AVAPRO- irbesartan tablet
Bryant Ranch Prepack
----------
AVAPRO®
(irbesartan)
Tablets
WARNING: FETAL TOXICITY
- When pregnancy is detected, discontinue AVAPRO as soon as possible.
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus. See WARNINGS: Fetal Toxicity.
DESCRIPTION
AVAPRO®* (irbesartan) is an angiotensin II receptor (AT1 subtype) antagonist.
*Registered trademark
Irbesartan is a non-peptide compound, chemically described as a 2-butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4.4]non-1-en-4-one.
Its empirical formula is C25H28N6O, and the structural formula:

Irbesartan is a white to off-white crystalline powder with a molecular weight of 428.5. It is a nonpolar compound with a partition coefficient (octanol/water) of 10.1 at pH of 7.4. Irbesartan is slightly soluble in alcohol and methylene chloride and practically insoluble in water.
AVAPRO is available for oral administration in unscored tablets containing 75 mg, 150 mg, or 300 mg of irbesartan. Inactive ingredients include: lactose, microcrystalline cellulose, pregelatinized starch, croscarmellose sodium, poloxamer 188, silicon dioxide, and magnesium stearate.
CLINICAL PHARMACOLOGY
Mechanism of Action
Angiotensin II is a potent vasoconstrictor formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system (RAS) and also stimulates aldosterone synthesis and secretion by adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system, and smooth muscle cell growth. Irbesartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively binding to the AT1 angiotensin II receptor. There is also an AT2 receptor in many tissues, but it is not involved in cardiovascular homeostasis.
Irbesartan is a specific competitive antagonist of AT1 receptors with a much greater affinity (more than 8500-fold) for the AT1 receptor than for the AT2 receptor and no agonist activity.
Blockade of the AT1 receptor removes the negative feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and circulating angiotensin II do not overcome the effects of irbesartan on blood pressure.
Irbesartan does not inhibit ACE or renin or affect other hormone receptors or ion channels known to be involved in the cardiovascular regulation of blood pressure and sodium homeostasis. Because irbesartan does not inhibit ACE, it does not affect the response to bradykinin; whether this has clinical relevance is not known.
Pharmacokinetics
Irbesartan is an orally active agent that does not require biotransformation into an active form. The oral absorption of irbesartan is rapid and complete with an average absolute bioavailability of 60% to 80%. Following oral administration of AVAPRO, peak plasma concentrations of irbesartan are attained at 1.5 to 2 hours after dosing. Food does not affect the bioavailability of AVAPRO.
Irbesartan exhibits linear pharmacokinetics over the therapeutic dose range.
The terminal elimination half-life of irbesartan averaged 11 to 15 hours. Steady-state concentrations are achieved within 3 days. Limited accumulation of irbesartan (<20%) is observed in plasma upon repeated once-daily dosing.
Metabolism and Elimination
Irbesartan is metabolized via glucuronide conjugation and oxidation. Following oral or intravenous administration of 14C-labeled irbesartan, more than 80% of the circulating plasma radioactivity is attributable to unchanged irbesartan. The primary circulating metabolite is the inactive irbesartan glucuronide conjugate (approximately 6%). The remaining oxidative metabolites do not add appreciably to irbesartan’s pharmacologic activity.
Irbesartan and its metabolites are excreted by both biliary and renal routes. Following either oral or intravenous administration of 14C-labeled irbesartan, about 20% of radioactivity is recovered in the urine and the remainder in the feces, as irbesartan or irbesartan glucuronide.
In vitro studies of irbesartan oxidation by cytochrome P450 isoenzymes indicated irbesartan was oxidized primarily by 2C9; metabolism by 3A4 was negligible. Irbesartan was neither metabolized by, nor did it substantially induce or inhibit, isoenzymes commonly associated with drug metabolism (1A1, 1A2, 2A6, 2B6, 2D6, 2E1). There was no induction or inhibition of 3A4.
