FOSINOPRIL SODIUM AND HYDROCHLOROTHIAZIDE - fosinopril sodium and hydrochlorothiazide tablet
Rising Pharma Holdings, Inc.
----------
Fosinopril Sodium and Hydrochlorothiazide Tablets, USP
Rx only
WARNING: FETAL TOXICITY
- When pregnancy is detected, discontinue fosinopril and hydrochlorothiazide as soon as possible.
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus. See WARNINGS: Fetal Toxicity
DESCRIPTION
Fosinopril sodium, USP is a white to almost white powder, soluble (>100 mg/mL) in water, in ethanol, and in methanol, and slightly soluble in hexane. Fosinopril sodium is designated chemically as L-proline, 4-cyclohexyl-1-[[[2-methyl-1-(1-oxopropoxy)propoxy](4-phenylbutyl)phosphinyl]acetyl]-, sodium salt, trans-; its structural formula is:
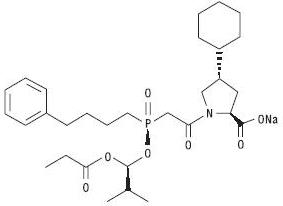
Its molecular formula is C30H45NNaO7P, and its molecular weight is 585.65.
Fosinoprilat, the active metabolite of fosinopril, is a non-sulfhydryl angiotensin-converting enzyme inhibitor. Fosinopril is converted to fosinoprilat by hepatic cleavage of the ester group.
Hydrochlorothiazide, USP is a white or practically white, practically odorless, crystalline powder. It is slightly soluble in water; freely soluble in sodium hydroxide solution, in n-butylamine, and in dimethylformamide; sparingly soluble in methanol; and insoluble in ether, in chloroform, and in dilute mineral acids. Hydrochlorothiazide is designated chemically as 6-chloro-3,4-dihydro-2H-1,2,4-benzothiadiazine-7-sulfonamide 1,1-dioxide; its structural formula is:
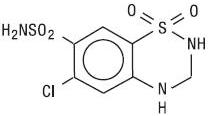
Its molecular formula is C7H8ClN3O4S2, and its molecular weight is 297.73. Hydrochlorothiazide is a thiazide diuretic.
Fosinopril sodium and hydrochlorothiazide tablets, USP are a combination of fosinopril sodium, USP and hydrochlorothiazide, USP. They are available for oral use in two tablet strengths: fosinopril sodium and hydrochlorothiazide tablets, USP 10 mg/12.5 mg, containing 10 mg of fosinopril sodium, USP and 12.5 mg of hydrochlorothiazide, USP; and fosinopril sodium and hydrochlorothiazide tablets, USP 20 mg/12.5 mg, containing 20 mg of fosinopril sodium, USP and 12.5 mg of hydrochlorothiazide, USP. The inactive ingredients of the tablets include lactose anhydrous, ferric oxide red, ferric oxide yellow, croscarmellose sodium, povidone, isopropyl alcohol, glyceryl distearate, and sodium lauryl sulfate.
CLINICAL PHARMACOLOGY
Mechanism of Action
Fosinopril and fosinoprilat inhibit angiotensin-converting enzyme (ACE) in human subjects and in animals. ACE is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor substance, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex. Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. The latter decrease may result in a small increase of serum potassium. Hypertensive patients treated with fosinopril alone for an average of 8 weeks had elevations of serum potassium of approximately 0.1 mEq/L. Similar patients treated with hydrochlorothiazide alone had a mean reduction in serum potassium of 0.15 mEq/L, while patients who received combined treatment with 10/12.5 mg or 20/12.5 mg of fosinopril and hydrochlorothiazide had reductions of 0.07 and 0.03 mEq/L, respectively. (See PRECAUTIONS.)
Removal of angiotensin II negative feedback on renin secretion leads to increased plasma renin activity.
ACE is identical to kininase, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effects of fosinopril sodium and hydrochlorothiazide tablets remains to be elucidated.
While the mechanism through which fosinopril lowers blood pressure is believed to be primarily suppression of the renin-angiotensin-aldosterone system, fosinopril has an antihypertensive effect even in patients with low-renin hypertension.
Hydrochlorothiazide is a thiazide diuretic. Thiazides affect the renal tubular mechanisms of electrolyte reabsorption, directly increasing excretion of sodium and chloride in approximately equivalent amounts. Indirectly, the diuretic action of hydrochlorothiazide reduces plasma volume, with consequent increases in plasma renin activity, increases in aldosterone secretion, increases in urinary potassium loss, and decreases in serum potassium. The renin-aldosterone link is mediated by angiotensin, so coadministration of an ACE inhibitor tends to reverse the potassium loss associated with these diuretics.
The mechanism of the antihypertensive effect of thiazides is unknown.
Pharmacokinetics and Metabolism
The absolute absorption of fosinopril averages 36% of an oral dose. The primary site of absorption is the proximal small intestine. While the rate of absorption may be slowed by the presence of food in the gastrointestinal tract, the extent of absorption of fosinopril is essentially unaffected.
Following oral administration of hydrochlorothiazide, peak plasma concentrations are achieved in 1 to 2.5 hours, and the extent of absorption is 50 to 80%. The reported studies of food effects on hydrochlorothiazide absorption have been inconclusive. The absorption of hydrochlorothiazide is increased by agents that reduce gastrointestinal motility. It is reported to be decreased by 50% in patients with congestive heart failure.
Cleavage of the ester group (primarily in the liver) converts fosinopril to its active metabolite, fosinoprilat. The time to peak plasma concentrations of fosinoprilat is about 3 hours, independent of the administered dose of fosinopril. In patients with hepatic dysfunction due to cirrhosis, conversion of fosinopril to fosinoprilat may be slowed, but the extent of this conversion is unchanged.
