DORIBAX- doripenem powder, for solution
Shionogi Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to useDORIBAX® safely and effectively. See full prescribing information for DORIBAX®.
DORIBAX® (doripenem for injection), for intravenous use Initial U.S. Approval: 2007 INDICATIONS AND USAGEDORIBAX ® is a penem antibacterial indicated in the treatment of the following infections caused by designated susceptible bacteria:
To reduce the development of drug-resistant bacteria and maintain the effectiveness of DORIBAX ® and other antibacterial drugs, DORIBAX ® should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.3) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS250 mg single use vial or 500 mg single use vial of sterile doripenem powder for reconstitution ( 3) CONTRAINDICATIONSPatients with known serious hypersensitivity to doripenem or to other drugs in the same class or patients who have demonstrated anaphylactic reactions to beta-lactams ( 4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (≥ 5%) are headache, nausea, diarrhea, rash and phlebitis.( 6)
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 5/2019 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Complicated Intra-Abdominal Infections
DORIBAX ® (doripenem for injection) is indicated as a single agent for the treatment of complicated intra-abdominal infections caused by Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Bacteroides caccae, Bacteroides fragilis, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides vulgatus, Streptococcus intermedius, Streptococcus constellatus and Peptostreptococcus micros.
1.2 Complicated Urinary Tract Infections, Including Pyelonephritis
DORIBAX ® (doripenem for injection) is indicated as a single agent for the treatment of complicated urinary tract infections, including pyelonephritis caused by Escherichia coli including cases with concurrent bacteremia, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, and Acinetobacter baumannii.
1.3 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of DORIBAX ® and other antibacterial drugs, DORIBAX ® should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting and modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of DORIBAX ® is 500 mg administered every 8 hours by intravenous infusion over one hour in patients ≥18 years of age. The recommended dosage and administration by infection is described in Table 1:
| Infection | Dosage | Frequency | Infusion Time (hours) | Duration |
| Complicated intra-abdominal infection | 500 mg | every 8 hours | 1 | 5–14 days * |
| Complicated UTI, including pyelonephritis | 500 mg | every 8 hours | 1 | 10 days * † |
2.2 Patients with Renal Impairment
|
|
| Estimated CrCl (mL/min) | Recommended Dosage Regimen of DORIBAX® |
| > 50 | No dosage adjustment necessary |
| ≥ 30 to ≤ 50 | 250 mg * administered intravenously (over 1 hour) every 8 hours |
| > 10 to < 30 | 250 mg * administered intravenously (over 1 hour) every 12 hours |
The following formula may be used to estimate CrCl. The serum creatinine used in the formula should represent a steady state of renal function.
| Males: Creatinine clearance (mL/min) = |
weight (kg) × (140 -
age in years)
72 × serum creatinine (mg/dL) |
| Females: Creatinine clearance (mL/min) = | 0.85 × value calculated for males |
DORIBAX ® is hemodialyzable; however, there is insufficient information to make dose adjustment recommendations in patients on hemodialysis.
2.3 Preparation of Solutions
DORIBAX ® does not contain a bacteriostatic preservative. Aseptic technique must be followed in preparation of the infusion solution.
To prepare DORIBAX ® infusions in Baxter Minibag Plus™ infusion bags consult the infusion bag manufacturer's instructions.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to use whenever solution and container permit. DORIBAX ® infusions range from clear, colorless solutions to solutions that are clear and slightly yellow. Variations in color within this range do not affect the potency of the product.
Preparation of 500 mg DORIBAX® dose using the 500 mg vial
- Constitute the 500 mg vial with 10 mL of sterile water for injection or 0.9% sodium chloride injection (normal saline) and gently shake to form a suspension. The resultant concentration is approximately 50 mg/mL.
CAUTION: THE CONSTITUTED SUSPENSION IS NOT FOR DIRECT INJECTION.
- Withdraw the suspension using a syringe with a 21 gauge needle and add it to an infusion bag containing 100 mL of normal saline or 5% dextrose; gently shake until clear. The final infusion solution concentration is approximately 4.5 mg/mL.
Preparation of 250 mg DORIBAX® dose using the 250 mg vial
- Constitute the 250 mg vial with 10 mL of sterile water for injection or 0.9% sodium chloride injection (normal saline) and gently shake to form a suspension. The resultant concentration is approximately 25 mg/mL.
CAUTION: THE CONSTITUTED SUSPENSION IS NOT FOR DIRECT INJECTION.
- Withdraw the suspension using a syringe with a 21 gauge needle and add it to an infusion bag containing either 50 or 100 mL of normal saline or 5% dextrose; gently shake until clear. The final infusion solution concentration is approximately 4.2 mg/mL (50 mL infusion bag) or approximately 2.3 mg/mL (100 mL infusion bag).
Preparation of 250 mg DORIBAX® dose using the 500 mg vial
- Constitute the 500 mg vial with 10 mL of sterile water for injection or 0.9% sodium chloride injection (normal saline) and gently shake to form a suspension. The resultant concentration is approximately 50 mg/mL.
CAUTION: THE CONSTITUTED SUSPENSION IS NOT FOR DIRECT INJECTION.
- Withdraw the suspension using a syringe with a 21 gauge needle and add it to an infusion bag containing 100 mL of normal saline or 5% dextrose; gently shake until clear.
