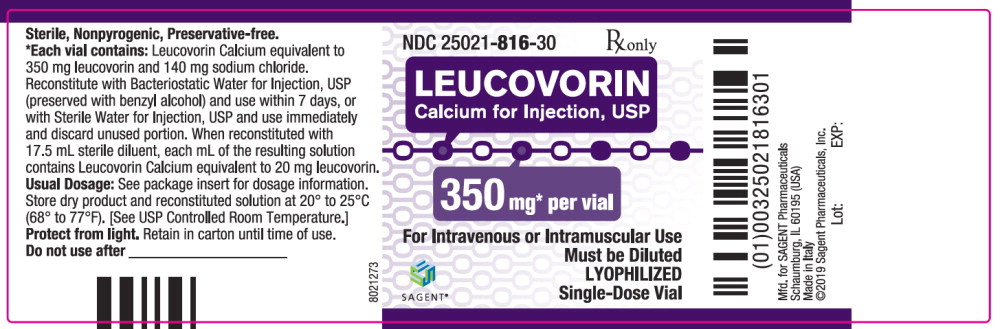
Label: LEUCOVORIN CALCIUM injection, powder, lyophilized, for solution




- NDC Code(s): 25021-813-10, 25021-814-30, 25021-815-30, 25021-816-30
- Packager: Sagent Pharmaceuticals
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated November 9, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Leucovorin is one of several active, chemically reduced derivatives of folic acid. It is useful as an antidote to drugs which act as folic acid antagonists.
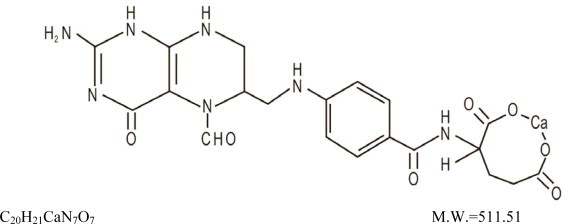
Also known as folinic acid, Citrovorum factor, or 5-formyl-5,6,7,8-tetrahydrofolic acid, this compound has the chemical designation of Calcium N-[p-[[[(6RS)-2-amino-5-formyl-5,6,7,8-tetrahydro-4-hydroxy-6-pteridinyl]methyl]amino]benzoyl]-L-glutamate (1:1). The structural formula of leucovorin calcium is:
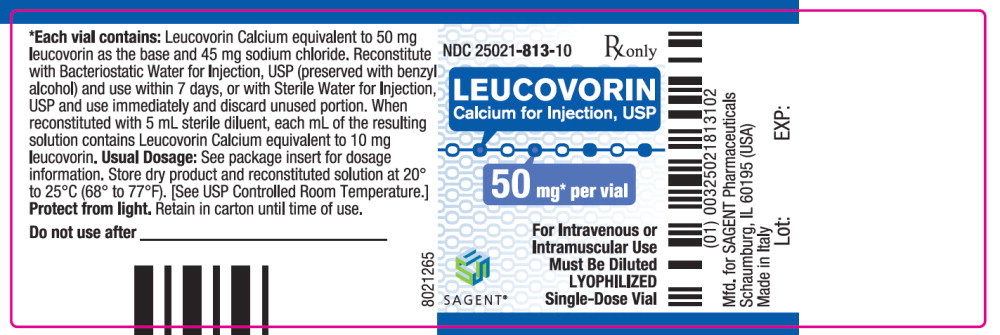
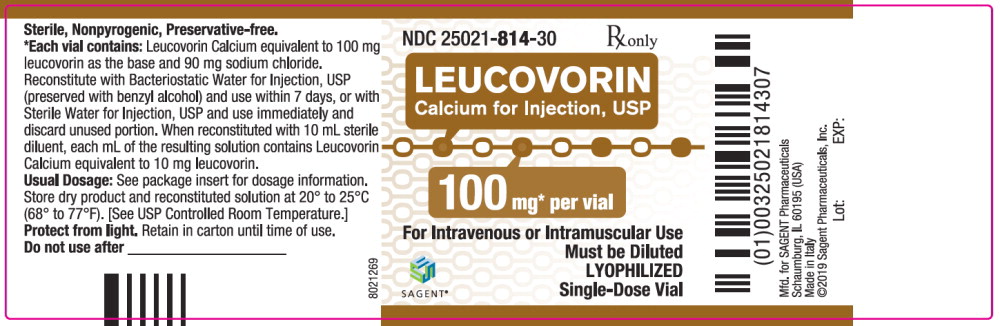
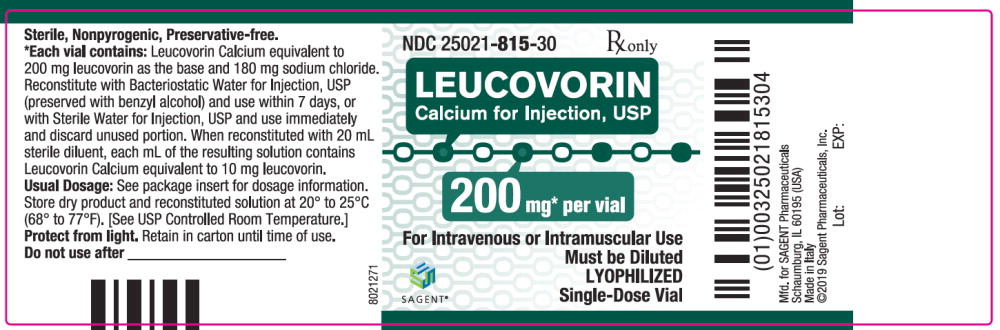
Leucovorin Calcium for Injection, USP is a sterile product indicated for intramuscular (IM) or intravenous (IV) administration and is supplied in 50 mg, 100 mg, 200 mg, and 350 mg vials.
Each 50 mg vial of Leucovorin Calcium for Injection, USP, when reconstituted with 5 mL of sterile diluent, contains leucovorin (as the calcium salt) 10 mg per mL.
Each 100 mg vial of Leucovorin Calcium for Injection, USP, when reconstituted with 10 mL of sterile diluent, contains leucovorin (as the calcium salt) 10 mg per mL.
Each 200 mg vial of Leucovorin Calcium for Injection, USP, when reconstituted with 20 mL of sterile diluent, contains leucovorin (as the calcium salt) 10 mg per mL.