Distribution
Irbesartan is 90% bound to serum proteins (primarily albumin and α1-acid glycoprotein) with negligible binding to cellular components of blood. The average volume of distribution is 53 liters to 93 liters. Total plasma and renal clearances are in the range of 157 mL/min to 176 mL/min and 3.0 mL/min to 3.5 mL/min, respectively. With repetitive dosing, irbesartan accumulates to no clinically relevant extent.
Studies in animals indicate that radiolabeled irbesartan weakly crosses the blood-brain barrier and placenta. Irbesartan is excreted in the milk of lactating rats.
Special Populations
Gender
No gender-related differences in pharmacokinetics were observed in healthy elderly (age 65-80 years) or in healthy young (age 18-40 years) subjects. In studies of hypertensive patients, there was no gender difference in half-life or accumulation, but somewhat higher plasma concentrations of irbesartan were observed in females (11-44%). No gender-related dosage adjustment is necessary.
Geriatric
In elderly subjects (age 65-80 years), irbesartan elimination half-life was not significantly altered, but AUC and Cmax values were about 20% to 50% greater than those of young subjects (age 18-40 years). No dosage adjustment is necessary in the elderly.
Race
In healthy black subjects, irbesartan AUC values were approximately 25% greater than whites; there were no differences in Cmax values.
Renal Insufficiency
The pharmacokinetics of irbesartan were not altered in patients with renal impairment or in patients on hemodialysis. Irbesartan is not removed by hemodialysis. No dosage adjustment is necessary in patients with mild to severe renal impairment unless a patient with renal impairment is also volume depleted. (See WARNINGS: Hypotension in Volume- or Salt-Depleted Patients and DOSAGE AND ADMINISTRATION.)
Pharmacodynamics
In healthy subjects, single oral irbesartan doses of up to 300 mg produced dose-dependent inhibition of the pressor effect of angiotensin II infusions. Inhibition was complete (100%) 4 hours following oral doses of 150 mg or 300 mg and partial inhibition was sustained for 24 hours (60% and 40% at 300 mg and 150 mg, respectively).
In hypertensive patients, angiotensin II receptor inhibition following chronic administration of irbesartan causes a 1.5- to 2-fold rise in angiotensin II plasma concentration and a 2- to 3-fold increase in plasma renin levels. Aldosterone plasma concentrations generally decline following irbesartan administration, but serum potassium levels are not significantly affected at recommended doses.
In hypertensive patients, chronic oral doses of irbesartan (up to 300 mg) had no effect on glomerular filtration rate, renal plasma flow, or filtration fraction. In multiple dose studies in hypertensive patients, there were no clinically important effects on fasting triglycerides, total cholesterol, HDL-cholesterol, or fasting glucose concentrations. There was no effect on serum uric acid during chronic oral administration, and no uricosuric effect.
Clinical Studies
Hypertension
The antihypertensive effects of AVAPRO (irbesartan) were examined in 7 major placebo-controlled 8 to 12 week trials in patients with baseline diastolic blood pressures of 95 mmHg to 110 mmHg. Doses of 1 mg to 900 mg were included in these trials in order to fully explore the dose-range of irbesartan. These studies allowed comparison of once- or twice-daily regimens at 150 mg/day, comparisons of peak and trough effects, and comparisons of response by gender, age, and race. Two of the 7 placebo-controlled trials identified above examined the antihypertensive effects of irbesartan and hydrochlorothiazide in combination.
The 7 studies of irbesartan monotherapy included a total of 1915 patients randomized to irbesartan (1-900 mg) and 611 patients randomized to placebo. Once-daily doses of 150 mg and 300 mg provided statistically and clinically significant decreases in systolic and diastolic blood pressure with trough (24 hours post-dose) effects after 6 to 12 weeks of treatment compared to placebo, of about 8-10/5-6 mmHg and 8-12/5-8 mmHg, respectively. No further increase in effect was seen at dosages greater than 300 mg. The dose-response relationships for effects on systolic and diastolic pressure are shown in Figures 1 and 2.
