GUANFACINE- guanfacine tablet
Precision Dose, Inc.
----------
Guanfacine Tablets, USP
DESCRIPTION
Guanfacine hydrochloride, USP is a centrally acting antihypertensive with α2-adrenoceptor agonist properties in tablet form for oral administration.
The chemical name of guanfacine hydrochloride, USP is N-amidino-2-(2,6-dichlorophenyl) acetamide hydrochloride and its molecular weight is 282.56. Its structural formula is:
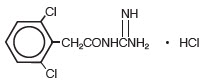
Guanfacine hydrochloride, USP is a white to off-white powder; sparingly soluble in water and alcohol and slightly soluble in acetone.
Each tablet, for oral administration, contains guanfacine hydrochloride, USP equivalent to 1 mg or 2 mg guanfacine. In addition, each tablet contains the following inactive ingredients: microcrystalline cellulose, pregelatinized starch, and stearic acid.
CLINICAL PHARMACOLOGY
Guanfacine hydrochloride is an orally active antihypertensive agent whose principal mechanism of action appears to be stimulation of central α2-adrenergic receptors. By stimulating these receptors, guanfacine reduces sympathetic nerve impulses from the vasomotor center to the heart and blood vessels. This results in a decrease in peripheral vascular resistance and a reduction in heart rate.
The dose-response relationship for blood pressure and adverse effects of guanfacine given once a day as monotherapy has been evaluated in patients with mild to moderate hypertension. In this study patients were randomized to placebo or to 0.5 mg, 1 mg, 2 mg, 3 mg or 5 mg of guanfacine. Results are shown in the following table. A useful effect was not observed overall until doses of 2 mg were reached, although responses in white patients were seen at 1 mg; 24 hour effectiveness of 1 mg to 3 mg doses was documented using 24 hour ambulatory monitoring. While the 5 mg dose added an increment of effectiveness, it caused an unacceptable increase in adverse reactions.
| Mean Change S/D* Seated | n=(range) | Placebo | 0.5 mg | 1 mg | 2 mg | 3 mg | 5 mg |
|---|---|---|---|---|---|---|---|
|
|||||||
| White Patients | 11 to 30 | -1/-5 | -6/-8 | -8/-9 | -12/-11 | -15/-12 | -18/-16 |
| Black Patients | 8 to 28 | -3/-5 | 0/-2 | -3/-5 | -7/-7 | -8/-9 | -19/-15 |
Controlled clinical trials in patients with mild to moderate hypertension who were receiving a thiazide-type diuretic have defined the dose-response relationship for blood pressure response and adverse reactions of guanfacine given at bedtime and have shown that the blood pressure response to guanfacine can persist for 24 hours after a single dose. In the 12-week placebo-controlled dose-response study, patients were randomized to placebo or to doses of 0.5, 1, 2 and 3 mg of guanfacine, in addition to 25 mg chlorthalidone, each given at bedtime. The observed mean changes from baseline, tabulated below, indicate the similarity of response for placebo and the 0.5 mg dose. Doses of 1, 2 and 3 mg resulted in decreased blood pressure in the sitting position with no real differences among the three doses. In the standing position, there was some increase in response with dose.
| Placebo | 0.5 mg | 1 mg | 2 mg | 3 mg | |
|---|---|---|---|---|---|
| Mean Change n = | 63 | 63 | 64 | 58 | 59 |
|
|||||
| S/D* Seated | -5/-7 | -5/-6 | -14/-13 | -12/-13 | -16/-13 |
| S/D* Standing | -3/-5 | -5/-4 | -11/-9 | -9/-10 | -15/-12 |
While most of the effectiveness of guanfacine in combination (and as monotherapy in white patients) was present at 1 mg, adverse reactions at this dose were not clearly distinguishable from those associated with placebo. Adverse reactions were clearly present at 2 and 3 mg (see ADVERSE REACTIONS).
