SCENESSE- afamelanotide implant
CSM Clinical Supplies Management Europe GmbH
Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use SCENESSE.
See full prescribing information for SCENESSE. Initial U.S. Approval: 2019 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATIONSCENESSE should be administered by a healthcare professional who is
DOSAGE FORMS AND STRENGTHSImplant: 16 mg of afamelanotide. (4) WARNINGS AND PRECAUTIONSSkin monitoring: May induce darkening of pre-existing nevi and ephelides due to its pharmacological effect. A regular full body skin examination (twice yearly) is recommended to monitor all nevi and other skin abnormalities. (6) ADVERSE REACTIONSThe most common adverse reactions (incidence > 2%) are implant site reaction, nausea, oropharyngeal pain, cough, fatigue, dizziness, skin hyperpigmentation, somnolence, melanocytic nevus, respiratory tract infection, non-acute porphyria, and skin irritation. (7) (7) To report suspected adverse reactions, contact CLINUVEL INC. at 1-888-288-2031 or FDA at 1-800-332-1088 or www.fda.gov/medwatch. (7) Revised: 10/2020 |
FULL PRESCRIBING INFORMATION: CONTENTS*
These highlights do not include all the information needed to use SCENESSE .
|
FULL PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SCENESSE .
See full prescribing information for SCENESSE.
Initial U.S. Approval: 2019
SCENESSE® is indicated to increase pain free light exposure in adult patients with a history of phototoxic reactions from erythropoietic protoporphyria (EPP).
Important Dosage and Administration Information
SCENESSE should be administered by a health care professional. All healthcare professionals should be proficient in the
subcutaneous implantation procedure and have completed the training program provided by CLINUVEL prior to
administration of the SCENESSE implant [see Dosage and Administration (2.2)]. Additional information, including a
video, is available at http://www.clinuvel.com/US-HCP. The additional information has not been evaluated or approved by
the FDA.
A single SCENESSE implant is inserted subcutaneously above the anterior supra-iliac crest every 2 months.
Use the SFM Implantation Cannula to implant SCENESSE. Contact CLINUVEL INC. for other implantation devices that
have been determined by the manufacturer to be suitable for implantation of SCENESSE.
Maintain sun and light protection measures during treatment with SCENESSE to prevent phototoxic reactions related to
EPP.
Instructions for Implantation of SCENESSE
Insert a single SCENESSE implant (containing 16 mg of afamelanotide) subcutaneously above the anterior supra-iliac
crest.
Implant SCENESSE observing an aseptic technique. The following equipment is needed for the implant insertion:
• SCENESSE implant
• SFM Implantation Cannula; use of a device that has not been determined to be suitable could result in damage to
the SCENESSE implant [see Dosage and Administration (2.1)].
• Sterile gloves
• Local anesthetic, needle and syringe
• Blunt forceps suitable for removing the SCENESSE implant from the glass vial and placement of the SCENESSE
implant
• Sterile gauze, adhesive bandage, pressure bandage
Step 1
• Take the carton containing SCENESSE out of the refrigerator to allow the product to gradually warm up to
ambient temperature.
• Remove the seal and stopper from the glass vial containing SCENESSE. Remove the implant from the vial
using the blunt forceps under aseptic conditions and place the implant on a sterile gauze.
Step 2
Put the patient in a comfortable reclined supine position.
Identify the insertion site 3-4 cm above the anterior suprailiac
crest and disinfect the skin surface.

Step 3 (optional)
Anesthetize the area of insertion (puncture) if deemed
necessary and in consultation with the patient
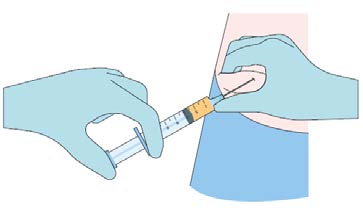
Step 4
While pinching the skin of the insertion site, insert the
cannula with the bevel facing upwards (away from the
abdomen) at a 30-45° angle into the subcutaneous layer.
Advance the cannula 2 cm into the subcutaneous layer.