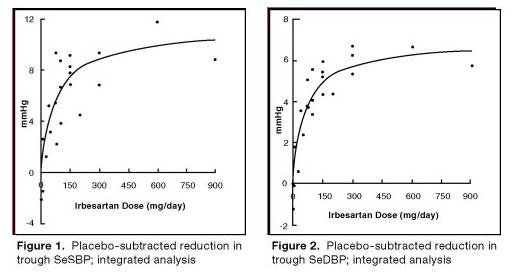
Once-daily administration of therapeutic doses of irbesartan gave peak effects at around 3 to 6 hours and, in one ambulatory blood pressure monitoring study, again around 14 hours. This was seen with both once-daily and twice-daily dosing. Trough-to-peak ratios for systolic and diastolic response were generally between 60% to 70%. In a continuous ambulatory blood pressure monitoring study, once-daily dosing with 150 mg gave trough and mean 24-hour responses similar to those observed in patients receiving twice-daily dosing at the same total daily dose.
In controlled trials, the addition of irbesartan to hydrochlorothiazide doses of 6.25 mg, 12.5 mg, or 25 mg produced further dose-related reductions in blood pressure similar to those achieved with the same monotherapy dose of irbesartan. HCTZ also had an approximately additive effect.
Analysis of age, gender, and race subgroups of patients showed that men and women, and patients over and under 65 years of age, had generally similar responses. Irbesartan was effective in reducing blood pressure regardless of race, although the effect was somewhat less in blacks (usually a low-renin population).
The effect of irbesartan is apparent after the first dose, and it is close to its full observed effect at 2 weeks. At the end of an 8-week exposure, about 2/3 of the antihypertensive effect was still present one week after the last dose. Rebound hypertension was not observed. There was essentially no change in average heart rate in irbesartan-treated patients in controlled trials.
Nephropathy in Type 2 Diabetic Patients
The Irbesartan Diabetic Nephropathy Trial (IDNT) was a randomized, placebo- and active-controlled, double-blind, multicenter study conducted worldwide in 1715 patients with type 2 diabetes, hypertension (SeSBP >135 mmHg or SeDBP >85 mmHg), and nephropathy (serum creatinine 1.0 to 3.0 mg/dL in females or 1.2 to 3.0 mg/dL in males and proteinuria ≥900 mg/day). Patients were randomized to receive AVAPRO 75 mg, amlodipine 2.5 mg, or matching placebo once-daily. Patients were titrated to a maintenance dose of AVAPRO 300 mg, or amlodipine 10 mg, as tolerated. Additional antihypertensive agents (excluding ACE inhibitors, angiotensin II receptor antagonists and calcium channel blockers) were added as needed to achieve blood pressure goal (≤135/85 or 10 mmHg reduction in systolic blood pressure if higher than 160 mmHg) for patients in all groups.
The study population was 66.5% male, 72.9% below 65 years of age and 72% White, (Asian/Pacific Islander 5.0%, Black 13.3%, Hispanic 4.8%). The mean baseline seated systolic and diastolic blood pressures were 159 mmHg and 87 mmHg, respectively. The patients entered the trial with a mean serum creatinine of 1.7 mg/dL and mean proteinuria of 4144 mg/day.
The mean blood pressure achieved was 142/77 mmHg for AVAPRO, 142/76 mmHg for amlodipine, and 145/79 mmHg for placebo. Overall, 83.0% of patients received the target dose of irbesartan more than 50% of the time. Patients were followed for a mean duration of 2.6 years.
The primary composite endpoint was the time to occurrence of any one of the following events: doubling of baseline serum creatinine, end-stage renal disease (ESRD; defined by serum creatinine ≥6 mg/dL, dialysis, or renal transplantation) or death. Treatment with AVAPRO resulted in a 20% risk reduction versus placebo (p=0.0234) (see Figure 3 and Table 1). Treatment with AVAPRO also reduced the occurrence of sustained doubling of serum creatinine as a separate endpoint (33%), but had no significant effect on ESRD alone and no effect on overall mortality (see Table 1).