Each 350 mg vial of Leucovorin Calcium for Injection, USP, when reconstituted with 17.5 mL of sterile diluent, contains leucovorin (as the calcium salt) 20 mg per mL.
In each dosage form, one milligram of leucovorin calcium contains 0.002 mmol of leucovorin and 0.002 mmol of calcium.
These lyophilized products contain no preservative. The inactive ingredient is Sodium Chloride, USP, added to adjust tonicity. Sodium hydroxide and / or hydrochloric acid may be used to adjust the pH. The pH is between 6.5 and 8.5. Reconstitute with Bacteriostatic Water for Injection, USP, which contains benzyl alcohol (see WARNINGS section), or with Sterile Water for Injection, USP.
-
CLINICAL PHARMACOLOGY
Leucovorin is a mixture of the diastereoisomers of the 5-formyl derivative of tetrahydrofolic acid (THF). The biologically active compound of the mixture is the (-)-l-isomer, known as Citrovorum factor or (-)-folinic acid. Leucovorin does not require reduction by the enzyme dihydrofolate reductase in order to participate in reactions utilizing folates as a source of “one-carbon” moieties. l-Leucovorin (l-5-formyltetrahydrofolate) is rapidly metabolized (via 5, 10-methenyltetrahydrofolate then 5, 10-methylenetetrahydrofolate) to l,5-methyltetrahydrofolate. l,5-Methyltetrahydrofolate can in turn be metabolized via other pathways back to 5, 10-methylenetetrahydrofolate, which is converted to 5-methyltetrahydrofolate by an irreversible, enzyme catalyzed reduction using the cofactors FADH2 and NADPH.
Administration of leucovorin can counteract the therapeutic and toxic effects of folic acid antagonists such as methotrexate, which act by inhibiting dihydrofolate reductase.
