MAKENA- hydroxyprogesterone caproate injection
AMAG Pharmaceuticals, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use MAKENA safely and effectively. See full prescribing information for MAKENA.
MAKENA® (hydroxyprogesterone caproate injection) for intramuscular or subcutaneous use Initial U.S. Approval: 1956 RECENT MAJOR CHANGESINDICATIONS AND USAGEMakena is a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth (1). The effectiveness of Makena is based on improvement in the proportion of women who delivered < 37 weeks of gestation (14). There are no controlled trials demonstrating a direct clinical benefit, such as improvement in neonatal mortality and morbidity. Limitation of use: Makena is not intended for use in women with multiple gestations or other risk factors for preterm birth. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS1.1 mL single-use auto-injector for subcutaneous use contains 275 mg of hydroxyprogesterone caproate (250 mg/mL) (3) 1 mL single-dose vial for intramuscular use contains 250 mg of hydroxyprogesterone caproate. (3) 5 mL multi-dose vial for intramuscular use contains 1250 mg of hydroxyprogesterone caproate (250 mg/mL). (3) CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact AMAG Pharmaceuticals at 1-877-411-2510 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 12/2022 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Makena is a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. The effectiveness of Makena is based on improvement in the proportion of women who delivered < 37 weeks of gestation. There are no controlled trials demonstrating a direct clinical benefit, such as improvement in neonatal mortality and morbidity.
Limitation of use: While there are many risk factors for preterm birth, safety and efficacy of Makena has been demonstrated only in women with a prior spontaneous singleton preterm birth. It is not intended for use in women with multiple gestations or other risk factors for preterm birth.
2 DOSAGE AND ADMINISTRATION
2.1 Dosing
- •
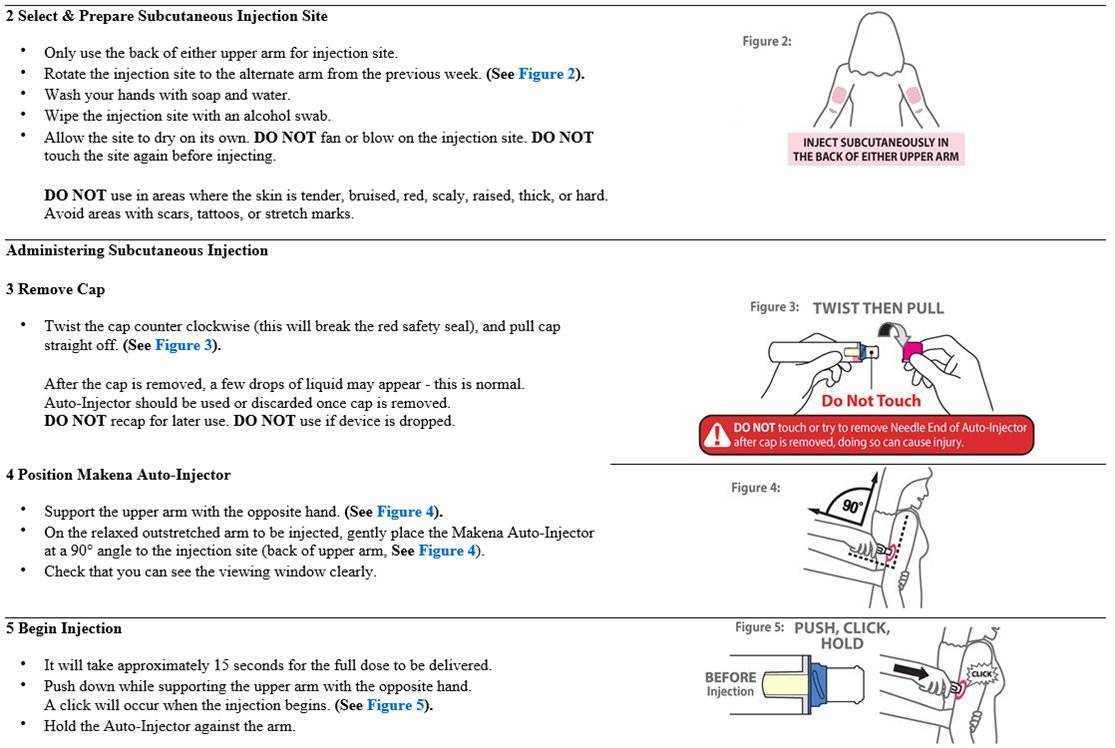
- Makena auto-injector: Administer subcutaneously using auto-injector at a dose of 275 mg (1.1 mL) once weekly (every 7 days) in the back of either upper arm by a healthcare provider
- •
- Makena (single- and multi-dose vials): Administer intramuscularly at a dose of 250 mg (1 mL) once weekly (every 7 days) in the upper outer quadrant of the gluteus maximus by a healthcare provider
- •
- Begin treatment between 16 weeks, 0 days and 20 weeks, 6 days of gestation
- •
- Continue administration once weekly until week 37 (through 36 weeks, 6 days) of gestation or delivery, whichever occurs first
2.2 Preparation and Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Makena is a clear, yellow solution. The solution must be clear at the time of use; replace vial if visible particles or crystals are present.