Fosinoprilat is highly protein bound (95%), but has negligible binding to cellular components of blood. The peak serum concentration and the area under the concentration-time curve of fosinoprilat is directly proportional to the administered dose of fosinopril.
After an oral dose of radiolabeled fosinopril, 75% of radioactivity in plasma was present as active fosinoprilat, 20 to 30% as a glucuronide conjugate of fosinoprilat, and 1 to 5% as a p-hydroxy metabolite of fosinoprilat. Since fosinoprilat is not biotransformed after intravenous administration, fosinopril, not fosinoprilat, appears to be the precursor for the glucuronide and p-hydroxy metabolites. In rats, the p-hydroxy metabolite of fosinoprilat is as potent an inhibitor of ACE as fosinoprilat; the glucuronide conjugate is devoid of ACE inhibitory activity.
Studies in animals indicate that fosinopril and fosinoprilat do not cross the blood-brain barrier, but fosinoprilat does cross the placenta of pregnant animals. In humans, hydrochlorothiazide crosses the placenta freely, and levels in umbilical-cord blood are similar to those in the maternal circulation.
Hydrochlorothiazide is not metabolized. Its apparent volume of distribution is 3.6 to 7.8 L/kg, and its measured plasma protein binding is 67.9%. The drug also accumulates in red blood cells, so that whole blood levels are 1.6 to 1.8 times those measured in plasma.
After intravenous administration, fosinoprilat is eliminated approximately equally by the liver and kidney. After oral administration of radiolabeled fosinopril, approximately half of the absorbed dose is excreted in the urine and the remainder is excreted in the feces. In two studies involving healthy subjects, the mean body clearance of intravenous fosinoprilat was between 26 and 39 mL/min.
In hypertensive patients with normal renal and hepatic function, the effective half-life of accumulation of fosinoprilat following multiple dosing of fosinopril sodium is 11.5 hours. Thus, steady-state concentrations of fosinoprilat should be reached after 2 or 3 doses of fosinopril sodium and hydrochlorothiazide given once daily.
In patients with renal insufficiency (creatinine clearance <80 mL/min/1.73 m2), the total body clearance of fosinoprilat is approximately one half of that in patients with normal renal function, while absorption, bioavailability, and protein binding are not appreciably altered. The clearance of fosinoprilat does not differ appreciably with the degree of renal insufficiency, because the diminished renal elimination is offset by increased hepatobiliary elimination. A modest increase in plasma AUC levels (less than two times that in normals) was observed in patients with various degrees of renal insufficiency, including end-stage renal failure (creatinine clearance <10 mL/min/1.73 m2). (See DOSAGE AND ADMINISTRATION.)
Fosinopril is not well dialyzed. Clearance of fosinoprilat by hemodialysis and peritoneal dialysis averages 2% and 7%, respectively, of urea clearances.
In patients with hepatic insufficiency (alcoholic or biliary cirrhosis), the apparent total body clearance of fosinoprilat is approximately one half of that in patients with normal hepatic function.
In elderly (male) subjects (65 to 74 years old) with clinically normal renal and hepatic function, there appear to be no significant differences in pharmacokinetic parameters for fosinoprilat compared to those of younger subjects (20 to 35 years old).
Thiazide diuretics are eliminated by the kidney, with a terminal half-life of 5 to 15 hours. In a study of patients with impaired renal function (mean creatinine clearance of 19 mL/min), the half-life of hydrochlorothiazide elimination was lengthened to 21 hours.
When fosinopril and hydrochlorothiazide are administered concomitantly, the pharmacokinetics of hydrochlorothiazide are essentially unaffected. Serum levels of fosinoprilat are increased after several weeks of coadministration of hydrochlorothiazide and fosinopril, but the increase is not sufficient to warrant any change in dosing.
Pharmacodynamics
After single doses of 10 to 40 mg of fosinopril, serum ACE activity was inhibited by at least 90% from 2 to 12 hours after dosing. At 24 hours, serum ACE activity remains suppressed by 85 to 93%.
Administration of fosinopril to patients with mild to moderate hypertension results in a reduction of both supine and standing blood pressure to about the same extent with no compensatory tachycardia. In studies in hypertensive patients after three months of fosinopril monotherapy, hemodynamic responses to various stimuli (isometric exercise, 45° head-up tilt, mental challenges) were unchanged compared to baseline, suggesting that fosinopril did not affect the activity of the sympathetic nervous system. Instead, fosinopril-induced reduction in blood pressure appears to be mediated by a reduction in peripheral vascular resistance without reflex cardiac effects. In similar studies, renal, splanchnic, cerebral, and skeletal-muscle blood flows were all unchanged compared to baseline, as was glomerular filtration rate. Symptomatic postural hypotension is infrequent, although it can occur in patients who are salt- and/or volume-depleted (see WARNINGS).
Following oral administration of single doses of 10 to 40 mg, fosinopril lowered blood pressure within one hour, with peak reductions achieved 2 to 6 hours after dosing. The antihypertensive effect of a single dose persisted for 24 hours. Following four weeks of monotherapy in placebo-controlled trials in patients with mild to moderate hypertension, once-daily doses of 20 to 80 mg lowered supine or seated blood pressures (systolic/diastolic) 24 hours after dosing by an average of 8 to 9/6 to 7 mmHg more than placebo. The trough effect was about 50 to 60% of the peak diastolic response and about 80% of the peak systolic response.