In a second 12-week placebo-controlled study of 1, 2 or 3 mg of guanfacine hydrochloride administered with 25 mg of chlorthalidone once daily, a significant decrease in blood pressure was maintained for a full 24 hours after dosing. While there was no significant difference between the 12 and 24 hour blood pressure readings, the fall in blood pressure at 24 hours was numerically smaller, suggesting possible escape of blood pressure in some patients and the need for individualization of therapy.
In a double-blind, randomized trial, either guanfacine or clonidine was given at recommended doses with 25 mg chlorthalidone for 24 weeks and then abruptly discontinued. Results showed equal degrees of blood pressure reduction with the two drugs and there was no tendency for blood pressures to increase despite maintenance of the same daily dose of the two drugs. Signs and symptoms of rebound phenomena were infrequent upon discontinuation of either drug. Abrupt withdrawal of clonidine produced a rapid return of diastolic and especially systolic blood pressure to approximately pretreatment levels, with occasional values significantly greater than baseline, whereas guanfacine withdrawal produced a more gradual increase to pretreatment levels, but also with occasional values significantly greater than baseline.
Pharmacodynamics
Hemodynamic studies in man showed that the decrease in blood pressure observed after single-dose or long-term oral treatment with guanfacine was accompanied by a significant decrease in peripheral resistance and a slight reduction in heart rate (5 beats/min). Cardiac output under conditions of rest or exercise was not altered by guanfacine.
Guanfacine hydrochloride lowered elevated plasma renin activity and plasma catecholamine levels in hypertensive patients, but this does not correlate with individual blood-pressure responses.
Growth hormone secretion was stimulated with single oral doses of 2 and 4 mg of guanfacine. Long-term use of guanfacine had no effect on growth hormone levels.
Guanfacine had no effect on plasma aldosterone. A slight but insignificant decrease in plasma volume occurred after one month of guanfacine therapy. There were no changes in mean body weight or electrolytes.
Pharmacokinetics
Relative to an intravenous dose of 3 mg, the absolute oral bioavailability of guanfacine is about 80%. Peak plasma concentrations occur from 1 to 4 hours with an average of 2.6 hours after single oral doses or at steady-state.
The area under the concentration-time curve (AUC) increases linearly with the dose.
In individuals with normal renal function, the average elimination half-life is approximately 17 hr (range 10 to 30 hr). Younger patients tend to have shorter elimination half-lives (13 to 14 hr) while older patients tend to have half-lives at the upper end of the range. Steady-state blood levels were attained within 4 days in most subjects.
In individuals with normal renal function, guanfacine and its metabolites are excreted primarily in the urine. Approximately 50% (40 to 75%) of the dose is eliminated in the urine as unchanged drug; the remainder is eliminated mostly as conjugates of metabolites produced by oxidative metabolism of the aromatic ring.
The guanfacine-to-creatinine clearance ratio is greater than 1, which would suggest that tubular secretion of drug occurs.
The drug is approximately 70% bound to plasma proteins, independent of drug concentration.
The whole body volume of distribution is high (a mean of 6.3 L/kg), which suggests a high distribution of drug to the tissues.
The clearance of guanfacine in patients with varying degrees of renal insufficiency is reduced, but plasma levels of drug are only slightly increased compared to patients with normal renal function. When prescribing for patients with renal impairment, the low end of the dosing range should be used. Patients on dialysis also can be given usual doses of guanfacine hydrochloride as the drug is poorly dialyzed.
INDICATIONS AND USAGE
Guanfacine tablets, USP are indicated in the management of hypertension. Guanfacine may be given alone or in combination with other antihypertensive agents, especially thiazide-type diuretics.
CONTRAINDICATIONS
Guanfacine tablets, USP are contraindicated in patients with known hypersensitivity to guanfacine hydrochloride, USP.
PRECAUTIONS
General
Like other antihypertensive agents, guanfacine hydrochloride should be used with caution in patients with severe coronary insufficiency, recent myocardial infarction, cerebrovascular disease, or chronic renal or hepatic failure.
Sedation
Guanfacine, like other orally active central α2-adrenergic agonists, causes sedation or drowsiness, especially when beginning therapy. These symptoms are dose-related (see ADVERSE REACTIONS). When guanfacine is used with other centrally active depressants (such as phenothiazines, barbiturates, or benzodiazepines), the potential for additive sedative effects should be considered.