Specific instructions for administration by dosage form:
Makena single-dose or multi-dose vials (intramuscular use only)
Makena single-dose or multi-dose vials are only for intramuscular injection with a syringe into the upper outer quadrant of the gluteus maximus, rotating the injection site to the alternate side from the previous week, using the following preparation and administration procedure:
- 1.
- Clean the vial top with an alcohol swab before use.
- 2.
- Draw up 1 mL of drug into a 3 mL syringe with an 18 gauge needle.
- 3.
- Change the needle to a 21 gauge 1½ inch needle.
- 4.
- After preparing the skin, inject in the upper outer quadrant of the gluteus maximus. The solution is viscous and oily. Slow injection (over one minute or longer) is recommended.
- 5.
- Applying pressure to the injection site may minimize bruising and swelling.
If the 5 mL multi-dose vial is used, discard any unused product 5 weeks after first use.
Makena auto-injector (subcutaneous use only)
Makena auto-injector is a single-use, pre-filled, disposable device containing a 27 gauge, 0.5 inch needle that delivers one dose subcutaneously in the back of the upper arm.
Because Makena auto-injector is preservative-free, once the cap is removed the device should be used immediately or discarded.
Rotate the injection site to the alternate arm from the previous week. Do not use in areas where the skin is tender, bruised, red, scaly, raised, thick, or hard. Avoid areas with scars, tattoos, or stretch marks.
The solution is viscous and oily. The auto-injector takes approximately 15 seconds to deliver the dose; when the viewing window is fully blocked (completely orange), the full dose has been administered.
The “Instructions for Use” contains detailed steps for administering the subcutaneous injection using the auto-injector [see Dosage and Administration (2.3)]. Read the “Instructions for Use” carefully before administering Makena auto-injector.
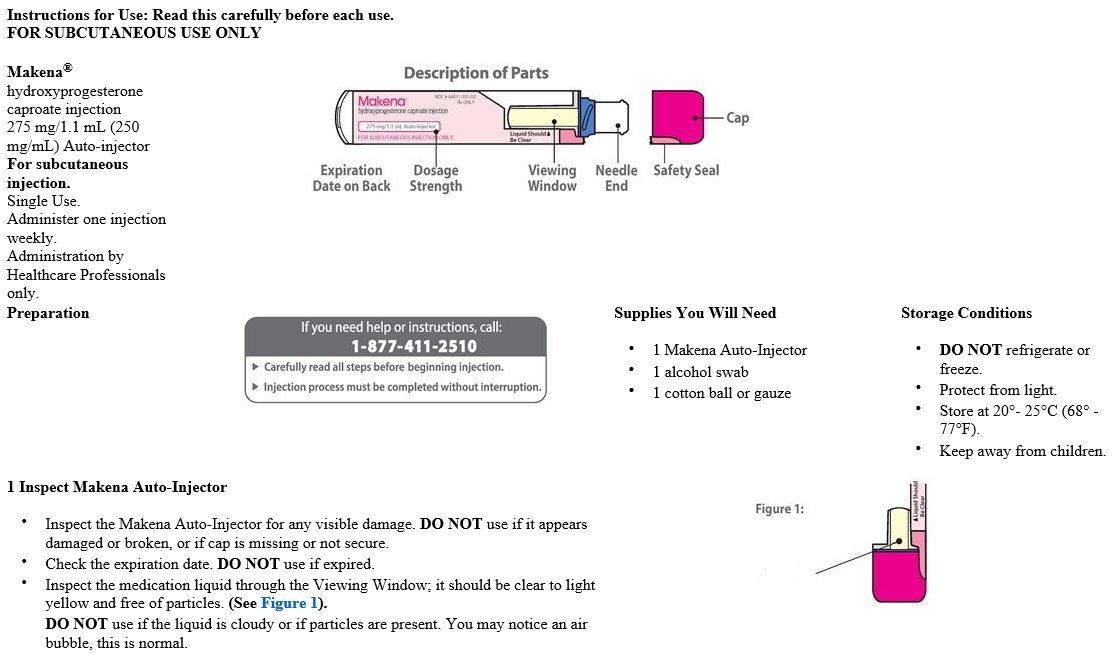

3 DOSAGE FORMS AND STRENGTHS
Subcutaneous injection: 275 mg/1.1 mL clear yellow solution in single-use auto-injector.
Intramuscular injection: 250 mg/mL clear yellow solution in single-dose vials.
Intramuscular injection: 1250 mg/5 mL (250 mg/mL) clear yellow solution in multiple-dose vials.