- Remove 55 mL of this solution from the bag and discard.
- Infuse the remaining solution, which contains 250 mg (approximately 4.5 mg/mL).
2.4 Compatibility
The compatibility of DORIBAX ® with other drugs has not been established. DORIBAX ® should not be mixed with or physically added to solutions containing other drugs.
2.5 Storage of Constituted Solutions
Upon constitution with sterile water for injection or 0.9% sodium chloride (normal saline) injection, DORIBAX ® suspension in the vial may be held for 1-hour prior to transfer and dilution in the infusion bag.
Following dilution of the suspension with normal saline or 5% dextrose, DORIBAX ® infusions stored at room temperature or under refrigeration should be completed according to the times in Table 3.
| Infusion prepared in | Stability Time at Room Temp. (includes room temperature storage and infusion time) | Stability time at 2–8°C (Refrigeration) (includes refrigerator storage and infusion time) |
| Normal saline | 12 hours | 72 hours |
| 5% Dextrose | 4 hours | 24 hours |
Constituted DORIBAX ® suspension or DORIBAX ® infusion should not be frozen. This storage information applies also to DORIBAX ® diluted in Baxter Minibag Plus™.
3 DOSAGE FORMS AND STRENGTHS
Single use clear glass vials containing 250 mg or 500 mg (on an anhydrous basis) of sterile doripenem powder for reconstitution.
4 CONTRAINDICATIONS
DORIBAX ® is contraindicated in patients with known serious hypersensitivity to doripenem or to other drugs in the same class or in patients who have demonstrated anaphylactic reactions to beta-lactams.
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality in Ventilator-Associated Bacterial Pneumonia
In a clinical trial of patients with ventilator-associated bacterial pneumonia comparing doripenem to imipenem, more subjects receiving doripenem died 23% (31/135) compared to those receiving imipenem 16.7% (22/132) based on 28-day all-cause mortality in the intent-to-treat (ITT) population. Clinical response rates were also lower in the doripenem arm. Doripenem is not approved for the treatment of ventilator-associated bacterial pneumonia.
5.2 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) and serious skin reactions have been reported in patients receiving beta-lactam antibiotics. These reactions are more likely to occur in individuals with a history of sensitivity to multiple allergens. Before therapy with DORIBAX ® is instituted, careful inquiry should be made to determine whether the patient has had a previous hypersensitivity reaction to other carbapenems, cephalosporins, penicillins or other allergens. If this product is to be given to a penicillin- or other beta-lactam-allergic patient, caution should be exercised because cross-reactivity among beta-lactam antibiotics has been clearly documented.
If an allergic reaction to DORIBAX ® occurs, discontinue the drug. Serious acute hypersensitivity (anaphylactic) reactions require emergency treatment, as clinically indicated.
5.3 Seizures
Seizures have been reported during treatment with doripenem (see section 6.2). In clinical trials, doripenem-treated patients with pre-existing central nervous system (CNS) disorders (e.g. stroke or history of seizures), patients with compromised renal function and patients given doses greater than 500 mg every 8 hours appear to be at greater risk for developing seizures.
5.4 Interaction with Valproic Acid
Due to a drug interaction, patients with seizure disorders controlled with valproic acid or sodium valproate will be at an increased risk for breakthrough seizures when treated with DORIBAX ® concomitantly. Reduction in serum valproic acid concentrations to below the therapeutic concentration range (50 to 100 mcg/mL) was observed by 12 hours after the initiation of doripenem in healthy subjects co-administered both drugs. A similar drug interaction involving other carbapenem antibacterials and valproic acid has been described in published case reports. In some of these reports, increasing the dose of valproic acid or sodium valproate did not result in increased valproic acid serum concentrations. Alternative antibacterial therapies should be considered for patients receiving valproic acid or sodium valproate. If administration of DORIBAX ® is necessary, supplemental anti-convulsant therapy should be considered. [ see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]
5.5 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported with nearly all antibacterial agents and may range in severity from mild diarrhea to fatal colitis.
Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated. [ see Adverse Reactions (6.1)]
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of labeling:
- Anaphylaxis and serious hypersensitivity reactions [
see Warnings and Precautions (5.2)]
-
Seizures [
see Warnings and Precautions (5.3)]
- Interaction with sodium valproate
[
see Warnings and Precautions (5.4) and Drug Interactions (7.1)]
-
Clostridium difficile-associated diarrhea
[
see Warnings and Precautions (5.5)]
- Development of drug-resistant bacteria [
see Warnings and Precautions (5.6)]
- Pneumonitis with inhalational use [ see Warnings and Precautions (5.7)]
6.1 Adverse Reactions from Clinical Trials
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be compared directly to rates from clinical trials of another drug and may not reflect rates observed in practice.