Rebound
Abrupt cessation of therapy with orally active central α2-adrenergic agonists may be associated with increases (from depressed on-therapy levels) in plasma and urinary catecholamines, symptoms of "nervousness and anxiety" and, less commonly, increases in blood pressure to levels significantly greater than those prior to therapy.
Information for Patients
Patients who receive guanfacine should be advised to exercise caution when operating dangerous machinery or driving motor vehicles until it is determined that they do not become drowsy or dizzy from the medication. Patients should be warned that their tolerance for alcohol and other CNS depressants may be diminished. Patients should be advised not to discontinue therapy abruptly.
Laboratory Tests
In clinical trials, no clinically relevant laboratory test abnormalities were identified as causally related to drug during short-term treatment with guanfacine hydrochloride.
Drug Interactions
The potential for increased sedation when guanfacine is given with other CNS-depressant drugs should be appreciated.
The administration of guanfacine concomitantly with a known microsomal enzyme inducer (phenobarbital or phenytoin) to two patients with renal impairment reportedly resulted in significant reductions in elimination half-life and plasma concentration. In such cases, therefore, more frequent dosing may be required to achieve or maintain the desired hypotensive response. Further, if guanfacine is to be discontinued in such patients, careful tapering of the dosage may be necessary in order to avoid rebound phenomena (see Rebound above).
Anticoagulants
Ten patients who were stabilized on oral anticoagulants were given guanfacine, 1 to 2 mg/day, for 4 weeks. No changes were observed in the degree of anticoagulation.
In several well-controlled studies, guanfacine was administered together with diuretics with no drug interactions reported. In the long-term safety studies, guanfacine was given concomitantly with many drugs without evidence of any interactions. The principal drugs given (number of patients in parentheses) were: cardiac glycosides (115), sedatives and hypnotics (103), coronary vasodilators (52), oral hypoglycemics (45), cough and cold preparations (45), NSAIDs (38), antihyperlipidemics (29), antigout drugs (24), oral contraceptives (18), bronchodilators (13), insulin (10) and beta blockers (10).
Drug/Laboratory Test Interactions
No laboratory test abnormalities related to the use of guanfacine hydrochloride have been identified.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenic effect was observed in studies of 78 weeks in mice at doses more than 150 times the maximum recommended human dose and 102 weeks in rats at doses more than 100 times the maximum recommended human dose. In a variety of test models, guanfacine was not mutagenic.
No adverse effects were observed in fertility studies in male and female rats.
Pregnancy Category B
Administration of guanfacine to rats at 70 times the maximum recommended human dose and to rabbits at 20 times the maximum recommended human dose resulted in no evidence of harm to the fetus. Higher doses (100 and 200 times the maximum recommended human dose in rabbits and rats respectively) were associated with reduced fetal survival and maternal toxicity. Rat experiments have shown that guanfacine crosses the placenta.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Labor and Delivery
Guanfacine hydrochloride is not recommended in the treatment of acute hypertension associated with toxemia of pregnancy. There is no information available on the effects of guanfacine on the course of labor and delivery.
Nursing Mothers
It is not known whether guanfacine hydrochloride is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when guanfacine is administered to a nursing woman. Experiments with rats have shown that guanfacine is excreted in the milk.
Pediatric Use
Safety and effectiveness in children under 12 years of age have not been demonstrated. Therefore, the use of guanfacine in this age group is not recommended. There have been spontaneous postmarketing reports of mania and aggressive behavioral changes in pediatric patients with attention-deficit hyperactivity disorder (ADHD) receiving guanfacine. The reported cases were from a single center. All patients had medical or family risk factors for bipolar disorder. All patients recovered upon discontinuation of guanfacine HCl. Hallucinations have been reported in pediatric patients receiving guanfacine for treatment of attention-deficit hyperactivity disorder.
Geriatric Use
Clinical studies of guanfacine did not include sufficient numbers of subjects aged 65 and over to determine whether they responded differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy (see CLINICAL PHARMACOLOGY: Pharmacokinetics).
