manifestations encountered with phenytoin therapy are referable to this system and are usually dose-related. These include nystagmus, ataxia, slurred speech, decreased coordination, somnolence, and mental confusion. Dizziness, insomnia, transient nervousness, motor twitchings, paresthesia, and headaches have also been observed. There have also been rare reports of phenytoin induced dyskinesias, including chorea, dystonia, tremor and asterixis, similar to those induced by phenothiazine and other neuroleptic drugs.
A predominantly sensory peripheral polyneuropathy has been observed in patients receiving long-term phenytoin therapy.
Digestive System
Nausea, vomiting, constipation, enlargement of the lips, gingival hyperplasia, toxic hepatitis and liver damage.
Skin and Appendages
Dermatological manifestations sometimes accompanied by fever have included scarlatiniform or morbilliform rashes. A morbilliform rash(measles-like) is the most common; other types of dermatitis are seen more rarely. Other more serious forms which may be fatal have included bullous, exfoliative or purpuric dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis (see WARNINGS).
There have also been reports of hypertrichosis. Local irritation, inflammation, tenderness, necrosis, and sloughing have been reported with or without extravasation of intravenous phenytoin (see WARNINGS).
Hematologic and Lymphatic System
Hematopoietic complications, some fatal, have occasionally been reported in association with administration of phenytoin. These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression. While macrocytosis and megaloblastic anemia have occurred, these conditions usually respond to folic acid therapy. Lymphadenopathy, including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s Disease have been reported (see WARNINGS).
OVERDOSAGE
The lethal dose in pediatric patients is not known. The lethal dose in adults is estimated to be 2 to 5 grams. The initial symptoms are nystagmus, ataxia, and dysarthria. Other signs are tremor, hyperreflexia, lethargy, slurred speech, nausea, vomiting. The patient may become comatose and hypotensive. Death is due to respiratory and circulatory depression. There are marked variations among individuals with respect to phenytoin plasma levels where toxicity may occur. Nystagmus, on lateral gaze, usually appears at 20 mcg/mL, ataxia at 30 mcg/mL, dysarthria and lethargy appear when the plasma concentration is over 40 mcg/mL, but as high a concentration as 50 mcg/mL has been reported without evidence of toxicity. As much as 25 times the therapeutic dose has been taken to result in a serum concentration over 100 mcg/mL with complete recovery.
Treatment
Treatment is nonspecific since there is no known antidote.
The adequacy of the respiratory and circulatory systems should be carefully observed and appropriate supportive measures employed. Hemodialysis can be considered since phenytoin is not completely bound to plasma proteins. Total exchange transfusion has been used in the treatment of severe intoxication in pediatric patients.
In acute overdosage the possibility of other CNS depressants, including alcohol, should be borne in mind.
DOSAGE AND ADMINISTRATION
Because of the increased risk of adverse cardiovascular reactions associated with rapid administration, intravenous administration should not exceed 50 mg per minute in adults. In pediatric patients, the drug should be administered at a rate not exceeding 1-3 mg/kg/min or 50 mg per minute, whichever is slower. As non-emergency therapy, Phenytoin Sodium Injection should be administered more slowly as either a loading dose or by intermittent infusion. Because of the risks of cardiac and local toxicity associated with intravenous phenytoin, oral phenytoin should be used whenever possible. Because adverse cardiovascular reactions have occurred during and after infusions, careful cardiac monitoring is needed during and after the administration of intravenous Phenytoin Sodium Injection. Reduction in rate of administration or discontinuation of dosing may be needed.
Because of the risk of local toxicity, intravenous Phenytoin Sodium Injection should be administered directly into a large peripheral or central vein through a large-gauge catheter. Prior to the administration, the patency of the IV catheter should be tested with a flush of sterile saline. Each injection of parenteral Phenytoin Sodium Injection should then be followed by a flush of sterile saline through the same catheter to avoid local venous irritation due to the alkalinity of the solution.
Phenytoin Sodium Injection can be given diluted with normal saline. The addition of parenteral Phenytoin Sodium Injection to dextrose and dextrose-containing solutions should be avoided due to lack of solubility and resultant precipitation.