In clinical studies of various combinations that included 0 to 40 mg of fosinopril and 0 to 37.5 mg of hydrochlorothiazide, the antihypertensive effects were increased with increasing dose of either component. Peak blood pressure reductions were achieved 2 to 6 hours after dosing. The mean reductions in seated blood pressure (systolic/diastolic) associated with fosinopril sodium and hydrochlorothiazide tablets 10/12.5 after 24 hours were 9 to 18/5 to 7 mmHg greater than those associated with placebo; those associated with fosinopril sodium and hydrochlorothiazide 20/12.5 after 24 hours were 12 to 17/8 to 10 mmHg greater than those associated with placebo. These trough effects were 60 to 90% of the corresponding peak effects.
Although hydrochlorothiazide tends to be more effective in low-renin hypertensive patients (mainly blacks), and fosinopril—like other ACE inhibitors—tends to be more effective in high-renin patients (mainly non-blacks), the effectiveness of fosinopril sodium and hydrochlorothiazide is independent of race, age, and gender.
INDICATIONS AND USAGE
Fosinopril sodium and hydrochlorothiazide tablets are indicated for the treatment of hypertension.
These fixed dose combinations are not indicated for initial therapy. (See DOSAGE AND ADMINISTRATION.)
In using fosinopril sodium and hydrochlorothiazide, consideration should be given to the fact that another angiotensin-converting enzyme inhibitor, captopril, has caused agranulocytosis, particularly in patients with renal impairment or collagen-vascular disease. Available data are insufficient to show that fosinopril does not have a similar risk (see WARNINGS: Neutropenia/Agranulocytosis).
ACE inhibitors (for which adequate data are available) cause a higher rate of angioedema in black than in non-black patients (see WARNINGS: Head and Neck Angioedema and Intestinal Angioedema).
CONTRAINDICATIONS
Fosinopril sodium and hydrochlorothiazide tablets are contraindicated in patients who are anuric. Fosinopril sodium and hydrochlorothiazide is also contraindicated in patients who are hypersensitive to fosinopril, to any other ACE inhibitor, to hydrochlorothiazide, or other sulfonamide-derived drugs, or any other ingredient or component in the formulation. Hypersensitivity reactions are more likely to occur in patients with a history of allergy or bronchial asthma.
WARNINGS
Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors (including fosinopril sodium and hydrochlorothiazide) may be subject to a variety of adverse reactions, some of them serious.
Head and Neck Angioedema
Angioedema of the face, extremities, lips, tongue, glottis, and larynx has been reported in patients treated with angiotensin-converting enzyme inhibitors. Angioedema associated with laryngeal edema can be fatal. If laryngeal stridor or angioedema of the face, tongue, or glottis occurs, treatment with fosinopril sodium and hydrochlorothiazide should be discontinued and appropriate therapy instituted immediately. When involvement of the tongue, glottis, or larynx appears likely to cause airway obstruction, appropriate therapy, e.g., subcutaneous epinephrine injection 1:1000 (0.3 to 0.5 mL) should be promptly administered (see PRECAUTIONS and ADVERSE REACTIONS).
Intestinal Angioedema
Intestinal angioedema has been reported in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor. Intestinal angioedema should be included in the differential diagnosis of patients on ACE inhibitors presenting with abdominal pain.
Anaphylactoid Reactions During Desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions were avoided when ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Anaphylactoid Reactions During Membrane Exposure
Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
Hypotension
Fosinopril sodium and hydrochlorothiazide can cause symptomatic hypotension. Like other ACE inhibitors, fosinopril has been only rarely associated with hypotension in uncomplicated hypertensive patients. Symptomatic hypotension is most likely to occur in patients who have been volume- and/or salt-depleted as a result of prolonged diuretic therapy, dietary salt restriction, dialysis, diarrhea, or vomiting. Volume and/or salt depletion should be corrected before initiating therapy with fosinopril sodium and hydrochlorothiazide.
Fosinopril sodium and hydrochlorothiazide tablets should be used cautiously in patients receiving concomitant therapy with other antihypertensives. The thiazide component of fosinopril sodium and hydrochlorothiazide may potentiate the action of other antihypertensive drugs, especially ganglionic or peripheral adrenergic-blocking drugs. The antihypertensive effects of the thiazide component may also be enhanced in the post-sympathectomy patient.
In patients with congestive heart failure, with or without associated renal insufficiency, ACE inhibitor therapy may cause excessive hypotension, which may be associated with oliguria, azotemia, and (rarely) with acute renal failure and death. In such patients, fosinopril sodium and hydrochlorothiazide therapy should be started under close medical supervision; they should be followed closely for the first 2 weeks of treatment and whenever the dose of fosinopril or diuretic is increased.
If hypotension occurs, the patient should be placed in a supine position, and, if necessary, treated with intravenous infusion of physiological saline. Fosinopril sodium and hydrochlorothiazide treatment usually can be continued following restoration of blood pressure and volume.
Impaired Renal Function
Fosinopril sodium and hydrochlorothiazide should be used with caution in patients with severe renal disease. Thiazides may precipitate azotemia in such patients, and the effects of repeated dosing may be cumulative.
When the renin-angiotensin-aldosterone system is inhibited by ACE inhibitors, changes in renal function may be anticipated in susceptible individuals. In patients with severe congestive heart failure, whose renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with angiotensin-converting enzyme inhibitors (including fosinopril) may be associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death.