During clinical investigations, 1338 adult patients were treated with DORIBAX ® (1076 patients received doripenem 500 mg administered over 1 hour every 8 hours and 262 patients received doripenem 500 mg administered over 4 hours every 8 hours); in some patients parenteral therapy was followed by a switch to an oral antimicrobial. [ see Clinical Studies (14)]. The median age of patients treated with DORIBAX ® was 54 years (range 18–90) in the comparative complicated urinary tract infections (cUTI) study, 46 years (range 18–94) in the pooled comparative complicated intra-abdominal infections (cIAI) studies, and 56 years (range 18-94) in the other Phase 3 trials. There was a female predominance (62%) in the comparative cUTI study and a male predominance (63% and 75%) in the comparative cIAI and other Phase 3 trials, respectively. The patients treated with DORIBAX ® were predominantly Caucasian (79%) in the five comparator-controlled Phase 3 studies.
The most common adverse drug reactions (≥ 5%) observed in the five DORIBAX ® comparator-controlled Phase 3 clinical trials were anemia, headache, nausea, diarrhea, rash, phlebitis, and elevated hepatic enzymes. During clinical trials, adverse events led to discontinuation of DORIBAX ® in 4.1% (55 of 1338) of patients compared to 4.3% (58 of 1325) of comparator-treated patients.
Adverse reactions due to DORIBAX ® 500 mg every 8 hours that occurred at a rate ≥ 1 % are listed in Table 4. Hypersensitivity reactions related to intravenous study drug occurred at a rate of less than 1%.
|
||||||
|
Complicated Urinary Tract
Infections (one trial) |
Complicated Intra-Abdominal
Infections (two trials) |
Other Phase 3 Trials
(two trials) |
||||
| System organ class | DORIBAX
®
500 mg administered every 8 hours (n =376 ) | Levofloxacin
250 mg administered IV every 24 hours (n = 372) | DORIBAX
®
500 mg administered every 8 hours (n = 477) | Meropenem
1 g administered every 8 hours (n = 469) | DORIBAX
®
500 mg administered every 8 hours (n =485 ) | Comparator
*
(n=484) |
| Nervous system disorders | ||||||
| Headache | 16 | 15 | 4 | 5 | 3 | 3 |
| Vascular disorders | ||||||
| Phlebitis | 4 | 4 | 8 | 6 | 2 | 1 |
| Gastro-intestinal disorders | ||||||
| Nausea | 4 | 6 | 12 | 9 | 7 | 7 |
| Diarrhea | 6 | 10 | 11 | 11 | 12 | 14 |
| C. difficile colitis | <1 | 0 | <1 | 0 | 1 | 2 |
| Blood and Lymphatic System disorders | ||||||
| Anemia | 2 | 1 | 10 | 5 | 5 | 6 |
| Skin and subcutaneous disorders | ||||||
| Pruritus | 1 | 1 | 3 | 2 | 1 | 1 |
| Rash | 1 | 1 | 4 | 2 | 6 | 5 |
| Investigations | ||||||
| Hepatic Enzyme elevation † | 2 | 4 | 2 | 4 | 7 | 6 |
| Infections and Infestations | ||||||
| Oral candidiasis | 1 | 0 | 1 | 2 | 3 | 1 |
| Vulvomycotic infection | 2 | 1 | 1 | <1 | 0 | <1 |
In a Phase 1 study of healthy subjects receiving doripenem doses greater than the approved dose of 500 mg every 8 hours for 10 to 14 days, the incidence of rash was higher than that observed in subjects who received 500 mg every 8 hours. The rash resolved within 10 days after doripenem administration was discontinued.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of doripenem. Because these reactions were reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hematologic: Leukopenia, Neutropenia, Thrombocytopenia
Immune System: Anaphylaxis
Nervous System: Seizure
Renal: Renal impairment/failure
Respiratory: Interstitial pneumonia
Skin: Toxic epidermal necrolysis, Stevens-Johnson Syndrome
7 DRUG INTERACTIONS
7.1 Valproic Acid
Co-administration of DORIBAX ® with valproic acid causes the serum concentrations of valproic acid to fall below the therapeutic range, increasing the risk for breakthrough seizures. Although the mechanism of this interaction is not fully understood, data from in vitro and animal studies suggest that doripenem may inhibit the hydrolysis of valproic acid's glucuronide metabolite (VPA-g) back to valproic acid, thus decreasing the plasma concentrations of valproic acid. This is consistent with case reports for other carbapenems, where serum concentrations of valproic acid were reduced upon co-administration with a carbapenem. If administration of DORIBAX ® is necessary, supplemental anti-convulsant therapy should be considered. The pharmacokinetics of doripenem were unaffected by the co-administration of valproic acid. [ see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)]
7.2 Probenecid
Probenecid interferes with the active tubular secretion of doripenem, resulting in increased plasma concentrations of doripenem. [ see Clinical Pharmacology (12.3)] Coadministration of probenecid with DORIBAX ® is not recommended.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Category B: Doripenem was not teratogenic and did not produce effects on ossification, developmental delays or fetal weight following intravenous administration during organogenesis at doses as high as 1 g/kg/day in rats and 50 mg/kg/day in rabbits (based on AUC, at least 2.4 and 0.8 times the exposure to humans dosed at 500 mg administered every 8 hours, respectively). There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when DORIBAX ® is administered to a nursing woman.
8.5 Geriatric Use
Of the total number of subjects in clinical studies of DORIBAX ®, 28% were 65 and over, while 12% were 75 and over. Clinical cure rates in complicated intra-abdominal and complicated urinary tract infections were slightly lower in patients ≥ 65 years of age and also in the subgroup of patients ≥ 75 years of age versus patients < 65. These results were similar between doripenem and comparator treatment groups.