Figure 3: IDNT: Kaplan-Meier Estimates Of Primary Endpoint (Doubling of Serum Creatinine, End-Stage Renal Disease or All-Cause Mortality
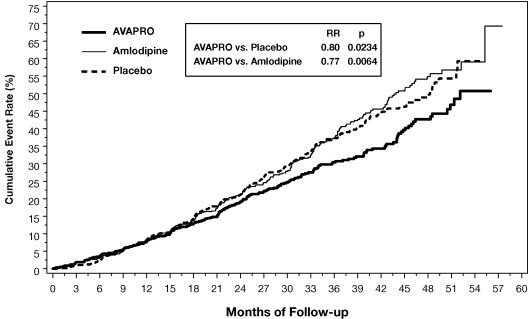
The percentages of patients experiencing an event during the course of the study can be seen in Table 1 below:
| Comparison With Placebo | Comparison With Amlodipine | ||||||
|---|---|---|---|---|---|---|---|
| AVAPRO N=579 (%) | Placebo N=569 (%) | Hazard Ratio | 95% CI | Amlodipine N=567 (%) | Hazard Ratio | 95% CI | |
| Primary Composite Endpoint | 32.6 | 39.0 | 0.80 | 0.66-0.97 (p=0.0234) | 41.1 | 0.77 | 0.63-0.93 |
| Breakdown of first occurring event contributing to primary endpoint | |||||||
| 2x creatinine | 14.2 | 19.5 | --- | --- | 22.8 | --- | --- |
| ESRD | 7.4 | 8.3 | --- | --- | 8.8 | --- | --- |
| Death | 11.1 | 11.2 | --- | --- | 9.5 | --- | ---- |
| Incidence of total events over entire period of follow-up | |||||||
| 2x creatinine | 16.9 | 23.7 | 0.67 | 0.52-0.87 | 25.4 | 0.63 | 0.49-0.81 |
| ESRD | 14.2 | 17.8 | 0.77 | 0.57-1.03 | 18.3 | 0.77 | 0.57-1.03 |
| Death | 15.0 | 16.3 | 0.92 | 0.69-1.23 | 14.6 | 1.04 | 0.77-1.40 |
The secondary endpoint of the study was a composite of cardiovascular mortality and morbidity (myocardial infarction, hospitalization for heart failure, stroke with permanent neurological deficit, amputation). There were no statistically significant differences among treatment groups in these endpoints. Compared with placebo, AVAPRO significantly reduced proteinuria by about 27%, an effect that was evident within 3 months of starting therapy. AVAPRO significantly reduced the rate of loss of renal function (glomerular filtration rate), as measured by the reciprocal of the serum creatinine concentration, by 18.2%.
Table 2 presents results for demographic subgroups. Subgroup analyses are difficult to interpret and it is not known whether these observations represent true differences or chance effects. For the primary endpoint, AVAPRO’s favorable effects were seen in patients also taking other antihypertensive medications (angiotensin II receptor antagonists, angiotensin-converting-enzyme inhibitors and calcium channel blockers were not allowed), oral hypoglycemic agents, and lipid-lowering agents.
| Comparison With Placebo | ||||
|---|---|---|---|---|
| Baseline Factors | AVAPRO N=579 (%) | Placebo N=569 (%) | Hazard Ratio | 95% Cl |
| Gender | ||||
| Male | 27.5 | 36.7 | 0.68 | 0.53-0.88 |
| Female | 42.3 | 44.6 | 0.98 | 0.72-1.34 |
| Race | ||||
| White | 29.5 | 37.3 | 0.75 | 0.60-0.95 |
| Non-White | 42.6 | 43.5 | 0.95 | 0.67-1.34 |
| Age (years) | ||||
| <65 | 31.8 | 39.9 | 0.77 | 0.62-0.97 |
| ≥65 | 35.1 | 36.8 | 0.88 | 0.61-1.29 |
INDICATIONS AND USAGE
Hypertension
AVAPRO (irbesartan) is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.