In some studies of hypertensive patients with unilateral or bilateral renal artery stenosis, treatment with ACE inhibitors has been associated with increases in blood urea nitrogen and serum creatinine; these increases were reversible upon discontinuation of ACE inhibitor therapy, concomitant diuretic therapy, or both. When such patients are treated with fosinopril sodium and hydrochlorothiazide, renal function should be monitored during the first few weeks of therapy.
Some ACE-inhibitor-treated hypertensive patients with no apparent preexisting renal vascular disease have developed increases in blood urea nitrogen and serum creatinine, usually minor and transient, especially when the ACE inhibitor has been given concomitantly with a diuretic. Dosage reduction of fosinopril sodium and hydrochlorothiazide may be required. Evaluation of the hypertensive patient should always include assessment of renal function (see DOSAGE AND ADMINISTRATION).
Neutropenia/Agranulocytosis
Another angiotensin-converting enzyme inhibitor, captopril, has been shown to cause agranulocytosis and bone marrow depression, rarely in uncomplicated patients (incidence probably less than once per 10,000 exposures), but more frequently (incidence possibly as great as once per 1,000 exposures) in patients with renal impairment, especially those who also have a collagen-vascular disease such as systemic lupus erythematosus or scleroderma. Available data from clinical trials of fosinopril are insufficient to show that fosinopril does not cause agranulocytosis at similar rates. Monitoring of white blood cell counts should be considered in patients with collagen-vascular disease, especially if the disease is associated with impaired renal function.
Neutropenia/agranulocytosis has also been associated with thiazide diuretics.
Fetal Toxicity
Pregnancy Category D
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue fosinopril and hydrochlorothiazide as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimester of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue fosinopril and hydrochlorothiazide, unless it is considered lifesaving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to fosinopril and hydrochlorothiazide for hypotension, oliguria, and hyperkalemia (see PRECAUTIONS, Pediatric Use).
Intrauterine exposure to thiazide diuretics is associated with fetal or neonatal jaundice, thrombocytopenia, and possibly other adverse reactions that occurred in adults.
No teratogenic effects of fosinopril were seen in studies of pregnant rats and rabbits. On a mg/kg basis, the doses used were up to 180 times (in rats) and one time (in rabbits) the maximum recommended human dose. No teratogenic effects of fosinopril and hydrochlorothiazide were seen in studies of pregnant rats and rabbits. On a mg/kg (fosinopril and hydrochlorothiazide) basis, the doses used were up to 188/94 times (in rats) and 0.6/0.3 times (in rabbits) the maximum recommended human dose.
Impaired Hepatic Function
Rarely, ACE inhibitors have been associated with a syndrome that begins with cholestatic jaundice and progresses to fulminant hepatic necrosis and (sometimes) death. The mechanism of this syndrome is not understood. A patient receiving fosinopril sodium and hydrochlorothiazide who develops jaundice or marked elevation of hepatic enzymes should discontinue fosinopril sodium and hydrochlorothiazide tablets and receive appropriate medical follow-up.
Fosinopril sodium and hydrochlorothiazide should be used with caution in patients with impaired hepatic function or progressive liver disease, since minor alterations of fluid and electrolyte balance may precipitate hepatic coma. Also, since the metabolism of fosinopril to fosinoprilat is normally dependent upon hepatic esterases, patients with impaired liver function could develop elevated plasma levels of fosinopril. In a study of patients with alcoholic or biliary cirrhosis the rate (but not the extent) of hydrolysis to fosinoprilat was reduced. In these patients the clearance of fosinoprilat was reduced, and the area under the fosinoprilat-time curve was approximately doubled.
PRECAUTIONS
General
Derangements of Serum Electrolytes
In clinical trials of fosinopril monotherapy, hyperkalemia (serum potassium at least 10% greater than the upper limit of normal) occurred in approximately 2.6% of hypertensive patients receiving fosinopril. In most cases, these were isolated values which resolved despite continued therapy. Risk factors for the development of hyperkalemia included renal insufficiency, diabetes mellitus, and the concomitant use of potassium-sparing diuretics, potassium supplements, and/or potassium-containing salt substitutes.
Conversely, treatment with thiazide diuretics has been associated with hypokalemia, hyponatremia, and hypochloremic alkalosis. These disturbances have sometimes been manifest as one or more of dryness of mouth, thirst, weakness, lethargy, drowsiness, restlessness, muscle pains or cramps, muscular fatigue, hypotension, oliguria, tachycardia, nausea, and vomiting. Hypokalemia can also sensitize or exaggerate the response of the heart to the toxic effects of digitalis. The risk of hypokalemia is greatest in patients with cirrhosis of the liver, in patients experiencing a brisk diuresis, in patients who are receiving inadequate oral intake of electrolytes, and in patients receiving concomitant therapy with corticosteroids or ACTH.
The opposite effects of fosinopril and hydrochlorothiazide on serum potassium will approximately balance each other in many patients, so that no net effect upon serum potassium will be seen. In other patients, one or the other effect may be dominant. Initial and periodic determinations of serum electrolytes to detect possible electrolyte imbalance should be performed at appropriate intervals.
Chloride deficits are generally mild and require specific treatment only under extraordinary circumstances (e.g., in liver disease or renal disease). Dilutional hyponatremia may occur in edematous patients; appropriate therapy is water restriction rather than administration of salt, except in rare instances when the hyponatremia is life-threatening. In actual salt depletion, appropriate replacement is the therapy of choice.
Calcium excretion is decreased by thiazides. In a few patients on prolonged thiazide therapy, pathological changes in the parathyroid gland have been observed, with hypercalcemia and hypophosphatemia. More serious complications of hyperparathyroidism (renal lithiasis, bone resorption, and peptic ulceration) have not been seen.