In contrast, leucovorin can enhance the therapeutic and toxic effects of fluoropyrimidines used in cancer therapy, such as 5-fluorouracil. Concurrent administration of leucovorin does not appear to alter the plasma pharmacokinetics of 5-fluorouracil. 5-Fluorouracil is metabolized to fluorodeoxyuridylic acid, which binds to and inhibits the enzyme thymidylate synthase (an enzyme important in DNA repair and replication).
Leucovorin is readily converted to another reduced folate, 5,10-methylenetetrahydrofolate, which acts to stabilize the binding of fluorodeoxyuridylic acid to thymidylate synthase and thereby enhances the inhibition of this enzyme.
The pharmacokinetics after intravenous, intramuscular and oral administration of a 25 mg dose of leucovorin were studied in male volunteers. After intravenous administration, serum total reduced folates (as measured by Lactobacillus casei assay) reached a mean peak of 1259 ng/mL (range 897 to 1625). The mean time to peak was 10 minutes. This initial rise in total reduced folates was primarily due to the parent compound 5-formyl-THF (measured by Streptococcus faecalis assay) which rose to 1206 ng/mL at 10 minutes. A sharp drop in parent compound followed and coincided with the appearance of the active metabolite 5-methyl-THF which became the predominant circulating form of the drug.
The mean peak of 5-methyl-THF was 258 ng/mL and occurred at 1.3 hours. The terminal half-life for total reduced folates was 6.2 hours. The area under the concentration versus time curves (AUCs) for l-leucovorin, d-leucovorin and 5-methyltetrahydrofolate were 28.4 ± 3.5, 956 ± 97 and 129 ± 12 (mg/min/L ± S.E.). When a higher dose of d,l-leucovorin (200 mg/m2) was used, similar results were obtained. The d-isomer persisted in plasma at concentrations greatly exceeding those of the l-isomer.
After intramuscular injection, the mean peak of serum total reduced folates was 436 ng/mL (range 240 to 725) and occurred at 52 minutes. Similar to IV administration, the initial sharp rise was due to the parent compound. The mean peak of 5-formyl-THF was 360 ng/mL and occurred at 28 minutes. The level of the metabolite 5-methyl-THF increased subsequently over time until at 1.5 hours it represented 50% of the circulating total folates. The mean peak of 5-methyl-THF was 226 ng/mL at 2.8 hours. The terminal half-life of total reduced folates was 6.2 hours. There was no difference of statistical significance between IM and IV administration in the AUC for total reduced folates, 5-formyl-THF, or 5-methyl-THF.
After oral administration of leucovorin reconstituted with aromatic elixir, the mean peak concentration of serum total reduced folates was 393 ng/mL (range 160 to 550). The mean time to peak was 2.3 hours and the terminal half-life was 5.7 hours. The major component was the metabolite 5-methyltetrahydrofolate to which leucovorin is primarily converted in the intestinal mucosa. The mean peak of 5-methyl-THF was 367 ng/mL at 2.4 hours. The peak level of the parent compound was 51 ng/mL at 1.2 hours. The AUC of total reduced folates after oral administration of the 25 mg dose was 92% of the AUC after intravenous administration.
Following oral administration, leucovorin is rapidly absorbed and expands the serum pool of reduced folates. At a dose of 25 mg, almost 100% of the l-isomer but only 20% of the d-isomer is absorbed. Oral absorption of leucovorin is saturable at doses above 25 mg. The apparent bioavailability of leucovorin was 97% for 25 mg, 75% for 50 mg, and 37% for 100 mg.
In a randomized clinical study conducted by the Mayo Clinic and the North Central Cancer Treatment Group (Mayo/NCCTG) in patients with advanced metastatic colorectal cancer three treatment regimens were compared: Leucovorin (LV) 200 mg/m2 and 5-fluorouracil (5-FU) 370 mg/m2 versus LV 20 mg/m2 and 5-FU 425 mg/m2 versus 5-FU 500 mg/m2. All drugs were administered by slow intravenous infusion daily for 5 days repeated every 28 to 35 days. Response rates were 26% (p=0.04 versus 5-FU alone), 43% (p=0.001 versus 5-FU alone) and 10% for the high dose leucovorin, low dose leucovorin and 5-FU alone groups respectively. Respective median survival times were 12.2 months (p=0.037), 12 months (p=0.050), and 7.7 months. The low dose LV regimen gave a statistically significant improvement in weight gain of more than 5%, relief of symptoms, and improvement in performance status. The high dose LV regimen gave a statistically significant improvement in performance status and trended toward improvement in weight gain and in relief of symptoms but these were not statistically significant.