Nephropathy in Type 2 Diabetic Patients
AVAPRO is indicated for the treatment of diabetic nephropathy with an elevated serum creatinine and proteinuria (>300 mg/day) in patients with type 2 diabetes and hypertension. In this population, AVAPRO reduces the rate of progression of nephropathy as measured by the occurrence of doubling of serum creatinine or end-stage renal disease (need for dialysis or renal transplantation) (see CLINICAL PHARMACOLOGY: Clinical Studies).
CONTRAINDICATIONS
AVAPRO is contraindicated in patients who are hypersensitive to any component of this product.
Do not coadminister aliskiren with AVAPRO in patients with diabetes (see PRECAUTIONS: Drug Interactions).
WARNINGS
Fetal Toxicity
Pregnancy Category D
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue AVAPRO as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimesters of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue AVAPRO, unless it is considered lifesaving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to AVAPRO for hypotension, oliguria, and hyperkalemia (see PRECAUTIONS: Pediatric Use).
When pregnant rats were treated with irbesartan from day 0 to day 20 of gestation (oral doses of 50 mg/kg/day, 180 mg/kg/day, and 650 mg/kg/day), increased incidences of renal pelvic cavitation, hydroureter and/or absence of renal papilla were observed in fetuses at doses ≥50 mg/kg/day (approximately equivalent to the maximum recommended human dose [MRHD], 300 mg/day, on a body surface area basis). Subcutaneous edema was observed in fetuses at doses ≥180 mg/kg/day (about 4 times the MRHD on a body surface area basis). As these anomalies were not observed in rats in which irbesartan exposure (oral doses of 50, 150, and 450 mg/kg/day) was limited to gestation days 6 to 15, they appear to reflect late gestational effects of the drug. In pregnant rabbits, oral doses of 30 mg irbesartan/kg/day were associated with maternal mortality and abortion. Surviving females receiving this dose (about 1.5 times the MRHD on a body surface area basis) had a slight increase in early resorptions and a corresponding decrease in live fetuses. Irbesartan was found to cross the placental barrier in rats and rabbits.
Radioactivity was present in the rat and rabbit fetus during late gestation and in rat milk following oral doses of radiolabeled irbesartan.
Hypotension in Volume- or Salt-Depleted Patients
Excessive reduction of blood pressure was rarely seen (<0.1%) in patients with uncomplicated hypertension. Initiation of antihypertensive therapy may cause symptomatic hypotension in patients with intravascular volume- or sodium-depletion, eg, in patients treated vigorously with diuretics or in patients on dialysis. Such volume depletion should be corrected prior to administration of AVAPRO, or a low starting dose should be used (see DOSAGE AND ADMINISTRATION).
If hypotension occurs, the patient should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
PRECAUTIONS
Impaired Renal Function
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. In patients whose renal function may depend on the activity of the renin-angiotensin-aldosterone system (eg, patients with severe congestive heart failure), treatment with angiotensin-converting-enzyme inhibitors has been associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death. AVAPRO would be expected to behave similarly.
In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or BUN have been reported. There has been no known use of AVAPRO in patients with unilateral or bilateral renal artery stenosis, but a similar effect should be anticipated.
Drug Interactions
No significant drug-drug pharmacokinetic (or pharmacodynamic) interactions have been found in interaction studies with hydrochlorothiazide, digoxin, warfarin, and nifedipine.
In vitro studies show significant inhibition of the formation of oxidized irbesartan metabolites with the known cytochrome CYP 2C9 substrates/inhibitors sulphenazole, tolbutamide and nifedipine. However, in clinical studies the consequences of concomitant irbesartan on the pharmacodynamics of warfarin were negligible. Based on in vitro data, no interaction would be expected with drugs whose metabolism is dependent upon cytochrome P450 isoenzymes 1A1, 1A2, 2A6, 2B6, 2D6, 2E1, or 3A4.