Thiazides increase the urinary excretion of magnesium, and hypomagnesemia may result.
Other Metabolic Disturbances
Thiazide diuretics tend to reduce glucose tolerance and to raise serum levels of cholesterol, triglycerides, and uric acid. These effects are usually minor, but frank gout or overt diabetes may be precipitated in susceptible patients.
Cough
Presumably due to the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. ACE inhibitor-induced cough should be considered in the differential diagnosis of cough.
Surgery/Anesthesia
In patients undergoing surgery or during anesthesia with agents that produce hypotension, fosinopril will block the angiotensin II formation that could otherwise occur secondary to compensatory renin release. Hypotension that occurs as a result of this mechanism can be corrected by volume expansion.
Information for Patients
Angioedema
Angioedema, including laryngeal edema, can occur with treatment with ACE inhibitors, especially following the first dose. A patient receiving fosinopril sodium and hydrochlorothiazide should be told to report immediately any signs or symptoms suggesting angioedema (swelling of face, eyes, lips, or tongue, or difficulty in breathing) and to take no more drug until after consulting with the prescribing physician.
Pregnancy
Female patients of childbearing age should be told about the consequences of exposure to fosinopril and hydrochlorothiazide during pregnancy. Discuss treatment options with women planning to become pregnant. Patients should be asked to report pregnancies to their physicians as soon as possible.
Symptomatic Hypotension
A patient receiving fosinopril sodium and hydrochlorothiazide tablets should be cautioned that lightheadedness can occur, especially during the first days of therapy, and that it should be reported to the prescribing physician. The patients should be told that if syncope occurs, fosinopril sodium and hydrochlorothiazide should be discontinued until the physician has been consulted.
All patients should be cautioned that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
Hyperkalemia
A patient receiving fosinopril sodium and hydrochlorothiazide should be told not to use potassium supplements or salt substitutes containing potassium without consulting the prescribing physician.
Neutropenia
Patients should be told to promptly report any indication of infection (e.g., sore throat, fever), which could be a sign of neutropenia.
Non-melanoma Skin Cancer: Instruct patients taking hydrochlorothiazide to protect skin from the sun and undergo regular skin cancer screening.
Laboratory Tests
Therapy with fosinopril sodium and hydrochlorothiazide should be interrupted for a few days before carrying out tests of parathyroid function.
Fosinopril may cause false low measurement of serum digoxin levels when the Digi-Tab® (Nuclear Medical) RIA Kit is used. The accuracy of the Coat-A-Count® (Diagnostic Products Corporation) kit is not affected.
Drug Interactions
Potassium supplements and potassium-sparing diuretics
As noted above (“Derangements of Serum Electrolytes”), the net effect of fosinopril sodium and hydrochlorothiazide may be to elevate a patient’s serum potassium, to reduce it, or to leave it unchanged. Potassium-sparing diuretics (spironolactone, amiloride, triamterene, and others) or potassium supplements can increase the risk of hyperkalemia. If concomitant use of such agents is indicated, they should be given with caution, and the patient’s serum potassium should be monitored frequently.
Lithium
Increased serum lithium levels and symptoms of lithium toxicity have been reported in patients receiving ACE inhibitors during therapy with lithium. Because renal clearance of lithium is reduced by thiazides, the risk of lithium toxicity is presumably raised further when, as in therapy with fosinopril sodium and hydrochlorothiazide tablets, a thiazide diuretic is coadministered with the ACE inhibitor. Fosinopril sodium and hydrochlorothiazide and lithium should be coadministered with caution, and frequent monitoring of serum lithium levels is recommended.
Antacids
In a clinical pharmacology study, serum levels and urinary excretion of fosinoprilat were reduced when fosinopril was coadministered with an antacid (aluminum hydroxide, magnesium hydroxide, and simethicone) suggesting that antacids may impair absorption of fosinopril. If concomitant administration of these agents is indicated, dosing should be separated by 2 hours.
Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting, and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE Inhibitor therapy including fosinopril sodium and hydrochlorothiazide.
Other
The bioavailability of unbound fosinoprilat is not altered by coadministration of fosinopril with aspirin, chlorthalidone, cimetidine, digoxin, metoclopramide, nifedipine, propranolol, propantheline, or warfarin. Other ACE inhibitors have had less than additive effects with beta-adrenergic blockers, presumably because drugs of both classes lower blood pressure by inhibiting parts of the renin-angiotensin system.
Interaction studies with warfarin have failed to identify any clinically important effects of fosinopril on the serum concentration or clinical effects of the anticoagulant.
Insulin requirements in diabetic patients may be increased, decreased, or unchanged.
Thiazides may decrease arterial responsiveness to norepinephrine, but not enough to preclude effectiveness of the pressor agent for therapeutic use.
Thiazides may increase the responsiveness to tubocurarine.
The diuretic, natriuretic, and antihypertensive effects of thiazide diuretics may be reduced by concurrent administration of nonsteroidal anti-inflammatory agents; the effects (if any) of these agents on the antihypertensive effect of fosinopril sodium and hydrochlorothiazide have not been studied.
By alkalinizing the urine, hydrochlorothiazide may decrease the effectiveness of methenamine.
Cholestyramine and Colestipol Resins
Absorption of hydrochlorothiazide is impaired in the presence of anionic exchange resins. Single doses of either cholestyramine or colestipol resins bind the hydrochlorothiazide and reduce its absorption from the gastrointestinal tract by up to 85% and 43%, respectively.
Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Closely monitor blood pressure, renal function and electrolytes in patients on fosinopril and hydrochlorothiazide and other agents that affect the RAS.