This drug is known to be excreted substantially by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function or pre-renal azotemia. Because elderly patients are more likely to have decreased renal function or pre-renal azotemia, care should be taken in dose selection, and it may be useful to monitor renal function.
Elderly subjects had greater doripenem plasma concentrations relative to non-elderly subjects; however, this increase in exposure was mainly attributed to age-related changes in renal function. [ see Clinical Pharmacology (12.3)]
No overall differences in safety were observed between older and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Patients with Renal Impairment
Dosage adjustment is required in patients with moderately or severely impaired renal function. [ see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)] In such patients, renal function should be monitored.
10 OVERDOSAGE
In the event of overdose, DORIBAX ® should be discontinued and general supportive treatment given.
Doripenem can be removed by hemodialysis. In subjects with end-stage renal disease administered DORIBAX ® 500 mg, the mean total recovery of doripenem and doripenem-M1 in the dialysate following a 4-hour hemodialysis session was 259 mg (52% of the dose). However, no information is available on the use of hemodialysis to treat overdosage. [ see Clinical Pharmacology (12.3)]
11 DESCRIPTION
DORIBAX ®, doripenem monohydrate for injection vials contain 500 mg of doripenem on an anhydrous basis, a white to slightly-yellowish off-white sterile crystalline powder. All references to doripenem activity are expressed in terms of the active doripenem moiety. The powder is constituted for intravenous infusion. The pH of the infusion solution is between 4.5 and 5.5.
DORIBAX ® is not formulated with any inactive ingredients.
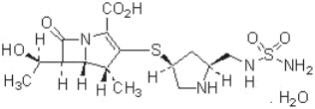
DORIBAX ® (doripenem monohydrate) is a synthetic broad-spectrum carbapenem antibiotic structurally related to beta-lactam antibiotics. The chemical name for doripenem monohydrate is (4 R,5 S,6 S)-3-[((3 S,5 S)-5-[[(aminosulfonyl)amino]methyl]-3-pyrrolidinyl) thio]-6-[(1 R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid monohydrate.
Its molecular weight is 438.52, and its chemical structure is:
12 CLINICAL PHARMACOLOGY
Doripenem is a carbapenem with in vitro antibacterial activity against aerobic and anaerobic Gram-positive and Gram-negative bacteria.
12.2 Pharmacodynamics
Similar to other beta-lactam antimicrobial agents, the time that unbound plasma concentration of doripenem exceeds the MIC of the infecting organism has been shown to best correlate with efficacy in animal models of infection. However, the pharmacokinetic/pharmacodynamic relationship for doripenem has not been evaluated in patients.
In a randomized, positive- and placebo-controlled crossover QT study, 60 healthy subjects were administered DORIBAX ® 500 mg IV every 8 hours × 4 doses and DORIBAX ® 1 g IV every 8 hours × 4 doses, placebo, and a single oral dose of positive control. At both the 500 mg and 1 g DORIBAX ® doses, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.
12.3 Pharmacokinetics
- Plasma Concentrations
Mean plasma concentrations of doripenem following a single 1-hour intravenous infusion of a 500 mg dose of DORIBAX ® to 24 healthy subjects are shown below in Figure 1. The mean (SD) plasma C max and AUC 0–∞ values were 23.0 (6.6) µg/mL and 36.3 (8.8) µg•hr/mL, respectively.
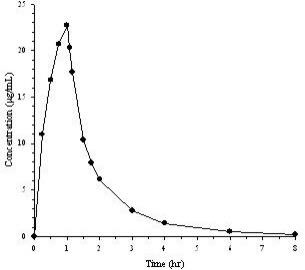
The pharmacokinetics of doripenem (C max and AUC) are linear over a dose range of 500 mg to 1 g when intravenously infused over 1 hour. There is no accumulation of doripenem following multiple intravenous infusions of either 500 mg or 1 g administered every 8 hours for 7 to 10 days in subjects with normal renal function.
- Distribution
The average binding of doripenem to plasma proteins is approximately 8.1% and is independent of plasma drug concentrations. The median (range) volume of distribution at steady state in healthy subjects is 16.8 L (8.09–55.5 L), similar to extracellular fluid volume (18.2 L).