In separate studies of patients receiving maintenance doses of warfarin, hydrochlorothiazide, or digoxin, irbesartan administration for 7 days had no effect on the pharmacodynamics of warfarin (prothrombin time) or pharmacokinetics of digoxin. The pharmacokinetics of irbesartan were not affected by coadministration of nifedipine or hydrochlorothiazide.
Concomitant use of potassium-sparing diuretics, potassium supplements, or salt substitutes containing potassium may lead to increases in serum potassium.
Non-Steroidal Anti-Inflammatory Agents Including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, coadministration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists, including irbesartan, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving irbesartan and NSAID therapy.
The antihypertensive effect of angiotensin II receptor antagonists, including irbesartan, may be attenuated by NSAIDs including selective COX-2 inhibitors.
Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin-receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Closely monitor blood pressure, renal function, and electrolytes in patients on AVAPRO and other agents that affect the RAS.
Do not coadminister aliskiren with AVAPRO in patients with diabetes. Avoid use of aliskiren with AVAPRO in patients with renal impairment (GFR <60 mL/min).
Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was observed when irbesartan was administered at doses of up to 500/1000 mg/kg/day (males/females, respectively) in rats and 1000 mg/kg/day in mice for up to 2 years. For male and female rats, 500 mg/kg/day provided an average systemic exposure to irbesartan (AUC0-24 hour, bound plus unbound) about 3 and 11 times, respectively, the average systemic exposure in humans receiving the maximum recommended dose (MRD) of 300 mg irbesartan/day, whereas 1000 mg/kg/day (administered to females only) provided an average systemic exposure about 21 times that reported for humans at the MRD. For male and female mice, 1000 mg/kg/day provided an exposure to irbesartan about 3 and 5 times, respectively, the human exposure at 300 mg/day.
Irbesartan was not mutagenic in a battery of in vitro tests (Ames microbial test, rat hepatocyte DNA repair test, V79 mammalian-cell forward gene-mutation assay). Irbesartan was negative in several tests for induction of chromosomal aberrations (in vitro-human lymphocyte assay; in vivo-mouse micronucleus study).
Irbesartan had no adverse effects on fertility or mating of male or female rats at oral doses ≤650 mg/kg/day, the highest dose providing a systemic exposure to irbesartan (AUC0-24 hour, bound plus unbound) about 5 times that found in humans receiving the maximum recommended dose of 300 mg/day.
Nursing Mothers
It is not known whether irbesartan is excreted in human milk, but irbesartan or some metabolite of irbesartan is secreted at low concentration in the milk of lactating rats. Because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Neonates with a history of in utero exposure to AVAPRO:
If oliguria or hypotension occurs, direct attention toward support of blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function.
Irbesartan, in a study at a dose of up to 4.5 mg/kg/day, once daily, did not appear to lower blood pressure effectively in pediatric patients ages 6 to 16 years.
AVAPRO has not been studied in pediatric patients less than 6 years old.
Geriatric Use
Of 4925 subjects receiving AVAPRO (irbesartan) in controlled clinical studies of hypertension, 911 (18.5%) were 65 years and over, while 150 (3.0%) were 75 years and over. No overall differences in effectiveness or safety were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out. (See CLINICAL PHARMACOLOGY: Pharmacokinetics, Special Populations, and Clinical Studies.)
ADVERSE REACTIONS
Hypertension
AVAPRO has been evaluated for safety in more than 4300 patients with hypertension and about 5000 subjects overall. This experience includes 1303 patients treated for over 6 months and 407 patients for 1 year or more. Treatment with AVAPRO was well-tolerated, with an incidence of adverse events similar to placebo. These events generally were mild and transient with no relationship to the dose of AVAPRO.