4 CONTRAINDICATIONS
Do not use Makena in women with any of the following conditions:
- •
- Current or history of thrombosis or thromboembolic disorders
- •
- Known or suspected breast cancer, other hormone-sensitive cancer, or history of these conditions
- •
- Undiagnosed abnormal vaginal bleeding unrelated to pregnancy
- •
- Cholestatic jaundice of pregnancy
- •
- Liver tumors, benign or malignant, or active liver disease
- •
- Uncontrolled hypertension
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Disorders
Discontinue Makena if an arterial or deep venous thrombotic or thromboembolic event occurs.
5.2 Allergic Reactions
Allergic reactions, including urticaria, pruritus and angioedema, have been reported with use of Makena or with other products containing castor oil. Consider discontinuing the drug if such reactions occur.
5.3 Decrease in Glucose Tolerance
A decrease in glucose tolerance has been observed in some patients on progestin treatment. The mechanism of this decrease is not known. Carefully monitor prediabetic and diabetic women while they are receiving Makena.
5.4 Fluid Retention
Because progestational drugs may cause some degree of fluid retention, carefully monitor women with conditions that might be influenced by this effect (e.g., preeclampsia, epilepsy, migraine, asthma, cardiac or renal dysfunction).
5.5 Depression
Monitor women who have a history of clinical depression and discontinue Makena if clinical depression recurs.
6 ADVERSE REACTIONS
For the most serious adverse reactions to the use of progestins, see Warnings and Precautions (5).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a vehicle (placebo)-controlled clinical trial of 463 pregnant women at risk for spontaneous preterm delivery based on obstetrical history, 310 received 250 mg of Makena and 153 received a vehicle formulation containing no drug by a weekly intramuscular injection beginning at 16 to 20 weeks of gestation and continuing until 37 weeks of gestation or delivery, whichever occurred first. [See Clinical Studies (14.1).]
Certain pregnancy-related fetal and maternal complications or events were numerically increased in the Makena-treated subjects as compared to control subjects, including miscarriage and stillbirth, admission for preterm labor, preeclampsia or gestational hypertension, gestational diabetes, and oligohydramnios (Tables 1 and 2).
| 1 N = Total number of subjects enrolled prior to 20 weeks 0 days 2 N = Total number of subjects at risk ≥ 20 weeks |
||
|
Pregnancy Complication |
Makena |
Control |
|
n/N |
n/N |
|
|
Miscarriage (< 20 weeks)1 |
5/209 |
0/107 |
|
Stillbirth (≥ 20 weeks)2 |
6/305 |
2/153 |
| 1 Other than delivery admission. | |||||
|
Pregnancy Complication |
Makena
|
Control
|
|||
|
Admission for preterm labor1 |
16.0 |
13.8 |
|||
|
Preeclampsia or gestational hypertension |
8.8 |
4.6 |
|||
|
Gestational diabetes |
5.6 |
4.6 |
|||
|
Oligohydramnios |
3.6 |
1.3 |
|||
Common Adverse Reactions:
The most common adverse reaction with intramuscular injection was injection site pain, which was reported after at least one injection by 34.8% of the Makena group and 32.7% of the control group. Table 3 lists adverse reactions that occurred in ≥ 2% of subjects and at a higher rate in the Makena group than in the control group.
|
Preferred Term |
Makena
|
Control
|
|
Injection site pain |
34.8 |
32.7 |
|
Injection site swelling |
17.1 |
7.8 |
|
Urticaria |
12.3 |
11.1 |
|
Pruritus |
7.7 |
5.9 |
|
Injection site pruritus |
5.8 |
3.3 |
|
Nausea |
5.8 |
4.6 |
|
Injection site nodule |
4.5 |
2.0 |
|
Diarrhea |
2.3 |
0.7 |
In the clinical trial using intramuscular injection, 2.2% of subjects receiving Makena were reported as discontinuing therapy due to adverse reactions compared to 2.6% of control subjects. The most common adverse reactions that led to discontinuation in both groups were urticaria and injection site pain/swelling (1% each).
Pulmonary embolus in one subject and injection site cellulitis in another subject were reported as serious adverse reactions in Makena-treated subjects.