Do not co-administer aliskiren with fosinopril and hydrochlorothiazide in patients with diabetes. Avoid use of aliskiren with fosinopril and hydrochlorothiazide in patients with renal impairment (GFR <60 mL/min).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Fosinopril-Hydrochlorothiazide
Reproductive studies and long-term carcinogenicity studies with fosinopril sodium and hydrochlorothiazide have not been conducted. The combination of fosinopril and hydrochlorothiazide was not mutagenic in the Ames microbial mutagen test, the mouse lymphoma forward mutation assay, or the Chinese hamster ovary cell cytogenetic assay. The combination was also not genotoxic in a mouse micronucleus test in vivo.
Fosinopril Sodium
No evidence of a carcinogenicity was found when fosinopril was given in the diet to rats and mice for up to 24 months at doses up to 400 mg/kg/day. On a body weight basis, the highest dose was about 250 times the maximum human dose of 80 mg, given to a 50 kg subject. On a body surface area basis, this dose is 20 (mice) to 40 (rats) times the maximum human dose.
Neither fosinopril nor the fosinoprilat moiety was mutagenic in the Ames microbial mutagen test, the mouse lymphoma forward mutation assay, or a mitotic gene conversion assay. Fosinopril was also not genotoxic in a mouse micronucleus test in vivo and a mouse bone marrow cytogenetic assay in vivo.
In Chinese hamster ovary cell cytogenetic assay, fosinopril increased the frequency of chromosomal aberrations when tested without metabolic activation at a concentration that was toxic to the cells. However, there was no increase in chromosomal aberrations at lower drug concentrations without metabolic activation or at any concentration with metabolic activation.
There were no adverse reproductive effects in male and female rats treated with up to 60 mg/kg daily. At doses 4 times higher, slight increases in pairing time were seen. This higher dose is about 125 (body surface area basis) or 600 (body-weight basis) times greater than the dose received by a 50 kg human receiving 20 mg a day.
Hydrochlorothiazide
Under the auspices of the National Toxicology Program, rats and mice received hydrochlorothiazide for two years at doses up to 100 (rats) and 600 (mice) mg/kg/day. On a body weight basis, these highest doses were about 2400 times (mice) or 400 times (rats) the fosinopril sodium and hydrochlorothiazide dose of 12.5 mg, given to a 50 kg subject. On a body surface area basis, these doses are 226 times (mice) and 82 times (rats) the fosinopril sodium and hydrochlorothiazide dose. These studies uncovered no evidence of carcinogenicity in rats or female mice, but there was equivocal evidence of hepatocarcinogenicity in male mice.
Hydrochlorothiazide was not genotoxic in in vitro assays using strains TA 98, TA 100, TA 1535, TA 1537, and TA 1538 of Salmonella typhimurium (Ames assay); in the Chinese Hamster Ovary (CHO) test for chromosomal aberrations; or in in vivo assays using mouse germinal cell chromosomes; Chinese Hamster bone-marrow chromosomes, and the Drosophila sex-linked recessive lethal trait gene. Using concentrations of hydrochlorothiazide of 43 to 1300 mg/mL, positive test results were obtained in the in vitro CHO Sister Chromatid Exchange (clastogenicity) test and in the Mouse Lymphoma Cell (mutagenicity) assays. Using an unspecified concentration of hydrochlorothiazide, positive test results were also obtained in the Aspergillus nidulans nondisjunction assay.
No adverse effects upon fertility were seen when rats and mice received dietary hydrochlorothiazide prior to mating and throughout gestation at doses up to 4 (rats) and 100 (mice) mg/kg/day. These doses are from 3.2 (body surface area basis in rats) to 400 (weight basis in mice) times greater than the dose received by a 50 kg human receiving 12.5 mg a day.
Nursing Mothers
Both fosinopril and hydrochlorothiazide are excreted in human milk. Because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue fosinopril sodium and hydrochlorothiazide, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of fosinopril sodium and hydrochlorothiazide did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pediatric Use
Neonates with a history of in utero exposure to fosinopril and hydrochlorothiazide:
If oliguria or hypotension occurs, direct attention toward support of blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function. Removal of fosinopril and hydrochlorothiazide, which crosses the placenta, from the neonatal circulation is not significantly accelerated by these means.
Safety and effectiveness in pediatric patients have not been established.
ADVERSE REACTIONS
Fosinopril sodium and hydrochlorothiazide tablets have been evaluated for safety in over 660 patients with hypertension; approximately 137 of these patients were treated for more than one year. The observed adverse events were generally mild, transient, and similar to those seen with fosinopril and hydrochlorothiazide taken separately. There was no relationship between the incidence of side effects and age.
In placebo-controlled clinical trials of fosinopril sodium and hydrochlorothiazide, the usual duration of therapy was two months. Adverse clinical or laboratory events led to discontinuation of therapy by 4.3% of 368 placebo-treated patients and by 3.5% of 660 fosinopril sodium and hydrochlorothiazide-treated patients.
The most common reasons for discontinuation of therapy with fosinopril sodium and hydrochlorothiazide in U.S. studies were headache (0.3%), cough (0.3%; see PRECAUTIONS), and fatigue (0.2%).