In a second Mayo/NCCTG randomized clinical study the 5-FU alone arm was replaced by a regimen of sequentially administered methotrexate (MTX), 5-FU, and LV. Response rates with LV 200 mg/m2 and 5-FU 370 mg/m2 versus LV 20 mg/m2 and 5-FU 425 mg/m2 versus sequential MTX and 5-FU and LV were respectively 31% (p=<0.01), 42% (p=<0.01), and 14%. Respective median survival times were 12.7 months (p=<0.04), 12.7 months (p=<0.01), and 8.4 months. No statistically significant difference in weight gain of more than 5% or in improvement in performance status was seen between the treatment arms.
The pharmacokinetics of 200 mg doses of leucovorin administered intravenously and orally (reconstituted powder, not tablets) have been evaluated in healthy male subjects. The serum clearance corrected for bioavailability, terminal half-life, and apparent volume of distribution of total folate were not significantly different between routes of administration. The oral bioavailability of the 200 mg dose was 31%. Eighty-three percent of the biologically active IV dose was recovered in the urine within 24 hours, 31% as 5-methyltetrahydrofolate. Twenty percent of the same oral dose was excreted in 24 hours, 16% as 5-methyltetrahydrofolate.
-
INDICATIONS AND USAGE
Leucovorin calcium rescue is indicated after high dose methotrexate therapy in osteosarcoma. Leucovorin Calcium for Injection is also indicated to diminish the toxicity and counteract the effects of impaired methotrexate elimination and of inadvertent overdosages of folic acid antagonists.
Leucovorin Calcium for Injection is indicated in the treatment of megaloblastic anemias due to folic acid deficiency when oral therapy is not feasible.
Leucovorin Calcium for Injection is also indicated for use in combination with 5-fluorouracil to prolong survival in the palliative treatment of patients with advanced colorectal cancer. Leucovorin Calcium for Injection should not be mixed in the same infusion as 5-fluorouracil because a precipitate may form.
- CONTRAINDICATIONS
-
WARNINGS
In the treatment of accidental overdosages of folic acid antagonists, intravenous leucovorin should be administered as promptly as possible. As the time interval between antifolate administration (e.g., methotrexate) and leucovorin rescue increases, leucovorin's effectiveness in counteracting toxicity decreases. In the treatment of accidental overdosages of intrathecally administered folic acid antagonists, do not administer leucovorin intrathecally. LEUCOVORIN MAY BE HARMFUL OR FATAL IF GIVEN INTRATHECALLY.
Monitoring of the serum methotrexate concentration is essential in determining the optimal dose and duration of treatment with leucovorin.
Delayed methotrexate excretion may be caused by a third space fluid accumulation (i.e., ascites, pleural effusion), renal insufficiency, or inadequate hydration. Under such circumstances, higher doses of leucovorin or prolonged administration may be indicated. Doses higher than those recommended for oral use must be given intravenously.
Because of the benzyl alcohol contained in certain diluents used for reconstituting leucovorin, when doses greater than 10 mg/m2 are administered, leucovorin should be reconstituted with Sterile Water for Injection, USP, and used immediately (see DOSAGE AND ADMINISTRATION).
Because of the calcium content of the leucovorin solution, no more than 160 mg of leucovorin should be injected intravenously per minute (16 mL of a 10 mg/mL, or 8 mL of a 20 mg/mL solution per minute).
Leucovorin enhances the toxicity of 5-fluorouracil. When these drugs are administered concurrently in the palliative therapy of advanced colorectal cancer, the dosage of 5-fluorouracil must be lower than usually administered. Although the toxicities observed in patients treated with the combination of leucovorin plus 5-fluorouracil are qualitatively similar to those observed in patients treated with 5-fluorouracil alone, gastrointestinal toxicities (particularly stomatitis and diarrhea) are observed more commonly and may be more severe and of prolonged duration in patients treated with the combination.