Two clinical studies were conducted in healthy post-menopausal women, comparing Makena administered via subcutaneous auto-injector to Makena administered as an intramuscular injection. In the first study, injection site pain occurred in 3/30 (10%) of subjects who used the subcutaneous auto-injector vs. 2/30 (7%) of subjects receiving intramuscular injection. In the second study, injection site pain occurred in 20/59 (34%) of subjects who used the subcutaneous auto-injector vs. 5/61 (8%) of subjects receiving intramuscular injection.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Makena. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Body as a whole: Local injection site reactions (including erythema, urticaria, rash, irritation, hypersensitivity, warmth); fatigue; fever; hot flashes/flushes
- •
- Digestive disorders: Vomiting
- •
- Infections: Urinary tract infection
- •
- Nervous system disorders: Headache, dizziness
- •
- Pregnancy, puerperium and perinatal conditions: Cervical incompetence, premature rupture of membranes
- •
- Reproductive system and breast disorders: Cervical dilation, shortened cervix
- •
- Respiratory disorders: Dyspnea, chest discomfort
- •
- Skin: Rash
7 DRUG INTERACTIONS
In vitro drug-drug interaction studies were conducted with Makena. Hydroxyprogesterone caproate has minimal potential for CYP1A2, CYP2A6, and CYP2B6 related drug-drug interactions at the clinically relevant concentrations. In vitro data indicated that therapeutic concentration of hydroxyprogesterone caproate is not likely to inhibit the activity of CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 [See Clinical Pharmacology (12.3).] No in vivo drug-drug interaction studies were conducted with Makena.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Makena is indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. Fetal, neonatal, and maternal risks are discussed throughout labeling. Data from the placebo-controlled clinical trial and the infant follow-up safety study [see Clinical Studies (14.1, 14.2)] did not show a difference in adverse developmental outcomes between children of Makena-treated women and children of control subjects. However, these data are insufficient to determine a drug-associated risk of adverse developmental outcomes as none of the Makena-treated women received the drug during the first trimester of pregnancy. In animal reproduction studies, intramuscular administration of hydroxyprogesterone caproate to pregnant rats during gestation at doses 5 times the human dose equivalent based on a 60-kg human was not associated with adverse developmental outcomes.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Reproduction studies of hydroxyprogesterone caproate administered to various animal species have been reported in the literature. In nonhuman primates, embryolethality was reported in rhesus monkeys administered hydroxyprogesterone caproate up to 2.4 and 24 times the human dose equivalent, but not in cynomolgus monkeys administered hydroxyprogesterone caproate at doses up to 2.4 times the human dose equivalent, every 7 days between days 20 and 146 of gestation. There were no teratogenic effects in either strain of monkey.
Reproduction studies have been performed in mice and rats at doses up to 95 and 5, respectively, times the human dose and have revealed no evidence of impaired fertility or harm to the fetus due to hydroxyprogesterone caproate.
8.2 Lactation
Risk Summary
Low levels of progestins are present in human milk with the use of progestin-containing products, including hydroxyprogesterone caproate. Published studies have reported no adverse effects of progestins on the breastfed child or on milk production.
8.4 Pediatric Use
Makena is not indicated for use in women under 16 years of age. Safety and effectiveness in patients less than 16 years of age have not been established. A small number of women under age 18 years were studied; safety and efficacy are expected to be the same in women aged 16 years and above as for users 18 years and older [See Clinical Studies (14)].
10 OVERDOSAGE
There have been no reports of adverse events associated with overdosage of Makena in clinical trials. In the case of overdosage, the patient should be treated symptomatically.
11 DESCRIPTION
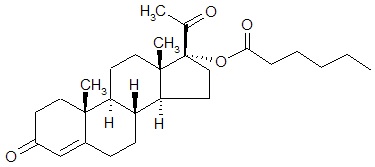
The active pharmaceutical ingredient in Makena is hydroxyprogesterone caproate, a progestin.
The chemical name for hydroxyprogesterone caproate is pregn-4-ene-3,20-dione, 17[(1-oxohexyl)oxy]. It has an empirical formula of C27H40O4 and a molecular weight of 428.60. Hydroxyprogesterone caproate exists as white to practically white crystals or powder with a melting point of 120°-124°C.
The structural formula is:
Makena is a clear, yellow, sterile, non-pyrogenic solution for intramuscular (vials) or subcutaneous (auto-injector) injection. Each 1.1 mL Makena auto-injector for subcutaneous use and each 1 mL single-dose vial for intramuscular use contains hydroxyprogesterone caproate USP, 250 mg/mL (25% w/v), in a preservative-free solution containing castor oil USP (30.6% v/v) and benzyl benzoate USP (46% v/v). Each 5 mL multi-dose vial contains hydroxyprogesterone caproate USP, 250 mg/mL (25% w/v), in castor oil USP (28.6%) and benzyl benzoate USP (46% v/v) with the preservative benzyl alcohol NF (2% v/v).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Hydroxyprogesterone caproate is a synthetic progestin. The mechanism by which hydroxyprogesterone caproate reduces the risk of recurrent preterm birth is not known.