ADVERSE REACTIONS
Adverse reactions noted with guanfacine hydrochloride are similar to those of other drugs of the central α2-adrenoreceptor agonist class: dry mouth, sedation (somnolence), weakness (asthenia), dizziness, constipation, and impotence. While the reactions are common, most are mild and tend to disappear on continued dosing.
Skin rash with exfoliation has been reported in a few cases; although clear cause and effect relationships to guanfacine could not be established, should a rash occur, guanfacine should be discontinued and the patient monitored appropriately.
In the dose-response monotherapy study described under CLINICAL PHARMACOLOGY, the frequency of the most commonly observed adverse reactions showed a dose relationship from 0.5 to 3 mg as follows:
| Adverse Reaction | Placebo n = 59 | 0.5 mg n = 60 | 1 mg n = 61 | 2 mg n = 60 | 3 mg n = 59 |
|---|---|---|---|---|---|
| Dry Mouth | 0% | 10% | 10% | 42% | 54% |
| Somnolence | 8% | 5% | 10% | 13% | 39% |
| Asthenia | 0% | 2% | 3% | 7% | 3% |
| Dizziness | 8% | 12% | 2% | 8% | 15% |
| Headache | 8% | 13% | 7% | 5% | 3% |
| Impotence | 0% | 0% | 0% | 7% | 3% |
| Constipation | 0% | 2% | 0% | 5% | 15% |
| Fatigue | 2% | 2% | 5% | 8% | 10% |
The percent of patients who dropped out because of adverse reactions are shown below for each dosage group.
| Placebo | 0.5 mg | 1 mg | 2 mg | 3 mg | |
|---|---|---|---|---|---|
| Percent dropouts | 0% | 2% | 5% | 13% | 32% |
The most common reasons for dropouts among patients who received guanfacine were dry mouth, somnolence, dizziness, fatigue, weakness and constipation.
In the 12-week, placebo-controlled, dose-response study of guanfacine administered with 25 mg chlorthalidone at bedtime, the frequency of the most commonly observed adverse reactions showed a clear dose relationship from 0.5 to 3 mg as follows:
| Adverse Reaction | Placebo n = 73 | 0.5 mg n = 72 | 1 mg n = 72 | 2 mg n = 72 | 3 mg n = 72 |
|---|---|---|---|---|---|
| Dry Mouth | 5 (7%) | 4 (5%) | 6 (8%) | 8 (11%) | 20 (28%) |
| Somnolence | 1 (1%) | 3 (4%) | 0 (0%) | 1 (1%) | 10 (14%) |
| Asthenia | 0 (0%) | 2 (3%) | 0 (0%) | 2 (2%) | 7 (10%) |
| Dizziness | 2 (2%) | 1 (1%) | 3 (4%) | 6 (8%) | 3 (4%) |
| Headache | 3 (4%) | 4 (3%) | 3 (4%) | 1 (1%) | 2 (2%) |
| Impotence | 1 (1%) | 1 (0%) | 0 (0%) | 1 (1%) | 3 (4%) |
| Constipation | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1%) | 1 (1%) |
| Fatigue | 3 (3%) | 2 (3%) | 2 (3%) | 5 (6%) | 3 (4%) |
There were 41 premature terminations because of adverse reactions in this study. The percent of patients who dropped out and the dose at which the dropout occurred were as follows:
| Dose: | Placebo | 0.5 mg | 1 mg | 2 mg | 3 mg |
|---|---|---|---|---|---|
| Percent dropouts | 6.9% | 4.2% | 3.2% | 6.9% | 8.3% |
Reasons for dropouts among patients who received guanfacine were: somnolence, headache, weakness, dry mouth, dizziness, impotence, insomnia, constipation, syncope, urinary incontinence, conjunctivitis, paresthesia and dermatitis.