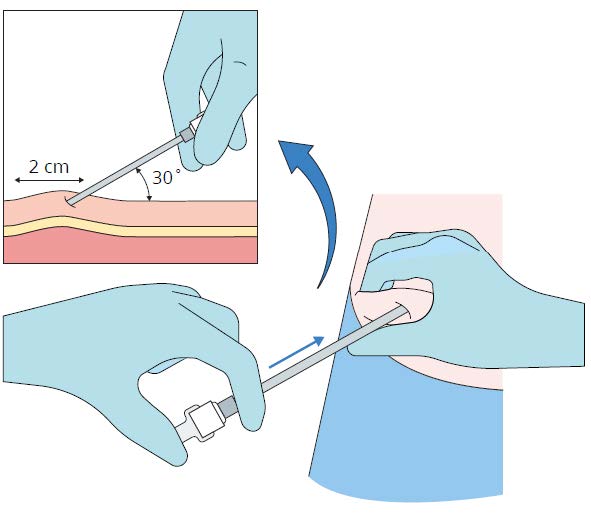
Step 5
• Remove the stylet (obturator) from the cannula
maintaining aseptic precautions
• Load the implant into the cannula
• Using the stylet (obturator) gently push the implant
down the full length of the cannula’s shaft
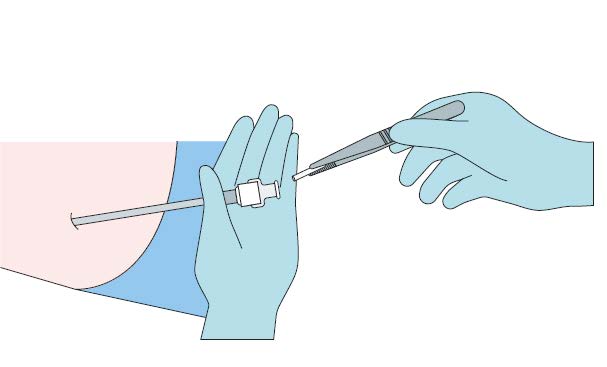
Step 6
Apply pressure to the site of the implant while removing the
stylet (obturator) and the cannula. Verify that no implant or
implant portion remains in the cannula.
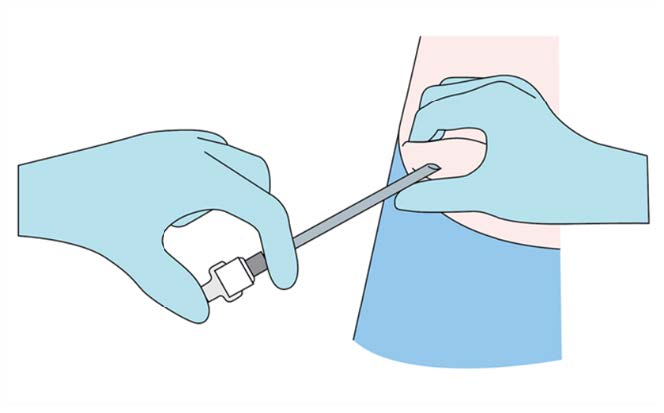
Step 7
Verify the correct insertion and placement of the implant by
palpating the skin overlying the implant.
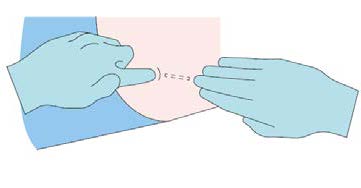
Step 8
Apply dressing to the insertion site. Leave dressing in place
for 24 hours.
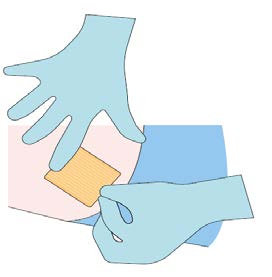
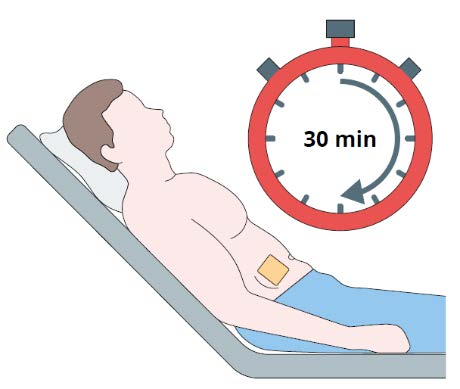
Step 9
Monitor the patient for 30 minutes after the implant
administration.
Implant: 16 mg of afamelanotide as a solid white to off-white, bioresorbable, sterile rod approximately 1.7 cm in length
and 1.45 mm in diameter.