Doripenem penetrates into several body fluids and tissues, including those at the site of infection for the approved indications. Doripenem concentrations in peritoneal and retroperitoneal fluid either match or exceed those required to inhibit most susceptible bacteria; however, the clinical relevance of this finding has not been established. Concentrations achieved in selected tissues and fluids following administration of DORIBAX ® are shown in Table 5:
| Tissue or Fluid | Dose
(mg) | Infusion Duration
(h) | Number of Samples or Subjects * | Sampling Period † | Concentration Range (µg/mL or µg/g) | Tissue- or Fluid-To-Plasma Concentration Ratio (%)
Mean (Range) |
|---|---|---|---|---|---|---|
| Retroperitoneal fluid | 250 | 0.5 | 9 ‡ | 30–90 min § | 3.15–52.4 | Range: 4.1(0.5–9.7) at 0.25 h
to 990 (173–2609) at 2.5 h |
| 500 | 0.5 | 4 ‡ | 90 min § | 9.53–13.9 | Range: 3.3 (0.0–8.1) at 0.25 h
to 516 (311–842) at 6.5 h |
|
| Peritoneal exudate | 250 | 0.5 | 5 ‡ | 30–150 min § | 2.36–5.17 | Range: 19.7 (0.00–47.3) at 0.5 h
to 160 (32.2–322) at 4.5 h |
| Gallbladder | 250 | 0.5 | 10 | 20–215 min | BQL–1.87 ¶ | 8.02 (0.00–44.4) |
| Bile | 250 | 0.5 | 10 | 20–215 min | BQL–15.4 # | 117 (0.00–611) |
| Urine | 500 | 1 | 110 | 0–4 hr | 601 (BQL #–3360) Þ | --- |
| 500 | 1 | 110 | 4–8 hr | 49.7 (BQL #–635) Þ | --- | |
- Metabolism
Metabolism of doripenem to a microbiologically inactive ring-opened metabolite (doripenem-M1) occurs primarily via dehydropeptidase-I. The mean (SD) plasma doripenem-M1-to-doripenem AUC ratio following single 500 mg and 1 g doses in healthy subjects is 18% (7.2%).
In pooled human liver microsomes, no in vitro metabolism of doripenem could be detected, indicating that doripenem is not a substrate for hepatic CYP450 enzymes.
- Excretion
Doripenem is primarily eliminated unchanged by the kidneys. The mean plasma terminal elimination half-life of doripenem in healthy non-elderly adults is approximately 1 hour and mean (SD) plasma clearance is 15.9 (5.3) L/hour. Mean (SD) renal clearance is 10.3 (3.5) L/hour. The magnitude of this value, coupled with the significant decrease in the elimination of doripenem with concomitant probenecid administration, suggests that doripenem undergoes both glomerular filtration and active tubular secretion. In healthy adults given a single 500 mg dose of DORIBAX ®, a mean of 71% and 15% of the dose was recovered in urine as unchanged drug and the ring-opened metabolite, respectively, within 48 hours. Following the administration of a single 500 mg dose of radiolabeled doripenem to healthy adults, less than 1% of the total radioactivity was recovered in feces after one week.
- Special Populations
Patients with Renal Impairment
Following a single 500 mg dose of DORIBAX ®, the mean AUC of doripenem in subjects with mild (CrCl 50–79 mL/min), moderate (CrCl 31–50 mL/min), and severe renal impairment (CrCl ≤ 30 mL/min) was 1.6-, 2.8-, and 5.1-times that of age-matched healthy subjects with normal renal function (CrCl ≥ 80 mL/min), respectively. Dosage adjustment is necessary in patients with moderate and severe renal impairment. [ seeDosage and Administration (2.2)]
A single 500 mg dose of DORIBAX ® was administered to subjects with end stage renal disease (ESRD) either one hour prior to or one hour after hemodialysis (HD). The mean doripenem AUC following the post-HD infusion was 7.8-times that of healthy subjects with normal renal function. The mean total recovery of doripenem and doripenem-M1 in the dialysate following a 4-hour HD session was 231 mg and 28 mg, respectively, or a total of 259 mg (52% of the dose). There is insufficient information to make dose adjustment recommendations in patients on hemodialysis.
Patients with Hepatic Impairment
The pharmacokinetics of doripenem in patients with hepatic impairment have not been established. As doripenem does not appear to undergo hepatic metabolism, the pharmacokinetics of doripenem are not expected to be affected by hepatic impairment.
Geriatric Patients
The impact of age on the pharmacokinetics of doripenem was evaluated in healthy male (n=6) and female (n=6) subjects ≥ 66 years of age. Mean doripenem AUC 0-∞ was 49% higher in elderly adults relative to non-elderly adults. This difference in exposure was mainly attributed to age-related changes in creatinine clearance. No dosage adjustment is recommended for elderly patients with normal (for their age) renal function.
Gender
The effect of gender on the pharmacokinetics of doripenem was evaluated in healthy male (n=12) and female (n=12) subjects. Doripenem C max and AUC were similar between males and females. No dose adjustment is recommended based on gender.
Race
The effect of race on doripenem pharmacokinetics was examined using a population pharmacokinetic analysis of data from phase 1 and 2 studies. No significant difference in mean doripenem clearance was observed across race groups and therefore, no dosage adjustment is recommended based on race.
- Drug Interactions
Administration of DORIBAX ® 500 mg every 8 hours x 4 doses to 23 healthy male subjects receiving valproic acid 500 mg every 12 hours for 7 days decreased the mean C max of valproic acid by 44.5% (from 86.1 mcg/mL to 47.8 mcg/mL) and the mean C min by 77.7% (from 55.7 mcg/mL to 12.4 mcg/mL) compared to administration of valproic acid alone. The mean AUC 0-tau of valproic acid also decreased by 63%. Conversely, the C max of the VPA-g metabolite was increased by 62.6% (from 5.19 mcg/mL to 8.44 mcg/mL) and the mean AUC 0-tau of VPA-g was increased by 50%. The pharmacokinetics of doripenem were unaffected by the co-administration of valproic acid. [ see Warnings and Precautions (5.4) and Drug Interactions (7.1)]
Probenecid interferes with the active tubular secretion of doripenem, resulting in increased plasma concentrations. Probenecid increased doripenem AUC by 75% and prolonged the plasma elimination half-life by 53%. [ see also Drug Interactions (7.2)]
In vitro studies in human liver microsomes and hepatocytes indicate that doripenem does not inhibit the major cytochrome P450 isoenzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, and CYP4A11). Therefore, DORIBAX ® is not expected to inhibit the clearance of drugs that are metabolized by these metabolic pathways in a clinically relevant manner.