In a second 12-week placebo-controlled combination therapy study in which the dose could be adjusted upward to 3 mg per day in 1 mg increments at 3-week intervals, i.e., a setting more similar to ordinary clinical use, the most commonly recorded reactions were: dry mouth, 47%; constipation, 16%; fatigue, 12%; somnolence, 10%; asthenia, 6%; dizziness, 6%; headache, 4%; and insomnia, 4%.
Reasons for dropouts among patients who received guanfacine were: somnolence, dry mouth, dizziness, impotence, constipation, confusion, depression and palpitations.
In the clonidine/guanfacine comparison described in CLINICAL PHARMACOLOGY, the most common adverse reactions noted were as follows:
| Adverse Reactions | Guanfacine (n = 279) | Clonidine (n = 278) |
|---|---|---|
| Dry mouth | 30% | 37% |
| Somnolence | 21% | 35% |
| Dizziness | 11% | 8% |
| Constipation | 10% | 5% |
| Fatigue | 9% | 8% |
| Headache | 4% | 4% |
| Insomnia | 4% | 3% |
Adverse reactions occurring in 3% or less of patients in the three controlled trials of guanfacine hydrochloride with a diuretic were:
| • Cardiovascular: | bradycardia, palpitations, substernal pain |
| • Gastrointestinal: | abdominal pain, diarrhea, dyspepsia, dysphagia, nausea |
| • CNS: | amnesia, confusion, depression, insomnia, libido decrease |
| • ENT disorders: | rhinitis, taste perversion, tinnitus |
| • Eye disorders: | conjunctivitis, iritis, vision disturbance |
| • Musculoskeletal: | leg cramps, hypokinesia |
| • Respiratory: | dyspnea |
| • Dermatologic: | dermatitis, pruritus, purpura, sweating |
| • Urogenital: | testicular disorder, urinary incontinence |
| • Other: | malaise, paresthesia, paresis |
Adverse reaction reports tend to decrease over time. In an open-label trial of one year's duration, 580 hypertensive subjects were given guanfacine, titrated to achieve goal blood pressure, alone (51%), with diuretic (38%), with beta blocker (3%), with diuretic plus beta blocker (6%), or with diuretic plus vasodilator (2%). The mean daily dose of guanfacine reached was 4.7 mg.
| Adverse Reaction | Incidence of adverse reactions at any time during the study | Incidence of adverse reactions at end of one year |
|---|---|---|
| n = 580 | n = 580 | |
| Dry mouth | 60% | 15% |
| Drowsiness | 33% | 6% |
| Dizziness | 15% | 1% |
| Constipation | 14% | 3% |
| Weakness | 5% | 1% |
| Headache | 4% | 0.2% |
| Insomnia | 5% | 0% |
There were 52 (8.9%) dropouts due to adverse effects in this 1-year trial. The causes were: dry mouth (n = 20), weakness (n = 12), constipation (n = 7), somnolence (n = 3), nausea (n = 3), orthostatic hypotension (n = 2), insomnia (n = 1), rash (n = 1), nightmares (n = 1), headache (n = 1) and depression (n = 1).
Postmarketing Experience
An open-label postmarketing study involving 21,718 patients was conducted to assess the safety of guanfacine hydrochloride 1 mg/day given at bedtime for 28 days. Guanfacine was administered with or without other antihypertensive agents. Adverse events reported in the postmarketing study at an incidence greater than 1% included dry mouth, dizziness, somnolence, fatigue, headache and nausea. The most commonly reported adverse events in this study were the same as those observed in controlled clinical trials.
Less frequent, possibly guanfacine-related events observed in the postmarketing study and/or reported spontaneously include:
| • Body as a Whole: | asthenia, chest pain, edema, malaise, tremor |
| • Cardiovascular: | bradycardia, palpitations, syncope, tachycardia |
| • Central Nervous System: | paresthesias, vertigo |
| • Eye Disorders: | blurred vision |
| • Gastrointestinal System: | abdominal pain, constipation, diarrhea, dyspepsia |
| • Liver and Biliary System: | abnormal liver function tests |
| • Musculo-Skeletal System: | arthralgia, leg cramps, leg pain, myalgia |
| • Psychiatric: | agitation, anxiety, confusion, depression, insomnia, nervousness |
| • Reproductive System, Male: | impotence |
| • Respiratory System: | dyspnea |
| • Skin and Appendages: | alopecia, dermatitis, exfoliative dermatitis, pruritus, rash |
| • Special Senses: | alterations in taste |
| • Urinary System: | nocturia, urinary frequency |
Rare, serious disorders with no definitive cause and effect relationship to guanfacine have been reported spontaneously and/or in the postmarketing study. These events include acute renal failure, cardiac fibrillation, cerebrovascular accident, congestive heart failure, heart block, and myocardial infarction.