12.3 Pharmacokinetics
Absorption: Female patients with a singleton pregnancy received intramuscular doses of 250 mg hydroxyprogesterone caproate for the reduction of preterm birth starting between 16 weeks 0 days and 20 weeks 6 days. All patients had blood drawn daily for 7 days to evaluate pharmacokinetics.
| Blood was drawn daily for 7 days (1) starting 24 hours after the first dose between Weeks 16-20 (Group 1), (2) after a dose between Weeks 24-28 (Group 2), or (3) after a dose between Weeks 32-36 (Group 3) | |||
| a Reported as median (range) | |||
| b t = 7 days | |||
|
Group (N) |
Cmax (ng/mL) |
Tmax (days)a |
AUC(0-t)b
|
|
Group 1 (N=6) |
5.0 (1.5) |
5.5 (2.0-7.0) |
571.4 (195.2) |
|
Group 2 (N=8) |
12.5 (3.9) |
1.0 (0.9-1.9) |
1269.6 (285.0) |
|
Group 3 (N=11) |
12.3 (4.9) |
2.0 (1.0-3.0) |
1268.0 (511.6) |
For all three groups, peak concentration (Cmax) and area under the curve (AUC(1-7 days)) of the mono-hydroxylated metabolites were approximately 3-8-fold lower than the respective parameters for the parent drug, hydroxyprogesterone caproate. While di-hydroxylated and tri-hydroxylated metabolites were also detected in human plasma to a lesser extent, no meaningful quantitative results could be derived due to the absence of reference standards for these multiple hydroxylated metabolites. The relative activity and significance of these metabolites are not known.
The elimination half-life of hydroxyprogesterone caproate, as evaluated from 4 patients in the study who reached full-term in their pregnancies, was 16.4 (±3.6) days. The elimination half-life of the mono-hydroxylated metabolites was 19.7 (±6.2) days.
In a single-dose, open-label, randomized, parallel design bioavailability study in 120 healthy post-menopausal women, comparable systemic exposure of hydroxyprogesterone caproate was seen when Makena was administered subcutaneously with the auto-injector (1.1 mL) in the back of the upper arm and when Makena was dosed intramuscularly (1 mL) in the upper outer quadrant of the gluteus maximus.
Distribution: Hydroxyprogesterone caproate binds extensively to plasma proteins including albumin and corticosteroid binding globulins.
Metabolism: In vitro studies have shown that hydroxyprogesterone caproate can be metabolized by human hepatocytes, both by phase I and phase II reactions. Hydroxyprogesterone caproate undergoes extensive reduction, hydroxylation and conjugation. The conjugated metabolites include sulfated, glucuronidated and acetylated products. In vitro data indicate that the metabolism of hydroxyprogesterone caproate is predominantly mediated by CYP3A4 and CYP3A5. The in vitro data indicate that the caproate group is retained during metabolism of hydroxyprogesterone caproate.
Excretion: Both conjugated metabolites and free steroids are excreted in the urine and feces, with the conjugated metabolites being prominent. Following intramuscular administration to pregnant women at 10-12 weeks gestation, approximately 50% of a dose was recovered in the feces and approximately 30% recovered in the urine.
Drug Interactions
Cytochrome P450 (CYP) enzymes: An in vitro inhibition study using human liver microsomes and CYP isoform-selective substrates indicated that hydroxyprogesterone caproate increased the metabolic rate of CYP1A2, CYP2A6, and CYP2B6 by approximately 80%, 150%, and 80%, respectively. However, in another in vitro study using human hepatocytes under conditions where the prototypical inducers or inhibitors caused the anticipated increases or decreases in CYP enzyme activities, hydroxyprogesterone caproate did not induce or inhibit CYP1A2, CYP2A6, or CYP2B6 activity. Overall, the findings indicate that hydroxyprogesterone caproate has minimal potential for CYP1A2, CYP2A6, and CYP2B6 related drug-drug interactions at the clinically relevant concentrations.
In vitro data indicated that therapeutic concentration of hydroxyprogesterone caproate is not likely to inhibit the activity of CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Hydroxyprogesterone caproate has not been adequately evaluated for carcinogenicity.
No reproductive or developmental toxicity or impaired fertility was observed in a multigenerational study in rats. Hydroxyprogesterone caproate administered intramuscularly, at gestational exposures up to 5 times the recommended human dose, had no adverse effects on the parental (F0) dams, their developing offspring (F1), or the latter offspring's ability to produce a viable, normal second (F2) generation.
14 CLINICAL STUDIES
14.1 Clinical Trial to Evaluate Reduction of Risk of Preterm Birth
In a multicenter, randomized, double-blind, vehicle (placebo)-controlled clinical trial, the safety and effectiveness of Makena for the reduction of the risk of spontaneous preterm birth was studied in women with a singleton pregnancy (age 16 to 43 years) who had a documented history of singleton spontaneous preterm birth (defined as delivery at less than 37 weeks of gestation following spontaneous preterm labor or premature rupture of membranes). At the time of randomization (between 16 weeks, 0 days and 20 weeks, 6 days of gestation), an ultrasound examination had confirmed gestational age and no known fetal anomaly. Women were excluded for prior progesterone treatment or heparin therapy during the current pregnancy, a history of thromboembolic disease, or maternal/obstetrical complications (such as current or planned cerclage, hypertension requiring medication, or a seizure disorder).