In placebo-controlled clinical trials, discontinuation of therapy due to a clinical adverse event was required in 3.3% of patients treated with AVAPRO, versus 4.5% of patients given placebo.
In placebo-controlled clinical trials, the following adverse event experiences reported in at least 1% of patients treated with AVAPRO (n=1965) and at a higher incidence versus placebo (n=641), excluding those too general to be informative and those not reasonably associated with the use of drug because they were associated with the condition being treated or are very common in the treated population, include: diarrhea (3% vs 2%), dyspepsia/heartburn (2% vs 1%), and fatigue (4% vs 3%).
The following adverse events occurred at an incidence of 1% or greater in patients treated with irbesartan, but were at least as frequent or more frequent in patients receiving placebo: abdominal pain, anxiety/nervousness, chest pain, dizziness, edema, headache, influenza, musculoskeletal pain, pharyngitis, nausea/vomiting, rash, rhinitis, sinus abnormality, tachycardia, and urinary tract infection.
Irbesartan use was not associated with an increased incidence of dry cough, as is typically associated with ACE inhibitor use. In placebo-controlled studies, the incidence of cough in irbesartan-treated patients was 2.8% versus 2.7% in patients receiving placebo.
The incidence of hypotension or orthostatic hypotension was low in irbesartan-treated patients (0.4%), unrelated to dosage, and similar to the incidence among placebo-treated patients (0.2%). Dizziness, syncope, and vertigo were reported with equal or less frequency in patients receiving irbesartan compared with placebo.
In addition, the following potentially important events occurred in less than 1% of the 1965 patients and at least 5 patients (0.3%) receiving irbesartan in clinical studies, and those less frequent, clinically significant events (listed by body system). It cannot be determined whether these events were causally related to irbesartan:
Body as a Whole: fever, chills, facial edema, upper extremity edema
Cardiovascular: flushing, hypertension, cardiac murmur, myocardial infarction, angina pectoris, arrhythmic/conduction disorder, cardio-respiratory arrest, heart failure, hypertensive crisis
Dermatologic: pruritus, dermatitis, ecchymosis, erythema face, urticaria
Endocrine/Metabolic/Electrolyte Imbalances: sexual dysfunction, libido change, gout
Gastrointestinal: constipation, oral lesion, gastroenteritis, flatulence, abdominal distention
Musculoskeletal/Connective Tissue: extremity swelling, muscle cramp, arthritis, muscle ache, musculoskeletal chest pain, joint stiffness, bursitis, muscle weakness
Nervous System: sleep disturbance, numbness, somnolence, emotional disturbance, depression, paresthesia, tremor, transient ischemic attack, cerebrovascular accident
Renal/Genitourinary: abnormal urination, prostate disorder
Respiratory: epistaxis, tracheobronchitis, congestion, pulmonary congestion, dyspnea, wheezing
Special Senses: vision disturbance, hearing abnormality, ear infection, ear pain, conjunctivitis, other eye disturbance, eyelid abnormality, ear abnormality
Nephropathy in Type 2 Diabetic Patients
In clinical studies in patients with hypertension and type 2 diabetic renal disease, the adverse drug experiences were similar to those seen in patients with hypertension with the exception of an increased incidence of orthostatic symptoms (dizziness, orthostatic dizziness, and orthostatic hypotension) observed in IDNT (proteinuria ≥900 mg/day, and serum creatinine ranging from 1.0-3.0 mg/dL). In this trial, orthostatic symptoms occurred more frequently in the AVAPRO group (dizziness 10.2%, orthostatic dizziness 5.4%, orthostatic hypotension 5.4%) than in the placebo group (dizziness 6.0%, orthostatic dizziness 2.7%, orthostatic hypotension 3.2%).
Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of AVAPRO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or to establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors: (1) seriousness of the reaction, (2) frequency of reporting, or (3) strength of causal connection to AVAPRO.
The following have been reported: urticaria; angioedema (involving swelling of the face, lips, pharynx, and/or tongue); increased liver function tests; jaundice; hepatitis; hyperkalemia, and thrombocytopenia.