DORIBAX ® is also not expected to have CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4/5, or UGT1A1 enzyme-inducing properties based on in vitro studies in cultured human hepatocytes.
12.4 Microbiology
- Mechanism of Action
Doripenem belongs to the carbapenem class of antimicrobials. Doripenem exerts its bactericidal activity by inhibiting bacterial cell wall biosynthesis. Doripenem inactivates multiple essential penicillin-binding proteins (PBPs) resulting in inhibition of cell wall synthesis with subsequent cell death. In E. coli and P. aeruginosa, doripenem binds to PBP 2, which is involved in the maintenance of cell shape, as well as to PBPs 3 and 4.
- Mechanism(s) of Resistance
Bacterial resistance mechanisms that affect doripenem include drug inactivation by carbapenem-hydrolyzing enzymes, mutant or acquired PBPs, decreased outer membrane permeability and active efflux. Doripenem is stable to hydrolysis by most beta-lactamases, including penicillinases and cephalosporinases produced by Gram-positive and Gram-negative bacteria, with the exception of carbapenem hydrolyzing beta-lactamases. Although cross-resistance may occur, some isolates resistant to other carbapenems may be susceptible to doripenem.
- Interaction with Other Antimicrobials
In vitro synergy tests with doripenem show doripenem has little potential to antagonize or be antagonized by other antibiotics (e.g., levofloxacin, amikacin, trimethoprim-sulfamethoxazole, daptomycin, linezolid, and vancomycin).
Doripenem has been shown to be active against most isolates of the following microorganisms, both
in vitro and in clinical infections. [
see Indications and Usage (1)]
Facultative Gram-negative microorganisms
Acinetobacter baumannii
Escherichia coli
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Facultative Gram-positive microorganisms
Streptococcus constellatus
Streptococcus intermedius
Anaerobic microorganisms
Bacteroides caccae
Bacteroides fragilis
Bacteroides thetaiotaomicron
Bacteroides uniformis
Bacteroides vulgatus
Peptostreptococcus micros
At least 90 percent of the following microorganisms exhibit an
in vitro minimal inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for doripenem of organisms of the same type shown in Table 6. The safety and efficacy of doripenem in treating clinical infections due to these microorganisms has not been established in adequate and well-controlled clinical trials.
Facultative Gram-positive microorganisms
Staphylococcus aureus (methicillin-susceptible isolates only)
Streptococcus agalactiae
Streptococcus pyogenes
Facultative Gram-negative microorganisms
Citrobacter freundii
Enterobacter cloacae
Enterobacter aerogenes
Klebsiella oxytoca
Morganella morganii
Serratia marcescens
- Susceptibility Test Methods
When available, the clinical microbiology laboratory should provide the results of in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas to the physician as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting the most effective antimicrobial.
Dilution Techniques
Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MICs should be determined using a standardized procedure. Standardized procedures are based on a dilution method (1,3) (broth or agar) or equivalent with standardized inoculum concentrations and standardized concentrations of doripenem powder. The MIC values should be interpreted according to the criteria provided in Table 6.
Diffusion Techniques
Quantitative methods that require measurement of zone diameters provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure (2,3) requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 10 µg of doripenem to test the susceptibility of microorganisms to doripenem. Results should be interpreted according to the criteria in Table 6.
Anaerobic Techniques
For anaerobic bacteria, the susceptibility to doripenem as MICs should be determined by standardized test methods (4). The MIC values obtained should be interpreted according to the criteria in Table 6.
|
||
| Minimum Inhibitory
Concentrations (µg/mL) | Disk Diffusion
(zone diameters in mm) |
|
| Pathogen | Susceptible * | Susceptible * |
| Enterobacteriaceae | ≤ 0.5 | ≥ 23 |
| Pseudomonas aeruginosa | ≤ 2 | ≥ 24 |
| Acinetobacter baumannii | ≤ 1 | ≥ 17 |
| Streptococcus anginosus group ( S. constellatus and S. intermedius) | ≤ 0.12 | ≥ 24 |
| Anaerobes | ≤ 1 | n/a |
n/a = not applicable
A report of Susceptible indicates that the antimicrobial is likely to inhibit growth of the pathogen if the antimicrobial compound in the blood reaches the concentrations usually achievable.