DRUG ABUSE AND DEPENDENCE
No reported abuse or dependence has been associated with the administration of guanfacine hydrochloride.
OVERDOSAGE
Signs and Symptoms
Drowsiness, lethargy, bradycardia and hypotension have been observed following overdose with guanfacine.
A 25-year-old female intentionally ingested 60 mg. She presented with severe drowsiness and bradycardia of 45 beats/minute. Gastric lavage was performed and an infusion of isoproterenol (0.8 mg in 12 hours) was administered. She recovered quickly and without sequelae.
A 28-year-old female who ingested 30 to 40 mg developed only lethargy, was treated with activated charcoal and a cathartic, was monitored for 24 hours, and was discharged in good health.
A 2-year-old male weighing 12 kg who ingested up to 4 mg of guanfacine developed lethargy. Gastric lavage (followed by activated charcoal and sorbitol slurry via NG tube) removed some tablet fragments within 2 hours after ingestion, and vital signs were normal.
During 24-hour observation in ICU, systolic pressure was 58 and heart rate 70 at 16 hours post-ingestion. No intervention was required, and child was discharged fully recovered the next day.
DOSAGE AND ADMINISTRATION
The recommended initial dose of guanfacine tablets, USP when given alone or in combination with another antihypertensive drug is 1 mg daily given at bedtime to minimize somnolence. If after 3 to 4 weeks of therapy 1 mg does not give a satisfactory result, a dose of 2 mg may be given, although most of the effect of guanfacine is seen at 1 mg (see CLINICAL PHARMACOLOGY). Higher daily doses have been used, but adverse reactions increase significantly with doses above 3 mg/day.
The frequency of rebound hypertension is low, but it can occur. When rebound occurs, it does so after 2 to 4 days, which is delayed compared with clonidine hydrochloride. This is consistent with the longer half-life of guanfacine. In most cases, after abrupt withdrawal of guanfacine, blood pressure returns to pretreatment levels slowly (within 2 to 4 days) without ill effects.
HOW SUPPLIED
Guanfacine tablets, USP are available in 1 tablet strength of guanfacine (as the hydrochloride salt) as follows:
1 mg: white, oval, flat-faced, beveled-edge tablet with "AN" on one side and "711" on the other side.
They are supplied as follows:
NDC 68094-019-62
Unit Dose Packages of 30 Tablets (3×10) per carton
For inquiries call Precision Dose, Inc. at 1-800-397-9228 or e-mail druginfo@precisiondose.com
Distributed by:
Amneal Pharmaceuticals
Bridgewater, NJ 08807
Packaged by:
Precision Dose, Inc.
South Beloit, IL 61080
LI1118
Rev. 05/19
PRINCIPAL DISPLAY PANEL - 1 mg Tablet Blister Pack Carton Label
PrecisionDose™
NDC 68094-019-62
Unit Dose
guanFACINE
Tablets, USP
1 mg
30 TABLETS
(3x10)
Each tablet contains:
Guanfacine hydrochloride, USP
equivalent to 1 mg guanfacine.
USUAL DOSE: SEE ENCLOSED INSERT.
Store at 20° to 25°C (68° to 77°F)
[See USP Controlled Room Temperature].
KEEP THIS AND ALL DRUGS
OUT OF REACH OF CHILDREN.
Rx Only
LC1120 R0
Packaged by:
Precision Dose, Inc.
South Beloit, IL 61080

| GUANFACINE
guanfacine tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Precision Dose, Inc. (035886746) |