A total of 463 pregnant women were randomized to receive either Makena (N=310) or vehicle (N=153) at a dose of 250 mg administered weekly by intramuscular injection starting between 16 weeks, 0 days and 20 weeks, 6 days of gestation, and continuing until 37 weeks of gestation or delivery. Demographics of the Makena-treated women were similar to those in the control group, and included: 59.0% Black, 25.5% Caucasian, 13.9% Hispanic and 0.6% Asian. The mean body mass index was 26.9 kg/m2.
The proportions of women in each treatment arm who delivered at < 37 (the primary study endpoint), < 35, and < 32 weeks of gestation are displayed in Table 5.
| 1 Four Makena-treated subjects were lost to follow-up. They were counted as deliveries at their gestational ages at time of last contact (184, 220, 343 and 364 weeks). 2 Adjusted for interim analysis. |
|||
|
Delivery Outcome |
Makena1
% |
Control
% |
Treatment difference and
|
|
<37 weeks |
37.1 |
54.9 |
-17.8% [-28.0%, -7.4%] |
|
<35 weeks |
21.3 |
30.7 |
-9.4% [-19.0%, -0.4%] |
|
<32 weeks |
11.9 |
19.6 |
-7.7% [-16.1%, -0.3%] |
Compared to controls, treatment with Makena reduced the proportion of women who delivered preterm at < 37 weeks. The proportions of women delivering at < 35 and < 32 weeks also were lower among women treated with Makena. The upper bounds of the confidence intervals for the treatment difference at < 35 and < 32 weeks were close to zero. Inclusion of zero in a confidence interval would indicate the treatment difference is not statistically significant. Compared to the other gestational ages evaluated, the number of preterm births at < 32 weeks was limited.
After adjusting for time in the study, 7.5% of Makena-treated subjects delivered prior to 25 weeks compared to 4.7% of control subjects; see Figure 1.
Figure 1 Proportion of Women Remaining Pregnant as a Function of Gestational Age
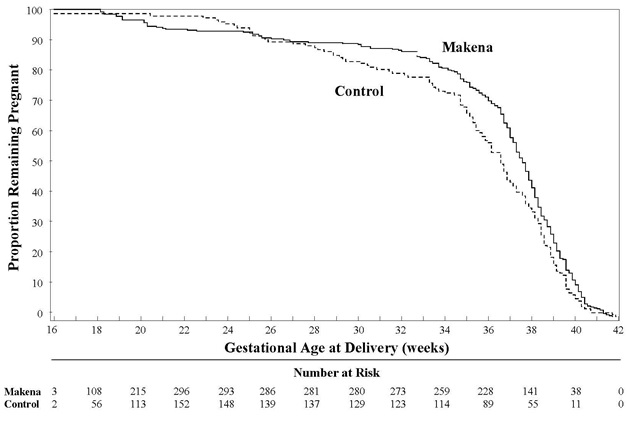
The rates of fetal losses and neonatal deaths in each treatment arm are displayed in Table 6. Due to the higher rate of miscarriages and stillbirths in the Makena arm, there was no overall survival difference demonstrated in this clinical trial.
| A Four of the 310 Makena-treated subjects were lost to follow-up and stillbirth or neonatal status could not be determined B Percentages are based on the number of enrolled subjects and not adjusted for time on drug C Percentage adjusted for the number of at risk subjects (n=209 for Makena, n=107 for control) enrolled at <20 weeks gestation. |
||||||
|
Complication |
Makena
|
Control
|
||||
|
Miscarriages <20 weeks gestation C |
5 (2.4) |
0 |
||||
|
Stillbirth |
6 (2.0) |
2 (1.3) |
||||
|
Antepartum stillbirth |
5 (1.6) |
1 (0.6) |
||||
|
Intrapartum stillbirth |
1 (0.3) |
1 (0.6) |
||||
|
Neonatal deaths |
8 (2.6) |
9 (5.9) |
||||
|
Total Deaths |
19 (6.2) |
11 (7.2) |
||||
A composite neonatal morbidity/mortality index evaluated adverse outcomes in live births. It was based on the number of neonates who died or experienced respiratory distress syndrome, bronchopulmonary dysplasia, grade 3 or 4 intraventricular hemorrhage, proven sepsis, or necrotizing enterocolitis. Although the proportion of neonates who experienced 1 or more events was numerically lower in the Makena arm (11.9% vs. 17.2%), the number of adverse outcomes was limited and the difference between arms was not statistically significant.