Skin Monitoring
SCENESSE may lead to generalized increased skin pigmentation and darkening of pre-existing nevi and ephelides because
of its pharmacologic effect. A full body skin examination (twice yearly) is recommended to monitor pre-existing and new
skin pigmentary lesions.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials
of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in
practice.
The safety of SCENESSE was evaluated in 3 randomized, multicenter, prospective, vehicle controlled clinical trials (Study
CUV029, Study CUV030, and Study CUV039) involving 244 adult subjects with erythropoietic protoporphyria (EPP)
without significant liver involvement. Subjects received subcutaneous SCENESSE implants containing 16 mg of
afamelanotide every 2 months. A total of 125 subjects received SCENESSE and 119 subjects received vehicle implants.
Table 1 summarizes the adverse reactions that occurred in more than 2% of subjects.
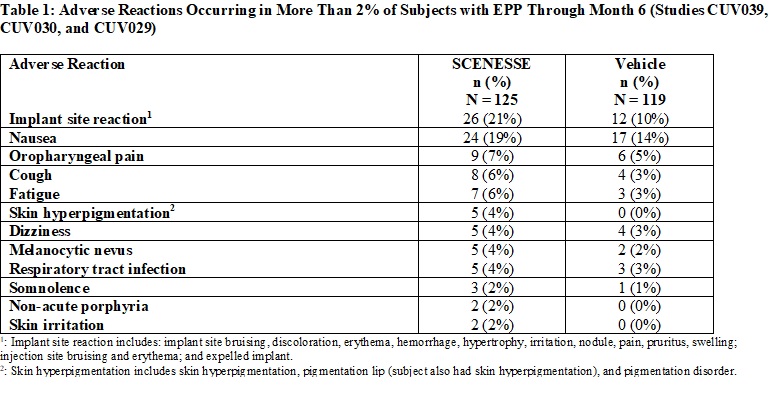
Specific Adverse Reactions
Implant Site Reactions: Implant site reactions were more common in the SCENESSE group (21%) compared to the vehicle
group (10%). In the SCENESSE group, the most common implant site reaction was implant site discoloration (10%).
To to report suspected adverse reactions, contact FDA at 1-800-332-1088 or www.fda.gov/medwatch.
Pregnancy
Risk Summary
There are no data on SCENESSE use in pregnant women to evaluate for any drug associated risk of major birth defects,
miscarriage, or adverse maternal or fetal outcome.
In animal reproductive and development toxicity studies, no adverse developmental effects were observed with
afamelanotide administration during the period of organogenesis to pregnant rats at subcutaneous doses up to 12 times the
maximum recommended human dose (MRHD) (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of
major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated
background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%,
respectively.
Data
Animal Data
In embryofetal development studies in Sprague Dawley and Lister Hooded rats, afamelanotide was administered
subcutaneously to pregnant rats at doses of 0.2, 2, or 20 mg/kg/day throughout the period of organogenesis. No adverse
embryofetal developmental effects were observed at doses up to 20 mg/kg/day (12 times the MRHD, based on a body
surface area comparison).
In pre- and post-natal development study in Sprague Dawley rats, afamelanotide was administered subcutaneously
at doses of 0.2, 2, or 20 mg/kg/day during the period of organogenesis through lactation. No treatment-related effects were
observed at doses up to 20 mg/kg/day (12 times the MRHD, based on a body surface area comparison).
Lactation
Risk Summary
There are no data on the presence of afamelanotide or any of its metabolites in human or animal milk, the effects on the
breastfed infant, or the effect on milk production. The developmental and health benefits of breastfeeding should be
considered along with the mother’s clinical need for SCENESSE and any potential adverse effects on the breastfed infant
from SCENESSE or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of SCENESSE have not been established in pediatric patients.
Geriatric Use
There were 10 subjects 65 years old and over in the clinical studies for EPP [see Clinical Studies (14)]. Of the 125
subjects treated with SCENESSE in these studies, 4 (3%) were 65 years of age and older. Clinical studies of SCENESSE
did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from
younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and
younger patients.
SCENESSE (afamelanotide) implant is a controlled-release dosage form for subcutaneous administration. Afamelanotide is
a melanocortin 1 receptor (MC1-R) agonist. The active ingredient afamelanotide acetate is a synthetic peptide containing
13 amino acids with molecular formula C78H111N21O19 •xC2H4O2 (3 ≤ x ≤ 4). The molecular weight of afamelanotide is
1646.85 (anhydrous free base). Afamelanotide acetate has the following structure:
Ac-Ser-Tyr-Ser-Nle-Glu-His-(D)Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2• xCH3COOH.