The side effects considered probably or possibly related to study drug that occurred in placebo-controlled trials in more than 2% of patients treated with fosinopril sodium and hydrochlorothiazide are shown in the table below.
| | Fosinopril Sodium and Hydrochlorothiazide (N=660) | Placebo (N=368) |
| % | % |
|
| Headache | 7 | 12.8 |
| Cough | 5.6 | 1.1 |
| Fatigue | 3.9 | 2.4 |
| Dizziness | 3.2 | 2.2 |
| Upper Respiratory Infection | 2.3 | 2.7 |
| Musculoskeletal Pain | 2 | 1.9 |
Other side effects considered possibly or probably related to study drug that occurred in controlled trials in 0.5% to <2% of patients treated with fosinopril sodium and hydrochlorothiazide, and rarer but clinically significant events regardless of causal relationship were:
General: Chest pain, weakness, fever, viral infection.
Cardiovascular: Orthostatic hypotension (seen in 1.8% of fosinopril sodium and hydrochlorothiazide patients and 0.3% of placebo patients; no patients discontinued therapy due to orthostatic hypotension), edema, flushing, rhythm disturbance, syncope.
Dermatologic: Pruritus, rash.
Endocrine/Metabolic: Sexual dysfunction, change in libido, breast mass.
Gastrointestinal: Nausea/vomiting, diarrhea, dyspepsia/heartburn, abdominal pain, gastritis/esophagitis.
Immunologic: Angioedema (see WARNINGS: Head and Neck Angioedema and Intestinal Angioedema).
Musculoskeletal: Myalgia/muscle cramps.
Neurologic/Psychiatric: Somnolence, depression, numbness/paresthesia.
Respiratory: Sinus congestion, pharyngitis, rhinitis.
Special Senses: Tinnitus.
Urogenital: Urinary tract infection, urinary frequency, dysuria.
Laboratory Test Abnormalities: Serum electrolytes, uric acid, glucose, magnesium, cholesterol, triglycerides, and calcium (see PRECAUTIONS). Neutropenia.
Antihypertensive monotherapy with fosinopril has been evaluated for safety in more than 1500 patients, of whom approximately 450 patients were treated for a year or more. The observed adverse events included events similar to those seen with fosinopril sodium and hydrochlorothiazide; in addition, the following others have also been reported with fosinopril:
Cardiovascular: Angina, myocardial infarction, cerebrovascular accident, hypertensive crisis, hypotension, claudication.
Dermatologic: Urticaria, photosensitivity.
Endocrine/Metabolic: Gout.
Gastrointestinal: Pancreatitis, hepatitis, dysphagia, abdominal distention, flatulence, appetite/weight change, dry mouth.
Hematologic: Lymphadenopathy.
Musculoskeletal: Arthralgia.
Neurologic/Psychiatric: Memory disturbance, tremor, confusion, mood change, sleep disturbance.
Respiratory: Bronchospasm, laryngitis/hoarseness, epistaxis, and (in two patients) a symptom-complex of cough, bronchospasm, and eosinophilia.
Special Senses: Vision disturbance, taste disturbance, eye irritation.
Urogenital: Renal insufficiency.
Laboratory Test Abnormalities: Elevations (usually transient and minor) of BUN and creatinine have been observed, but these have not been more frequent than in parallel patients treated with placebo. The hemoglobin in fosinopril-treated patients generally decreases by an average of 0.1 g/dL, but this nonprogressive change has never been symptomatic. Leukopenia and eosinophilia have also been reported.
Serum levels of liver function tests (transaminases, LDH, alkaline phosphatase and serum bilirubin) have occasionally been found to be elevated, and these elevations have lead to discontinuation of therapy in 0.7% of patients. Other risk factors for liver dysfunction have often been present in these cases; in any event the elevations generally have resolved after discontinuation of therapy with fosinopril.
Other Adverse Events Reported with ACE Inhibitors
Other adverse effects reported with ACE inhibitors include cardiac arrest; pancytopenia, hemolytic anemia; aplastic anemia; thrombocytopenia; bullous pemphigus, exfoliative dermatitis; and a syndrome that may include one or more of arthralgia/arthritis, vasculitis, serositis, myalgia, fever, rash or other dermopathy, positive ANA titer, leukocytosis, eosinophilia, and elevated ESR.
Hydrochlorothiazide has now been extensively prescribed for many years, but there has not been enough systematic collection of data to support an estimate of the frequency of the observed adverse reactions. Within organ-system groups, the reported reactions are listed here in decreasing order of severity, without regard to frequency.
Cardiovascular: Orthostatic hypotension (may be potentiated by alcohol, barbiturates, or narcotics).
Gastrointestinal: Orthostatic hypotension (may be potentiated by alcohol, barbiturates, or narcotics).
Pancreatitis, jaundice (intrahepatic cholestatic), sialadenitis, vomiting, diarrhea, cramping, nausea, gastric irritation, constipation, and anorexia.
Hematologic: Aplastic anemia, agranulocytosis, leukopenia, thrombocytopenia, and hemolytic anemia.
Immunologic: Necrotizing angiitis, Stevens-Johnson syndrome, respiratory distress (including pneumonitis and pulmonary edema), anaphylactic reactions, purpura, urticaria, rash, and photosensitivity.
Metabolic: Hyperglycemia, glycosuria, and hyperuricemia.
Musculoskeletal: Muscle spasm.
Neurologic: Vertigo, lightheadedness, transient blurred vision, headache, paresthesia, xanthopsia, weakness, and restlessness.
Non-melanoma Skin Cancer
Hydrochlorothiazide is associated with an increased risk of non-melanoma skin cancer. In a study conducted in the Sentinel System, increased risk was predominantly for squamous cell carcinoma (SCC) and in white patients taking large cumulative doses. The increased risk for SCC in the overall population was approximately 1 additional case per 16,000 patients per year, and for white patients taking a cumulative dose of ≥50,000mg the risk increase was approximately 1 additional SCC case for every 6,700 patients per year.