In the first Mayo/NCCTG controlled trial, toxicity, primarily gastrointestinal, resulted in 7% of patients requiring hospitalization when treated with 5-fluorouracil alone or 5-fluorouracil in combination with 200 mg/m2 of leucovorin and 20% when treated with 5-fluorouracil in combination with 20 mg/m2 of leucovorin. In the second Mayo/NCCTG trial, hospitalizations related to treatment toxicity also appeared to occur more often in patients treated with the low dose leucovorin/5-fluorouracil combination than in patients treated with the high dose combination — 11% versus 3%. Therapy with leucovorin and 5-fluorouracil must not be initiated or continued in patients who have symptoms of gastrointestinal toxicity of any severity, until those symptoms have completely resolved. Patients with diarrhea must be monitored with particular care until the diarrhea has resolved, as rapid clinical deterioration leading to death can occur. In an additional study utilizing higher weekly doses of 5-fluorouracil and leucovorin, elderly and/or debilitated patients were found to be at greater risk for severe gastrointestinal toxicity.
Seizures and/or syncope have been reported rarely in cancer patients receiving leucovorin, usually in association with fluoropyrimidine administration, and most commonly in those with CNS metastases or other predisposing factors, however, a causal relationship has not been established.
The concomitant use of leucovorin with trimethoprim-sulfamethoxazole for the acute treatment of Pneumocystis carinii pneumonia in patients with HIV infection was associated with increased rates of treatment failure and morbidity in a placebo-controlled study.
-
PRECAUTIONS
General
Parenteral administration is preferable to oral dosing if there is a possibility that the patient may vomit and not absorb the leucovorin. Leucovorin has no effect on non-hematologic toxicities of methotrexate such as the nephrotoxicity resulting from drug and/or metabolite precipitation in the kidney.
Since leucovorin enhances the toxicity of fluorouracil, leucovorin/5-fluorouracil combination therapy for advanced colorectal cancer should be administered under the supervision of a physician experienced in the use of antimetabolite cancer chemotherapy. Particular care should be taken in the treatment of elderly or debilitated colorectal cancer patients, as these patients may be at increased risk of severe toxicity.
Laboratory Tests
Patients being treated with the leucovorin/5-fluorouracil combination should have a CBC with differential and platelets prior to each treatment. During the first two courses a CBC with differential and platelets has to be repeated weekly and thereafter once each cycle at the time of anticipated WBC nadir. Electrolytes and liver function tests should be performed prior to each treatment for the first three cycles then prior to every other cycle. Dosage modifications of fluorouracil should be instituted as follows, based on the most severe toxicities:
Diarrhea and/or Stomatitis WBC/mm3
NadirPlatelets/mm3
Nadir5-FU Dose Moderate 1,000 to 1,900 25 to 75,000 decrease 20% Severe < 1,000 < 25,000 decrease 30% If no toxicity occurs, the 5-fluorouracil dose may increase 10%. Treatment should be deferred until WBCs are 4,000/mm3 and platelets 130,000/mm3. If blood counts do not reach these levels within two weeks, treatment should be discontinued. Patients should be followed up with physical examination prior to each treatment course and appropriate radiological examination as needed. Treatment should be discontinued when there is clear evidence of tumor progression.
Drug Interactions
Folic acid in large amounts may counteract the antiepileptic effect of phenobarbital, phenytoin and primidone, and increase the frequency of seizures in susceptible pediatric patients.
Preliminary animal and human studies have shown that small quantities of systemically administered leucovorin enter the CSF primarily as 5-methyltetrahydrofolate and, in humans, remain 1 to 3 orders of magnitude lower than the usual methotrexate concentrations following intrathecal administration. However, high doses of leucovorin may reduce the efficacy of intrathecally administered methotrexate.
Leucovorin may enhance the toxicity of 5-fluorouracil (see WARNINGS).