Afamelanotide is a white to off-white powder, freely soluble in water. Each SCENESSE implant contains 16 mg of
afamelanotide (equivalent to 18 mg of afamelanotide acetate), and 15.3-19.5 mg of poly (DL-lactide-co-glycolide).
SCENESSE implant is a single, solid white to off-white, bioresorbable and sterile rod approximately 1.7 cm in length and
1.45 mm in diameter. The implant core comprises of the drug substance admixed with a poly (DL-lactide-co-glycolide)
bioresorbable copolymer.
Mechanism of Action
Afamelanotide is a synthetic tridecapeptide and a structural analog of α-melanocyte stimulating hormone (α-MSH).
Afamelanotide is a melanocortin receptor agonist and binds predominantly to MC1-R.
Pharmacodynamics
Afamelanotide increases production of eumelanin in the skin independently of exposure to sunlight or artificial UV light
sources.
Pharmacokinetics
The pharmacokinetics of afamelanotide following administration of a single subcutaneous implant of SCENESSE were evaluated in 12 healthy adults. High variability was observed in the plasma concentrations of afamelanotide and for most subjects (9 out of 12), the last measurable afamelanotide concentration was at 96 hours post-dose. The mean ± SD C max and AUC 0-inf were 3.7 ± 1.3 ng/mL and 138.9 ± 42.6 hr*ng/mL, respectively.
Absorption
The median T max was 36 hr.
Elimination
The apparent half-life of afamelanotide is approximately 15 hr when administered subcutaneously in a controlled release implant.
Metabolism
Afamelanotide may undergo hydrolysis. However, its metabolic profile has not been fully characterized.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with SCENESSE.
Afamelanotide was negative in the Ames test, in vitro mouse lymphoma assay, and in vivo mouse bone marrow
micronucleus assay.
No effects on male or female fertility and reproductive performance were observed in rats at subcutaneous doses up to 20
mg/kg/day afamelanotide (12 times the MRHD, based on a body surface area comparison).
Three vehicle-controlled, parallel-group clinical trials of SCENESSE were conducted in subjects with EPP. Of these trials,
two trials (Study CUV039, NCT 01605136, and Study CUV029, NCT 00979745) were designed to assess exposure to
direct sunlight on days with no phototoxic pain. The two trials differed in the number of days of follow-up, the time
windows within a day in which time spent outdoors was recorded, and how the amount of time spent in direct sunlight on
each day was characterized. The subjects enrolled in these trials were primarily Caucasian (98%), the mean age was 40
years (range 18 to 74 years), and 53% of subjects were male and 47% were female.
Study CUV039 enrolled 93 subjects, of whom 48 received SCENESSE (16 mg of afamelanotide administered
subcutaneously every 2 months), 45 received vehicle. Subjects received three implants and were followed for 180 days.
On each study day, subjects recorded the number of hours spent in direct sunlight between 10 am and 6 pm, the number of
hours spent in shade between 10 am and 6 pm, and whether they experienced any phototoxic pain that day. The primary
endpoint was the total number of hours over 180 days spent in direct sunlight between 10 am and 6 pm on days with no
pain. The median total number of hours over 180 days spent in direct sunlight between 10 am and 6 pm on days with no
pain was 64.1 hours for subjects receiving SCENESSE and 40.5 hours for subjects receiving vehicle.
Study CUV029 enrolled 74 subjects, of whom 38 received SCENESSE (16 mg of afamelanotide administered
subcutaneously every 2 months), 36 received vehicle. Subjects received five implants and were followed for 270 days. On
each study day, subjects recorded the number of hours spent outdoors between 10 am and 3 pm, whether “most of the day”
was spent in direct sunlight, shade, or a combination of both, and whether they experienced any phototoxic pain that day.
The primary endpoint was the total number of hours over 270 days spent outdoors between 10 am and 3 pm on days with
no pain for which “most of the day” was spent in direct sunlight. This analysis does not include sun exposure on days for
which subjects reported spending time in a combination of both direct sunlight and shade. The median total number of
hours over 270 days spent outdoors between 10 am and 3 pm on days with no pain for which “most of the day” was spent
in direct sunlight was 6.0 hours for subjects in the SCENESSE group and 0.75 hours for subjects in the vehicle group.