Quality Control
Standardized susceptibility test procedures require the use of laboratory control microorganisms to monitor the performance of the supplies and reagents used in the assay, and the techniques of the individuals performing the test. Standard doripenem powder should provide the MIC values provided in Table 8. For the diffusion techniques using a 10 µg doripenem disk, the criteria noted in Table 7 should be achieved.
|
||
| QC Organism | Minimum Inhibitory
Concentrations (µg/mL) | Disk Diffusion
(zone diameters in mm) |
| Escherichia coli ATCC 25922 | 0.015–0.06 | 27–34 |
| Pseudomonas aeruginosa ATCC 27853 | 0.12–0.5 | 28–34 |
| Streptococcus pneumoniae ATCC 49619 * | 0.03–0.12 | 30–38 |
| Bacteroides fragilis ATCC 25285 | 0.12–0.5 | n/a |
| Bacteroides thetaiotaomicron ATCC 29741 | 0.12–1 | n/a |
n/a = not applicable
13 NON-CLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Because of the short duration of treatment and intermittent clinical use, long-term carcinogenicity studies have not been conducted with doripenem.
Doripenem did not show evidence of mutagenic activity in standard tests that included bacterial reverse mutation assay, chromosomal aberration assay with Chinese hamster lung fibroblast cells, and mouse bone marrow micronucleus assay.
Intravenous injection of doripenem had no adverse effects on general fertility of treated male and female rats or on postnatal development and reproductive performance of the offspring at doses as high as 1 g/kg/day (based on AUC, greater than 1.5 times the exposure to humans at the dose of 500 mg administered every 8 hours).
14 CLINICAL STUDIES
14.1 Complicated Intra-Abdominal Infections
A total of 946 adults with complicated intra-abdominal infections were randomized and received study medications in two identical multinational, multi-center, double-blind studies comparing DORIBAX ® (500 mg administered over 1 hour every 8 hours) to meropenem (1 g administered over 3–5 minutes every 8 hours). Both regimens allowed the option to switch to oral amoxicillin/clavulanate (875 mg/125 mg administered twice daily) after a minimum of 3 days of intravenous therapy for a total of 5–14 days of intravenous and oral treatment. Patients with complicated appendicitis, or other complicated intra-abdominal infections, including bowel perforation, cholecystitis, intra-abdominal or solid organ abscess and generalized peritonitis were enrolled.
DORIBAX ® was non-inferior to meropenem with regard to clinical cure rates in microbiologically evaluable (ME) patients, i.e., in patients with susceptible pathogens isolated at baseline and no major protocol deviations at test of cure (TOC) visit, 25–45 days after completing therapy. DORIBAX ® was also non-inferior to meropenem in microbiological modified intent-to-treat (mMITT) patients, i.e., patients with baseline pathogens isolated regardless of susceptibility. Clinical cure rates at TOC are displayed by patient populations in Table 8. Microbiological cure rates at TOC by pathogen in ME patients are presented in Table 9.
|
|||
| Analysis Populations | DORIBAX
®
*
n/N (%) † | Meropenem
‡
n/N (%) † | Treatment Difference
(2-sided 95% CI §) |
| Study 1: | |||
| ME ¶ | 130/157 (82.8) | 128/149 (85.9) | -3.1 (-11.3; 5.2) |
| mMITT # | 143/194 (73.7) | 149/191 (78.0) | -4.3 (-12.8; 4.3) |
| Study 2: | |||
| ME ¶ | 128/158 (81.0) | 119/145 (82.1) | -1.1 (-9.8; 7.8) |
| mMITT # | 143/199 (71.9) | 138/186 (74.2) | -2.3 (-11.2; 6.6) |
| Pathogen | DORIBAX® | Meropenem | ||||
| N* | n† | % | N* | n† | % | |
| Gram-positive, aerobic | ||||||
| Streptococcus constellatus | 10 | 9 | 90.0 | 7 | 5 | 71.4 |
| Streptococcus intermedius | 36 | 30 | 83.3 | 29 | 21 | 72.4 |
| Gram-positive, anaerobic | ||||||
| Peptostreptococcus micros | 13 | 11 | 84.6 | 14 | 11 | 78.6 |
| Gram-negative, aerobic | ||||||
| Enterobacteriaceae | 315 | 271 | 86.0 | 274 | 234 | 85.4 |
| Escherichia coli | 216 | 189 | 87.5 | 199 | 168 | 84.4 |
| Klebsiella pneumoniae | 32 | 25 | 78.1 | 20 | 19 | 95.0 |
| Non-fermenters | 51 | 44 | 86.3 | 39 | 28 | 71.8 |
| Pseudomonas aeruginosa | 40 | 34 | 85.0 | 32 | 24 | 75.0 |
| Gram-negative, anaerobic | ||||||
| Bacteroides fragilis group | 173 | 152 | 87.9 | 181 | 152 | 84.0 |
| Bacteroides caccae | 25 | 23 | 92.0 | 19 | 18 | 94.7 |
| Bacteroides fragilis | 67 | 56 | 83.6 | 68 | 54 | 79.4 |
| Bacteroides thetaiotaomicron | 34 | 30 | 88.2 | 36 | 32 | 88.9 |
| Bacteroides uniformis | 22 | 19 | 86.4 | 18 | 15 | 83.3 |
| Non-fragilis Bacteroides | 14 | 13 | 92.9 | 13 | 9 | 69.2 |
| Bacteroides vulgatus | 11 | 11 | 100.0 | 8 | 6 | 75.0 |
14.2 Complicated Urinary Tract Infections, Including Pyelonephritis
A total of 1171 adults with complicated urinary tract infections, including pyelonephritis (49 percent of microbiologically evaluable patients) were randomized and received study medications in two multi-center, multinational studies. Complicated pyelonephritis, i.e., pyelonephritis associated with predisposing anatomical or functional abnormality, comprised 17% of patients with pyelonephritis. One study was double-blind and compared DORIBAX ® (500 mg administered over 1 hour every 8 hours) to IV levofloxacin (250 mg administered every 24 hours). The second study was a non-comparative study but of otherwise similar design. Both studies permitted the option of switching to oral levofloxacin (250 mg administered every 24 hours) after a minimum of 3 days of IV therapy for a total of 10 days of treatment. Patients with confirmed concurrent bacteremia were allowed to receive 500 mg of IV levofloxacin (either IV or oral as appropriate) for a total of 10 to 14 days of treatment.