Treatment with Phenytoin Sodium Injection can be initiated either with a loading dose or an infusion:
Loading Dose: A loading dose of parenteral Phenytoin Sodium Injection should be injected slowly, not exceeding 50 mg per minute in adults and 1-3 mg/kg/min (or 50 mg per minute, whichever is slower) in pediatric patients.
Infusion: For infusion administration, parenteral Phenytoin Sodium Injection should be diluted in normal saline with the final concentration of phenytoin sodium in the solution no less than 5 mg/mL. Administration should commence immediately after the mixture has been prepared and must be completed within 1 to 4 hours (the infusion mixture should not be refrigerated). An in-line filter (0.22-0.55 microns) should be used.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution or container permit.
The diluted infusion mixture (Phenytoin Sodium Injection plus normal saline) should not be refrigerated. If the undiluted parenteral Phenytoin Sodium Injection is refrigerated or frozen, a precipitate might form: this will dissolve again after the solution is allowed to stand at room temperature. The product is still suitable for use. A faint yellow coloration may develop, however this has no effect on the potency of the solution.
Status Epilepticus
In adults, a loading dose of 10 to 15 mg/kg should be administered slowly intravenously, at a rate not exceeding 50 mg per minute (this will require approximately 20 minutes in a 70-kg patient).
The loading dose should be followed by maintenance doses of 100 mg orally or intravenously every 6-8 hours. In the pediatric population, a loading dose of 15-20 mg/kg of phenytoin sodium intravenously will usually produce plasma concentrations of phenytoin within the generally accepted therapeutic range (10-20 mcg/mL). The drug should be injected slowly intravenously at a rate not exceeding 1-3 mg/kg/min or 50mg per minute, whichever is slower.
Continuous monitoring of the electrocardiogram and blood pressure is essential. The patient should be observed for signs of respiratory depression.
Determination of phenytoin plasma levels is advised when using phenytoin in the management of status epilepticus and in the subsequent establishment of maintenance dosage.
Other measures, including concomitant administration of an intravenous benzodiazepine such as diazepam, or an intravenous short-acting barbiturate, will usually be necessary for rapid control of seizures because of the required slow rate of administration of phenytoin.
If administration of parenteral Phenytoin Sodium Injection does not terminate seizures, the use of other anticonvulsants, intravenous barbiturates, general anesthesia, and other appropriate measures should be considered. Intramuscular administration should not be used in the treatment of status epilepticus because the attainment of peak plasma levels may require up to 24 hours.
Nonemergent Loading and Maintenance Dosing
Because of the risks of cardiac and local toxicity associated with intravenous phenytoin, oral phenytoin should be used whenever possible. In adults, a loading dose of 10 to 15 mg/kg should be administered slowly. The rate of intravenous administration should not exceed 50mg per minute in adults and 1-3 mg/kg/min (or 50 mg per minute, whichever is slower) in pediatric patients. Slower administration rates are recommended to minimize the cardiovascular adverse reactions. Continuous monitoring of the electrocardiogram, blood pressure, and respiratory function is essential.
The loading dose should be followed by maintenance doses of oral or intravenous phenytoin every 6-8 hours. Ordinarily, Phenytoin Sodium Injection should not be given intramuscularly because of the risk of necrosis, abscess formation, and erratic absorption. If intramuscular administration is required, compensating dosage adjustments are necessary to maintain therapeutic plasma levels. An intramuscular dose 50% greater than the oral dose is necessary to maintain these levels. When returned to oral administration, the dose should be reduced by 50% of the original oral dose for one week to prevent excessive plasma levels due to sustained release from intramuscular tissue sites.
Monitoring plasma levels would help prevent a fall into the subtherapeutic range. Serum blood level determinations are especially helpful when possible drug interactions are suspected.
IV Substitution For Oral Phenytoin Therapy
When treatment with oral phenytoin is not possible, IV phenytoin can be substituted for oral phenytoin at the same total daily dose.