Impaired renal function, including cases of renal failure, has been reported.
Cases of increased CPK and rhabdomyolysis have been reported in patients receiving angiotensin II receptor blockers.
Laboratory Test Findings
Hypertension
In controlled clinical trials, clinically important differences in laboratory tests were rarely associated with administration of AVAPRO.
Creatinine, Blood Urea Nitrogen: Minor increases in blood urea nitrogen (BUN) or serum creatinine were observed in less than 0.7% of patients with essential hypertension treated with AVAPRO alone versus 0.9% on placebo. (See PRECAUTIONS: Impaired Renal Function.)
Hematologic: Mean decreases in hemoglobin of 0.2 g/dL were observed in 0.2% of patients receiving AVAPRO compared to 0.3% of placebo-treated patients. Neutropenia (<1000 cells/mm3) occurred at similar frequencies among patients receiving AVAPRO (0.3%) and placebo-treated patients (0.5%).
Nephropathy in Type 2 Diabetic Patients
Hyperkalemia: In IDNT (proteinuria ≥900 mg/day, and serum creatinine ranging from 1.0-3.0 mg/dL), the percent of patients with hyperkalemia (>6 mEq/L) was 18.6% in the AVAPRO group versus 6.0% in the placebo group. Discontinuations due to hyperkalemia in the AVAPRO group were 2.1% versus 0.4% in the placebo group.
OVERDOSAGE
No data are available in regard to overdosage in humans. However, daily doses of 900 mg for 8 weeks were well-tolerated. The most likely manifestations of overdosage are expected to be hypotension and tachycardia; bradycardia might also occur from overdose. Irbesartan is not removed by hemodialysis.
To obtain up-to-date information about the treatment of overdosage, a good resource is a certified regional Poison Control Center. Telephone numbers of certified Poison Control Centers are listed in the Physicians’ Desk Reference (PDR). In managing overdose, consider the possibilities of multiple-drug interactions, drug-drug interactions, and unusual drug kinetics in the patient.
Laboratory determinations of serum levels of irbesartan are not widely available, and such determinations have, in any event, no known established role in the management of irbesartan overdose.
Acute oral toxicity studies with irbesartan in mice and rats indicated acute lethal doses were in excess of 2000 mg/kg, about 25- and 50-fold the MRHD (300 mg) on a mg/m2 basis, respectively.
DOSAGE AND ADMINISTRATION
AVAPRO may be administered with other antihypertensive agents and with or without food.
Hypertension
The recommended initial dose of AVAPRO (irbesartan) is 150 mg once daily. Patients requiring further reduction in blood pressure should be titrated to 300 mg once daily.
A low dose of a diuretic may be added, if blood pressure is not controlled by AVAPRO alone. Hydrochlorothiazide has been shown to have an additive effect (see CLINICAL PHARMACOLOGY: Clinical Studies). Patients not adequately treated by the maximum dose of 300 mg once daily are unlikely to derive additional benefit from a higher dose or twice-daily dosing.
No dosage adjustment is necessary in elderly patients, or in patients with hepatic impairment or mild to severe renal impairment.
Nephropathy in Type 2 Diabetic Patients
The recommended target maintenance dose is 300 mg once daily. There are no data on the clinical effects of lower doses of AVAPRO on diabetic nephropathy (see CLINICAL PHARMACOLOGY: Clinical Studies).
Volume- and Salt-Depleted Patients
A lower initial dose of AVAPRO (75 mg) is recommended in patients with depletion of intravascular volume or salt (eg, patients treated vigorously with diuretics or on hemodialysis) (see WARNINGS: Hypotension in Volume- or Salt-Depleted Patients).

| AVAPRO
irbesartan tablet |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Bryant Ranch Prepack (171714327) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bryant Ranch Prepack | 171714327 | REPACK(63629-3373) , RELABEL(63629-3373) | |