DORIBAX ® was non-inferior to levofloxacin with regard to the microbiological eradication rates in microbiologically evaluable (ME) patients, i.e., patients with baseline uropathogens isolated, no major protocol deviations and urine cultures at test of cure (TOC) visit 5-11 days after completing therapy. DORIBAX ® was also non-inferior to levofloxacin in microbiological modified intent-to-treat (mMITT) patients, i.e., patients with pretreatment urine cultures. Overall microbiological eradication rates at TOC and the 95% CIs for the comparative study are displayed in Table 10. Microbiological eradication rates at TOC by pathogen in ME patients are presented in Table 11.
|
|||
| Analysis populations |
DORIBAX® *
n/N (%)† |
Levofloxacin‡
n/N (%)† |
Treatment Difference
(2-sided 95% CI§) |
| ME ¶ | 230/280 (82.1) | 221/265 (83.4) | -1.3 (-8.0, 5.5) |
| mMITT # | 259/327 (79.2) | 251/321 (78.2) | 1.0 (-5.6, 7.6) |
| Pathogen | DORIBAX®* | Levofloxacin | ||||
| N† | n‡ | % | N† | n‡ | % | |
| Gram-negative, aerobic | ||||||
| Escherichia coli | 357 | 313 | 87.7 | 211 | 184 | 87.2 |
| Klebsiella pneumoniae | 33 | 26 | 78.8 | 8 | 5 | 62.5 |
| Proteus mirabilis | 30 | 22 | 73.3 | 15 | 13 | 86.7 |
| Non-fermenters | 38 | 27 | 71.1 | 8 | 5 | 62.5 |
| Acinetobacter baumannii | 10 | 8 | 80.0 | 1 | 0 | 0.0 |
| Pseudomonas aeruginosa | 27 | 19 | 70.4 | 7 | 5 | 71.4 |
15 REFERENCES
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard – 7
th ed. CLSI Document M7-A7. CLSI, 940 West Valley Rd., Suite 1400, Wayne, PA 19087, 2006.
- CLSI. Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard – 9
th ed. CLSI Document M2-A9. CLSI, Wayne, PA 19087, 2006.
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; 17
th Informational Supplement. CLSI document M100-S17. CLSI, Wayne, PA 19087, 2007.
- CLSI. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard – 7 th ed. CLSI document M11-A7. CLSI, Wayne, PA 19087, 2007.
16 HOW SUPPLIED/STORAGE AND HANDLING
DORIBAX ® is supplied as single use type 1 clear glass vials containing 250 mg or 500 mg (on an anhydrous basis) of sterile doripenem powder. Vials are packaged individually and in cartons containing 10 vials.
- NDC: 59630-320-01 – 500 mg/vial, single vial
- NDC: 59630-320-10 – 500 mg/vial, 10 vials/carton
- NDC: 59630-309-01 – 250 mg/vial, single vial
- NDC: 59630-309-10 – 250 mg/vial, 10 vials/carton
Storage of DORIBAX® vials
DORIBAX ® should be stored at 25°C (77°F); excursions permitted to 15°–30°C (59° to 86°F) [refer to USP controlled room temperature].
17 PATIENT COUNSELING INFORMATION
- Patients should be advised that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. They should report any previous hypersensitivity reactions to DORIBAX
®, other carbapenems, beta-lactams or other allergens.
- Patients should be counseled that anti-bacterial drugs including DORIBAX
® should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When DORIBAX
® is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by DORIBAX
® or other antibacterial drugs in the future.
- Patients should be counseled to inform their physician
- if they have central nervous system disorders such as stroke or history of seizures. Seizures have been reported during treatment with DORIBAX ® and with closely related antibiotics
- if they are taking valproic acid or sodium valproate. Valproic acid concentrations in the blood will drop below the therapeutic range upon co-administration with DORIBAX ®. If treatment with DORIBAX ® is necessary and continued, alternative or supplemental anti-convulsant medication to prevent and/or treat seizures may be needed.
- Keep out of the reach of children.
MINI-BAG Plus is a trademark of Baxter International Inc.
Product of Japan
Manufactured by:
Shionogi & Co. Ltd.
Osaka 541-0045, Japan
Manufactured for:
Shionogi Inc.
Florham Park, NJ 07932
Rev. 08/2015


| DORIBAX
doripenem powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| DORIBAX
doripenem powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Shionogi Inc. (098241610) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Shionogi Pharma Co., Ltd. | 717959195 | manufacture(59630-320, 59630-309) , api manufacture(59630-320, 59630-309) , analysis(59630-320, 59630-309) | |