Phenytoin capsules are approximately 90% bioavailable by the oral route. Phenytoin is 100% bioavailable by the IV route. For this reason, plasma phenytoin concentrations may increase modestly when IV phenytoin is substituted for oral phenytoin sodium therapy.
The rate of administration for IV phenytoin should be no greater than 50 mg per minute in adults and 1-3 mg/kg/min (or 50 mg per minute, whichever is slower) in pediatric patients.
Serum concentrations should be monitored and care should be taken when switching a patient from the sodium salt to the free acid form. Phenytoin Sodium Injection is formulated with the sodium salt of phenytoin. Because there is approximately an 8% increase in drug content with the free acid form over that of the sodium salt, dosage adjustments and serum level monitoring may be necessary when switching from a product formulated with the free acid to a product formulated with the sodium salt and vice versa.
Dosing in Special Populations
Patients with Renal or Hepatic Disease: Due to an increased fraction of unbound phenytoin in patients with renal or hepatic disease, or in those with hypoalbuminemia, the interpretation of total phenytoin plasma concentrations should be made with caution. Unbound phenytoin concentrations may be more useful in these patient populations.
Elderly Patients: Phenytoin clearance is decreased slightly in elderly patients and lower or less frequent dosing may be required.
Pediatric: A loading dose of 15-20 mg/kg of Phenytoin Sodium Injection intravenously will usually produce plasma concentrations of phenytoin within the generally accepted therapeutic range (10-20 mcg/mL). The drug should be injected slowly intravenously at a rate not exceeding 1-3mg/kg/min or 50 mg per minute, whichever is slower.
HOW SUPPLIED
Phenytoin Sodium Injection, USP - 100 mg/2 mL (50 mg/mL) – in 2 mL vials (NDC 39822-4050-2) packaged in cartons of 25 (NDC 39822-4050-3)
Phenytoin Sodium Injection, USP - 250 mg/5 mL (50 mg/mL) – in 10 mL vials (NDC 39822-4050-5) packaged in cartons of 25 (NDC 39822-4050-6)
STORAGE
Store at 20° to 25°C (68° to 77°F), excursions permitted to 15° to 30°C (59° to 86°F) [See USP Controlled Room Temperature].
Manufactured by:
HIKMA FARMACÊUTICA (PORTUGAL), S.A.
Estrada do Rio da Mó, 8, 8A e 8B – Fervença – 2705-906 Terrugem SNT, PORTUGAL
Distributed by:
X-GEN Pharmaceuticals
Big Flats, NY 14814
Revised: 05/2013
PY-PI-03
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Phenytoin Sodium Injection, USP 100 mg/2 mL (50 mg/mL) in 2mL vial NDC 39822-4050-2
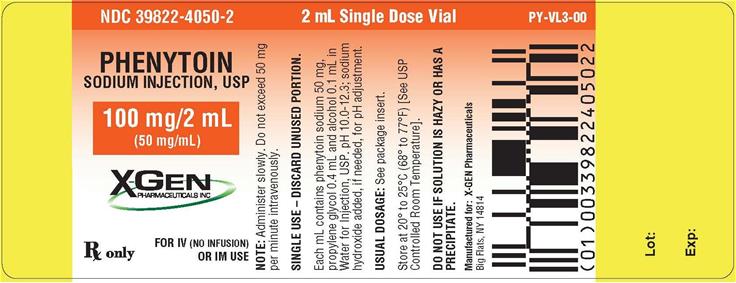
2 ml label
Phenytoin Sodium Injection, USP 100 mg/2 mL (50 mg/mL) in carton of 25 vials NDC 39822-4050-3

2 mL carton
Phenytoin Sodium Injection, USP 250 mg/5 mL (50 mg/mL) in 10mL vial NDC 39822-4050-5

5 ml vial
Phenytoin Sodium Injection, USP 250 mg/5 mL (50 mg/mL) in carton of 25 vials NDC 39822-4050-6

5 mL carton
| PHENYTOIN SODIUM
phenytoin sodium injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - X-GEN Pharmaceuticals, Inc. (790169531) |