Pregnancy
Teratogenic Effects
Pregnancy Category C
Adequate animal reproduction studies have not been conducted with leucovorin. It is also not known whether leucovorin can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Leucovorin should be given to a pregnant woman only if clearly needed.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when leucovorin is administered to a nursing mother.
Geriatric Use
Clinical studies of leucovorin calcium did not show differences in safety or effectiveness between subjects over 65 and younger subjects. Other clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older patients cannot be ruled out. This drug is known to be excreted by the kidney and the risk of toxic reactions to the drug may be greater in patients with impaired renal function. Because elder patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
ADVERSE REACTIONS
Allergic sensitization, including anaphylactoid reactions and urticaria, has been reported following the administration of both oral and parenteral leucovorin. No other adverse reactions have been attributed to the use of leucovorin per se.
The following table summarizes significant adverse events occurring in 316 patients treated with the leucovorin/5-fluorouracil combinations compared against 70 patients treated with 5-fluorouracil alone for advanced colorectal carcinoma. These data are taken from the Mayo/NCCTG large multicenter prospective trial evaluating the efficacy and safety of the combination regimen.
PERCENTAGE OF PATIENTS TREATED WITH LEUCOVORIN/FLUOROURACIL FOR ADVANCED COLORECTAL CARCINOMA REPORTING ADVERSE EXPERIENCES OR HOSPITALIZED FOR TOXICITY High LV = Leucovorin 200 mg/m2, Low LV = Leucovorin 20 mg/m2
Any = percentage of patients reporting toxicity of any severity
Grade 3+ = percentage of patients reporting toxicity of Grade 3 or higher
(High LV)/5-FU
(N=155)(Low LV)/5-FU
(N=161)5-FU Alone
(N=70)Any Grade 3+ Any Grade 3+ Any Grade 3+ (%) (%) (%) (%) (%) (%) Leukopenia 69 14 83 23 93 48 Thrombocytopenia 8 2 8 1 18 3 Infection 8 1 3 1 7 2 Nausea 74 10 80 9 60 6 Vomiting 46 8 44 9 40 7 Diarrhea 66 18 67 14 43 11 Stomatitis 75 27 84 29 59 16 Constipation 3 0 4 0 1 - Lethargy/Malaise/Fatigue 13 3 12 2 6 3 Alopecia 42 5 43 6 37 7 Dermatitis 21 2 25 1 13 - Anorexia 14 1 22 4 14 - Hospitalization for Toxicity 5% 15% 7% To report SUSPECTED ADVERSE REACTIONS, contact Sagent Pharmaceuticals, Inc. at 1-866-625-1618 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
Advanced Colorectal Cancer
Either of the following two regimens is recommended:
- Leucovorin calcium for injection is administered at 200 mg/m2 by slow intravenous injection over a minimum of 3 minutes, followed by 5-fluorouracil at 370 mg/m2 by intravenous injection.
- Leucovorin calcium for injection is administered at 20 mg/m2 by intravenous injection followed by 5-fluorouracil at 425 mg/m2 by intravenous injection.
5-Fluorouracil and leucovorin should be administered separately to avoid the formation of a precipitate.
Treatment is repeated daily for five days. This five-day treatment course may be repeated at 4 week (28-day) intervals, for 2 courses and then repeated at 4 to 5 week (28 to 35 day) intervals provided that the patient has completely recovered from the toxic effects of the prior treatment course.
In subsequent treatment courses, the dosage of 5-fluorouracil should be adjusted based on patient tolerance of the prior treatment course. The daily dosage of 5-fluorouracil should be reduced by 20% for patients who experienced moderate hematologic or gastrointestinal toxicity in the prior treatment course, and by 30% for patients who experienced severe toxicity (see PRECAUTIONS: Laboratory Tests). For patients who experienced no toxicity in the prior treatment course, 5-fluorouracil dosages may be increased by 10%. Leucovorin calcium for injection dosages are not adjusted for toxicity.
Leucovorin Rescue After High-Dose Methotrexate Therapy
The recommendations for leucovorin rescue are based on a methotrexate dose of 12 to 15 grams/m2 administered by intravenous infusion over 4 hours (see methotrexate package insert for full prescribing information).