OVERDOSAGE
To obtain up-to-date information about the treatment of overdose, a good resource is a certified Regional Poison Control Center. Telephone numbers of certified poison control centers are listed in the Physicians’ Desk Reference (PDR). In managing overdose, consider the possibilities of multiple-drug overdoses, drug-drug interactions, and unusual drug kinetics in your patient.
No specific information is available on the treatment of overdosage with fosinopril sodium and hydrochlorothiazide tablets; treatment should be symptomatic and supportive. Therapy with fosinopril sodium and hydrochlorothiazide should be discontinued, and the patient should be observed. Dehydration, electrolyte imbalance, and hypotension should be treated by established procedures.
In rats, single oral doses of 2600 mg/kg of fosinopril were associated with significant lethality. In single-dose studies of hydrochlorothiazide, most rats survived doses of up to 2750 mg/kg. Both doses are more than 6000 times (on a mg/kg basis) the maximum recommended daily dose of either fosinopril or hydrochlorothiazide in fosinopril sodium and hydrochlorothiazide.
Data from human overdoses of fosinopril are scanty, but the most common manifestation of human fosinopril overdosage is likely to be hypotension. In human hydrochlorothiazide overdose, the most common signs and symptoms observed have been those of dehydration and electrolyte depletion (hypokalemia, hypochloremia, hyponatremia). If digitalis has also been administered, hypokalemia may accentuate cardiac arrhythmias.
Laboratory determinations of serum levels of fosinopril and its metabolites are not widely available, and such determinations have, in any event, no established role in the management of fosinopril overdose. No data are available to suggest physiological maneuvers (e.g., maneuvers to change the pH of the urine) that might accelerate elimination of fosinopril and its metabolites. Fosinoprilat is poorly removed from the body by hemodialysis or peritoneal dialysis.
Angiotensin II could presumably serve as a specific antagonist-antidote in the setting of fosinopril overdose, but angiotensin II is essentially unavailable outside of scattered research facilities. Because the hypotensive effect of fosinopril is achieved through vasodilation and effective hypovolemia, it is reasonable to treat fosinopril overdose by infusion of normal saline solution.
DOSAGE AND ADMINISTRATION
Fosinopril is an effective treatment of hypertension in once-daily doses of 10 to 80 mg, while hydrochlorothiazide is effective in doses of 12.5 to 50 mg per day. In clinical trials of fosinopril/hydrochlorothiazide combination therapy using fosinopril doses of 2.5 to 40 mg and hydrochlorothiazide doses at 5 to 37.5 mg, the antihypertensive effects increased with increasing dose of either component.
The hazards (see WARNINGS) of fosinopril are generally rare and apparently independent of dose; those of hydrochlorothiazide are a mixture of dose-dependent phenomena (primarily hypokalemia) and dose-independent phenomena (e.g., pancreatitis), the former much more common than the latter. Therapy with any combination of fosinopril and hydrochlorothiazide will be associated with both sets of dose-independent hazards. To minimize dose-independent hazards, it is usually appropriate to begin combination therapy only after a patient has failed to achieve the desired effect with monotherapy.
Dose Titration by Clinical Effect
A patient whose blood pressure is not adequately controlled with fosinopril or hydrochlorothiazide monotherapy may be switched to combination therapy with fosinopril sodium and hydrochlorothiazide tablets. Dosage must be guided by clinical response; controlled clinical trials showed that the addition of 12.5 mg of hydrochlorothiazide to 10 to 20 mg of fosinopril will typically be associated with additional reduction in seated diastolic blood pressure at 24 hours after dosing. On average, the effect of the combination of 10 mg of fosinopril with 12.5 mg of hydrochlorothiazide was similar to the effect seen with monotherapy using either 40 mg of fosinopril or 37.5 mg of hydrochlorothiazide.
Use in Renal Impairment
In patients with severe renal impairment (creatinine clearance is <30 mL/min/1.73 m2, serum creatine roughly ≥3 mg/dL or 265 µmol/L), loop diuretics are preferred to thiazides, so fosinopril sodium and hydrochlorothiazide tablets are not recommended. In patients with lesser degrees of renal impairment, fosinopril sodium and hydrochlorothiazide tablets may be used with no change in dosage.
HOW SUPPLIED
Fosinopril Sodium and Hydrochlorothiazide Tablets USP, 10 mg/12.5 mg are peach colored, round biconvex, uncoated tablets debossed with ‘C 84’ on one side and plain on the other side.
Bottle of 30 NDC 57237-026-30
Bottle of 100 NDC 57237-026-01
Fosinopril Sodium and Hydrochlorothiazide Tablets USP, 20 mg/12.5 mg are peach colored, round biconvex, uncoated tablets debossed with ‘C 85’ on one side and “deep score line” on the other side.
Bottle of 30 NDC 57237-027-30
Bottle of 100 NDC 57237-027-01
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from moisture by keeping bottle tightly closed.
Distributed by:
Rising Health, LLC
Saddle Brook, NJ 07663
Made in India
Code: TS/DRUGS/19/1993
Revised: 04/2020


| FOSINOPRIL SODIUM AND HYDROCHLOROTHIAZIDE
fosinopril sodium and hydrochlorothiazide tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FOSINOPRIL SODIUM AND HYDROCHLOROTHIAZIDE
fosinopril sodium and hydrochlorothiazide tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Rising Pharma Holdings, Inc. (116880195) |
| Registrant - Aurobindo Pharma Limited (650082092) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Aurobindo Pharma Limited | 918917642 | ANALYSIS(57237-026, 57237-027) , MANUFACTURE(57237-026, 57237-027) | |