14.2 Infant Follow-Up Safety Study
Infants born to women enrolled in this study, and who survived to be discharged from the nursery, were eligible for participation in a follow-up safety study. Of 348 eligible offspring, 79.9% enrolled: 194 children of Makena-treated women and 84 children of control subjects. The primary endpoint was the score on the Ages & Stages Questionnaire (ASQ), which evaluates communication, gross motor, fine motor, problem solving, and personal/social parameters. The proportion of children whose scores met the screening threshold for developmental delay in each developmental domain was similar for each treatment group.
16 HOW SUPPLIED/STORAGE AND HANDLING
Makena auto-injector (for subcutaneous injection)
Makena auto-injector (NDC 64011-301-03) is supplied as 1.1 mL of a clear yellow sterile preservative-free solution in an auto-injector containing a pre-filled syringe. Each 1.1 mL auto-injector contains hydroxyprogesterone caproate USP, 250 mg/mL (25% w/v), in castor oil USP (30.6% v/v) and benzyl benzoate USP (46% v/v).
Single unit carton: Contains one 1.1 mL single-patient-use auto-injector of Makena containing 275 mg of hydroxyprogesterone caproate.
Store at 20° to 25°C (68° to 77°F). Do not refrigerate or freeze.
Caution: Protect auto-injector from light. Store auto-injector in its box.
Makena single- and multi-dose vials (for intramuscular injection)
Makena (NDC 64011-247-02) is supplied as 1 mL of a sterile preservative-free clear yellow solution in a single-dose glass vial.
Each 1 mL vial contains hydroxyprogesterone caproate USP, 250 mg/mL (25% w/v), in castor oil USP (30.6% v/v) and benzyl benzoate USP (46% v/v).
Single unit carton: Contains one 1 mL single-dose vial of Makena containing 250 mg of hydroxyprogesterone caproate.
Makena (NDC 64011-243-01) is supplied as 5 mL of a sterile clear yellow solution in a multi-dose glass vial.
Each 5 mL vial contains hydroxyprogesterone caproate USP, 250 mg/mL (25% w/v), in castor oil USP (28.6% v/v) and benzyl benzoate USP (46% v/v) with the preservative benzyl alcohol NF (2% v/v).
Single unit carton: Contains one 5 mL multi-dose vial of Makena (250 mg/mL) containing 1250 mg of hydroxyprogesterone caproate.
Store at 20° to 25°C (68° to 77°F). Do not refrigerate or freeze.
Use multi-dose vials within 5 weeks after first use.
Caution: Protect vial from light. Store vial in its box. Store upright.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Counsel patients that Makena injections may cause pain, soreness, swelling, itching or bruising. Inform the patient to contact her physician if she notices increased discomfort over time, oozing of blood or fluid, or inflammatory reactions at the injection site [see Adverse Reactions (6.1)].
Distributed by: AMAG Pharmaceuticals, Inc.
Waltham, MA 02451
02/2018
ver 1.2
| This Patient Information has been approved by the U.S. Food and Drug Administration |
| Revised: 02/2018 |
|
PATIENT INFORMATION
MAKENA (mah-KEE-na)
|
|
Read this Patient Information leaflet before you receive MAKENA. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. |
|
What is MAKENA? MAKENA is a prescription hormone medicine (progestin) used in women who are pregnant and who have delivered a baby too early (preterm) in the past. MAKENA is used in these women to help lower the risk of having a preterm baby again. It is not known if MAKENA reduces the number of babies who are born with serious medical conditions or die shortly after birth. MAKENA is for women who:
MAKENA is not intended for use to stop active preterm labor. It is not known if MAKENA is safe and effective in women who have other risk factors for preterm birth. MAKENA is not for use in women under 16 years of age. |
|
Who should not receive MAKENA? MAKENA should not be used if you have:
|
|
What should I tell my healthcare provider before receiving MAKENA? Before you receive MAKENA, tell your healthcare provider about all of your medical conditions, including if you have:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. MAKENA may affect the way other medicines work, and other medicines may affect how MAKENA works. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
How should I receive MAKENA?
|
|
What are the possible side effects of MAKENA? MAKENA may cause serious side effects, including:
The most common side effects of MAKENA include:
Call your healthcare provider if you have the following at your injection site:
Other side effects that may happen more often in women who receive MAKENA include:
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of MAKENA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store MAKENA?
Keep MAKENA and all medicines out of the reach of children. |
|
General information about the safe and effective use of MAKENA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use MAKENA for a condition for which it was not prescribed. Do not give MAKENA to other people, even if they have the same symptoms you have. It may harm them. This leaflet summarizes the most important information about MAKENA. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about MAKENA that is written for health professionals. |
|
What are the ingredients in MAKENA? Active ingredient: hydroxyprogesterone caproate Inactive ingredients: castor oil and benzyl benzoate. 5 mL multi-dose vials also contain benzyl alcohol (a preservative). Distributed by: AMAG Pharmaceuticals, Inc. |
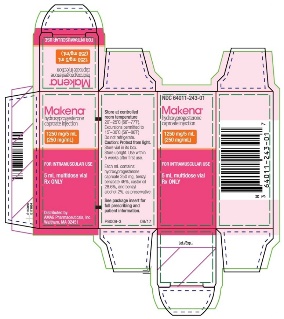
PRINCIPAL DISPLAY PANEL - NDC 64011-243-01 - 5 mL Carton Label
Makena®
hydroxyprogesterone caproate injection
1250 mg/5 mL
(250 mg/mL)
FOR INTRAMUSCULAR USE
5 mL multidose vial
Rx ONLY
Store at controlled room temperature
20°-25°C (68°-77°F).