Leucovorin rescue at a dose of 15 mg (approximately 10 mg/m2) every 6 hours for 10 doses starts 24 hours after the beginning of the methotrexate infusion. In the presence of gastrointestinal toxicity, nausea or vomiting, leucovorin calcium for injection should be administered parenterally. Do not administer leucovorin calcium for injection intrathecally.
Serum creatinine and methotrexate levels should be determined at least once daily. Leucovorin calcium for injection administration, hydration, and urinary alkalization (pH of 7.0 or greater) should be continued until the methotrexate level is below 5 x 10-8 M (0.05 micromolar). The leucovorin calcium for injection dose should be adjusted or leucovorin rescue extended based on the following guidelines:
GUIDELINES FOR LEUCOVORIN CALCIUM FOR INJECTION DOSAGE AND ADMINISTRATION DO NOT ADMINISTER LEUCOVORIN CALCIUM FOR INJECTION INTRATHECALLY Clinical
SituationLaboratory Findings Leucovorin Calcium for Injection Dosage and Duration Normal
Methotrexate EliminationSerum methotrexate level approximately 10 micromolar at 24 hours after administration, 1 micromolar at 48 hours, and less than 0.2 micromolar at 72 hours. 15 mg PO, IM, or IV q 6 hours for 60 hours (10 doses starting at 24 hours after start of methotrexate infusion). Delayed Late Methotrexate Elimination Serum methotrexate level remaining above 0.2 micromolar at 72 hours, and more than 0.05 micromolar at 96 hours after administration. Continue 15 mg PO, IM, or IV q 6 hours, until methotrexate level is less than 0.05 micromolar. Delayed Early Methotrexate Elimination and/or Evidence of Acute Renal Injury Serum methotrexate level of 50 micromolar or more at 24 hours, or 5 micromolar or more at 48 hours after administration, OR; a 100% or greater increase in serum creatinine level at 24 hours after methotrexate administration (e.g., an increase from 0.5 mg/dL to a level of 1 mg/dL or more). 150 mg IV q 3 hours, until methotrexate level is less than 1 micromolar; then 15 mg IV q 3 hours until methotrexate level is less than 0.05 micromolar. Patients who experience delayed early methotrexate elimination are likely to develop reversible renal failure. In addition to appropriate leucovorin therapy, these patients require continuing hydration and urinary alkalization, and close monitoring of fluid and electrolyte status, until the serum methotrexate level has fallen to below 0.05 micromolar and the renal failure has resolved.
Some patients will have abnormalities in methotrexate elimination or renal function following methotrexate administration, which are significant but less severe than abnormalities described in the table above. These abnormalities may or may not be associated with significant clinical toxicity. If significant clinical toxicity is observed, leucovorin rescue should be extended for an additional 24 hours (total of 14 doses over 84 hours) in subsequent courses of therapy. The possibility that the patient is taking other medications which interact with methotrexate (e.g., medications which may interfere with methotrexate elimination or binding to serum albumin) should always be reconsidered when laboratory abnormalities or clinical toxicities are observed.
Impaired Methotrexate Elimination or Inadvertent Overdosage
Leucovorin rescue should begin as soon as possible after an inadvertent overdosage and within 24 hours of methotrexate administration when there is delayed excretion (see WARNINGS). Leucovorin calcium for injection 10 mg/m2 should be administered IV, IM, or PO every 6 hours until the serum methotrexate level is less than 10-8 M. In the presence of gastrointestinal toxicity, nausea, or vomiting, leucovorin calcium for injection should be administered parenterally. Do not administer leucovorin calcium for injection intrathecally.
Serum creatinine and methotrexate levels should be determined at 24 hour intervals. If the 24 hour serum creatinine has increased 50% over baseline or if the 24 hour methotrexate level is greater than 5 x 10-6 M or the 48 hour level is greater than 9 x 10-7 M, the dose of leucovorin calcium for injection should be increased to 100 mg/m2 IV every 3 hours until the methotrexate level is less than 10-8 M.
Hydration (3 L/d) and urinary alkalization with sodium bicarbonate solution should be employed concomitantly. The bicarbonate dose should be adjusted to maintain the urine pH at 7.0 or greater.