SCENESSE (afamelanotide) implant, 16 mg, for subcutaneous administration (NDC 73372-0116-1) is supplied in a Type I
amber glass vial sealed with a PTFE coated rubber stopper. Each vial contains one afamelanotide implant and is packaged
individually in a cardboard box. SCENESSE implant is a solid white to off-white, bioresorbable and sterile rod
approximately 1.7 cm in length and 1.45 mm in diameter.
Store in a refrigerator at 2°C – 8°C (36°F-46°F). Protect from light.
SCENESSE implants are not supplied with an implantation device for subcutaneous administration [see Dosage and
Administration (2)].
Concomitant Measures
Advise patients to maintain sun and light protection measures during treatment with SCENESSE to prevent phototoxic
reactions related to EPP.
Skin Monitoring
Advise patients that darkening of pre-existing nevi and ephelides may occur with use of SCENESSE. A full body skin
examination is recommended twice yearly to monitor pre-existing and new skin pigmentary lesions.
Expelled Implant
Advise patients to contact their healthcare provider if the implant is expelled.
Dressing removal
Advise patients that the dressing can be removed after 24 hours.
Insertion Site Care and Monitoring
Advise patients to monitor the insertion site after dressing removal and to report any reaction observed at the site to their
healthcare provider.
Month/Day/Year
IMPORTANT PRESCRIBING INFORMATION
Re: Temporary changes to approved packaging and labeling of SCENESSE® (afamelanotide) implant.
Dear Health Care Provider,
CLINUVEL, INC. is issuing this letter to inform you of temporary changes to the approved packaging and labeling of SCENESSE®. This means that the appearance of SCENESSE® packaging will differ from that approved by the United States Food and Drug Administration (FDA). To meet U.S. demand, CLINUVEL, INC. is releasing batches ML1803 and ML1901 with the temporary changes to the packaging and labelling with the knowledge of the FDA. A summary of these changes is outlined below.
CLINUVEL will communicate updates to you on its ability to provide SCENESSE® in its final U.S. approved packaging.
CLINUVEL, INC. is the holder of the approved New Drug Application (NDA) for the medication SCENESSE® (afamelanotide) implant. SCENESSE® was approved by the U.S. FDA in October 2019 to increase pain-free light exposure in adult patients with a history of phototoxicity reactions from erythropoietic protoporphyria (EPP).
Summary of the temporary changes to packaging and labeling of SCENESSE®
SCENESSE® will be supplied in a single vial contained within a clear plastic pouch (transparent, zip-top bag), accompanied with the U.S. approved Prescribing Information, rather than in a cardboard carton. Both the pouch and label will feature the following information that is included in the approved label
Details on the contents of the packaging and ingredients:
“Each implant contains 16 mg of afamelanotide (equivalent to 18 mg of afamelanotide acetate) and 15.3-19.5mg of poly(DL-lactide-co-glycolide) bioresorbable copolymers”
NDC number
The statement “See Prescribing Information for dosage and administration information”
The statement “Rx only; for subcutaneous use only”
The lot number and expiration date of the SCENESSE® implant
Storage details
Manufacturer’s name
The pouch and vial will not feature a product identifier or any colored text or company logo. The pouch will also not feature a barcode.
The vial should not be removed from the plastic pouch until the product is ready to be administered to a patient.
Availability of SCENESSE®
SCENESSE® is only available to healthcare professionals trained to administer SCENESSE® and should only be administered to adult patients with a confirmed diagnosis of EPP.
SCENESSE® is only available by direct order from CLINUVEL and cannot be obtained through third party distributors or wholesalers.
Reporting adverse events and further information
A copy of the full prescribing information for SCENESSE® has been appended to this letter.
Health Care Providers are expected to report all adverse events experienced by EPP patients receiving SCENESSE® to CLINUVEL via email at safety@clinuvel.com or by calling CLINUVEL, INC. on 1(650) 733 2827. Adverse events or quality problems experienced with the use of this product may also be reported to the FDA’s MedWatch Adverse Event Reporting Program either online, by regular mail, or by fax:
Complete and submit the report Online: www.fda.gov/medwatch/report.htm
Regular mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178 (1-800-332-0178).
If you have any questions, you can call CLINUVEL, INC on 1-650-733-2827.
Yours sincerely,
Linda Teng
Director, Clinical Compliance
CLINUVEL INC.

| SCENESSE
afamelanotide implant |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - CSM Clinical Supplies Management Europe GmbH (313941179) |