Excursions permitted to
15°-30°C (59°-86°F)
Do not refrigerate.
Caution: Protect from light.
Store vial in its box. Store upright. Use within 5 weeks after first use.
Each mL contains: hydroxyprogesterone caproate 250 mg, benzyl benzoate 46%, castor oil 28.6%, and benzyl alcohol 2%, as preservative.
See package insert for full prescribing and patient information.
Distributed by:
AMAG Pharmaceuticals, Inc.
Waltham, MA 02451
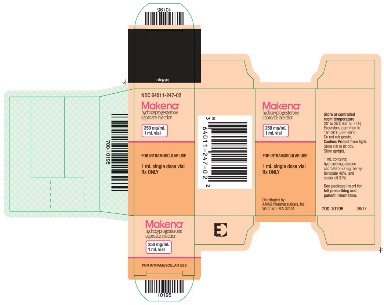
PRINCIPAL DISPLAY PANEL - NDC 64011-247-02 - 1 mL Carton Label
Makena®
hydroxyprogesterone caproate injection
250 mg/mL
1 mL vial
FOR INTRAMUSCULAR USE
1 mL single dose vial
Rx ONLY
Store at controlled room temperature
20° to 25°C (68° to 77°F).
Excursions permitted to
15°-30°C (59°-86°F).
Do not refrigerate.
Caution: Protect from light.
Store vial in its box.
Store upright.
1 mL contains:
hydroxyprogesterone caproate 250 mg, benzyl benzoate 46%, and castor oil 31%.
See package insert for full prescribing and patient information.
Distributed by:
AMAG Pharmaceuticals, Inc.
Waltham, MA 02451
PRINCIPAL DISPLAY PANEL - NDC 64011-301-03 - 1.1 mL Auto-Injector Carton Label
Rx ONLY
Makena®
hydroxyprogesterone caproate injection
275 mg/1.1 mL Auto-Injector
FOR SUBCUTANEOUS INJECTION ONLY.
Administration by Healthcare Professionals Only.
Read Instructions for Use before administration.
275 mg/1.1 mL
250 mg/mL
This carton contains:
1 single-use Makena Auto-Injector
Prescribing Information
and Instructions for Use
Storage & Handling:
Do not refrigerate or freeze | Protect from light (keep in carton until time of use) | Keep away from children
Store at 20°-25°C (68°-77° F)
Makena (hydroxyprogesterone caproate injection)
is a preservative-free solution.
Inactive ingredients: castor oil, benzyl benzoate.
Distributed by:
AMAG Pharmaceuticals, Inc.
Waltham, MA 02451

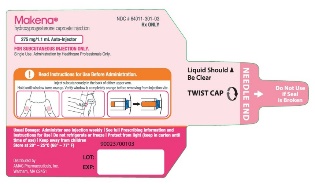
PRINCIPAL DISPLAY PANEL - NDC 64011-301-03 - 1.1 mL Auto-Injector Label
Rx ONLY
Makena®
hydroxyprogesterone caproate injection
275 mg/1.1 mL Auto-Injector
FOR SUBCUTANEOUS INJECTION ONLY.
Single Use. Administration by Healthcare Professionals Only.
Read Instructions for Use before administration.
Inject subcutaneously in the back of either upper arm.
Hold until window turns orange. Verify window is completely orange before removing from injection site.
Liquid Should
Be Clear
TWIST CAP
NEEDLE END
Do Not Use
if Seal
is Broken
Usual Dosage: Administer one injection weekly | See full Prescribing Information and
Instructions for Use | Do not refrigerate or freeze | Protect from light (keep in carton until
time of use) | Keep away from children
Store at 20° - 25°C (68° - 77°F)
Distributed by
AMAG Pharmaceuticals, Inc.
Waltham, MA 02451
| MAKENA
hydroxyprogesterone caproate injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| MAKENA
hydroxyprogesterone caproate injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| MAKENA
hydroxyprogesterone caproate injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - AMAG Pharmaceuticals, Inc. (017511155) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Piramal Pharma Solutions Inc. | 794520721 | MANUFACTURE(64011-247) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharmacia & Upjohn Company LLC | 618054084 | MANUFACTURE(64011-243, 64011-247) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fresenius Kabi Austria | 300206604 | MANUFACTURE(64011-301) | |