Megaloblastic Anemia Due to Folic Acid Deficiency
Up to 1 mg daily. There is no evidence that doses greater than 1 mg/day have greater efficacy than those of 1 mg; additionally, loss of folate in urine becomes roughly logarithmic as the amount administered exceeds 1 mg.
Each 50, 100, and 200 mg vial of leucovorin calcium for injection when reconstituted with 5, 10, and 20 mL, respectively, of sterile diluent yields a leucovorin concentration of 10 mg per mL. Each 350 mg vial of leucovorin calcium for injection when reconstituted with 17.5 mL of sterile diluent yields a leucovorin concentration of 20 mg per mL. Leucovorin calcium for injection contains no preservative. Reconstitute the lyophilized vial product with Bacteriostatic Water for Injection, USP (benzyl alcohol preserved), or Sterile Water for Injection, USP. When reconstituted with Bacteriostatic Water for Injection, USP, the resulting solution must be used within 7 days. If the product is reconstituted with Sterile Water for Injection, USP, use immediately and discard any unused portion.
Because of the benzyl alcohol contained in Bacteriostatic Water for Injection, USP, when doses greater than 10 mg/m2 are administered, leucovorin calcium for injection should be reconstituted with Sterile Water for Injection, USP, and used immediately (see WARNINGS). Because of the calcium content of the leucovorin solution, no more than 160 mg of leucovorin should be injected intravenously per minute (16 mL of a 10 mg/mL, or 8 mL of a 20 mg/mL solution per minute).
Parenteral products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Leucovorin calcium for injection should not be mixed in the same infusion as 5-fluorouracil, since this may lead to the formation of a precipitate.
-
HOW SUPPLIED
Leucovorin Calcium for Injection, USP is supplied as follows:
NDC Leucovorin Calcium for Injection, USP Package Factor 25021-813-10 50 mg Single-Dose Vial 10 vials per carton 25021-814-30 100 mg Single-Dose Vial 1 vial per carton 25021-815-30 200 mg Single-Dose Vial 1 vial per carton 25021-816-30 350 mg Single-Dose Vial 1 vial per carton Storage Conditions
Store dry product and reconstituted solution at 20° to 25°C (68° to 77°F); excursions permitted between 15° and 30°C (59° and 86°F). [See USP Controlled Room Temperature.]
Protect from light. Retain in carton until time of use.
Lyophilized.
Sterile, Nonpyrogenic, Preservative-free.
The container closure is not made with natural rubber latex.
SAGENT®
Mfd. for SAGENT Pharmaceuticals
Schaumburg, IL 60195 (USA)
Made in Italy
©2019 Sagent Pharmaceuticals, Inc.September 2019
SAGENT Pharmaceuticals®
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LEUCOVORIN CALCIUM
leucovorin calcium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25021-813 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength leucovorin calcium (UNII: RPR1R4C0P4) (leucovorin - UNII:Q573I9DVLP) leucovorin 50 mg in 5 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25021-813-10 10 in 1 CARTON 09/15/2012 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200753 09/15/2012 LEUCOVORIN CALCIUM
leucovorin calcium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25021-814 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength leucovorin calcium (UNII: RPR1R4C0P4) (leucovorin - UNII:Q573I9DVLP) leucovorin 100 mg in 10 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25021-814-30 1 in 1 CARTON 09/15/2012 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200753 09/15/2012 LEUCOVORIN CALCIUM
leucovorin calcium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25021-815 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength leucovorin calcium (UNII: RPR1R4C0P4) (leucovorin - UNII:Q573I9DVLP) leucovorin 200 mg in 20 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25021-815-30 1 in 1 CARTON 09/15/2012 1 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200753 09/15/2012 LEUCOVORIN CALCIUM
leucovorin calcium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25021-816 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength leucovorin calcium (UNII: RPR1R4C0P4) (leucovorin - UNII:Q573I9DVLP) leucovorin 350 mg in 17.5 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25021-816-30 1 in 1 CARTON 09/15/2012 1 17.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200855 09/15/2012 Labeler - Sagent Pharmaceuticals (796852890)