TAFINLAR- dabrafenib capsule
GlaxoSmithKline LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use TAFINLAR safely and effectively. See full prescribing information for TAFINLAR.
TAFINLAR (dabrafenib) capsules, for oral use Initial U.S. Approval: 2013 RECENT MAJOR CHANGESINDICATIONS AND USAGE
Limitation of Use: TAFINLAR is not indicated for treatment of patients with wild-type BRAF melanoma. (1.3, 5.2) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCapsules: 50 mg, 75 mg. (3) CONTRAINDICATIONSNone. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 11/2015 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma
TAFINLAR® is indicated as a single agent for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with TAFINLAR as a single agent [see Warnings and Precautions (5.2)]. Confirm the presence of BRAF V600E or V600K mutation in tumor specimens prior to initiation of treatment with TAFINLAR and trametinib. Information on FDA-approved tests for the detection of BRAF V600 mutations in melanoma is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosing
The recommended dosage regimen of TAFINLAR is 150 mg orally taken twice daily, approximately 12 hours apart as a single agent or with trametinib. Continue treatment until disease progression or unacceptable toxicity occurs.
Take TAFINLAR at least 1 hour before or 2 hours after a meal [see Clinical Pharmacology (12.3)]. Do not take a missed dose of TAFINLAR within 6 hours of the next dose of TAFINLAR. Do not open, crush, or break TAFINLAR capsules.
2.3 Dose Modifications
Review the Full Prescribing Information for trametinib for recommended dose modifications. Dose modifications are not recommended for TAFINLAR when administered with trametinib for the following adverse reactions of trametinib: retinal vein occlusion, retinal pigment epithelial detachment, interstitial lung disease/pneumonitis, and uncomplicated venous thromboembolism.
For New Primary Cutaneous Malignancies
No dose modifications are required.
For New Primary Non-Cutaneous Malignancies
Permanently discontinue TAFINLAR in patients who develop RAS mutation-positive non-cutaneous malignancies.
|
|
|
|
|
|
|
|
|
|
| Severity of Adverse
Reactiona | TAFINLARb |
|---|---|
|
Febrile Drug Reaction |
|
|
Withhold TAFINLAR until fever resolves. Then resume at same or lower dose level. |
|
Or
|
|
Cutaneous |
|
|
Withhold TAFINLAR for up to 3 weeks.
|
|
Cardiac |
|
|
Withhold TAFINLAR, if improved, then resume at the same dose. |
|
Uveitis |
|
|
If mild or moderate uveitis does not respond to ocular therapy, or for severe uveitis, withhold TAFINLAR for up to 6 weeks.
|
|
Other |
|
|
Withhold TAFINLAR.
|
|
Or
|
|
Permanently discontinue TAFINLAR. |
- a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0.
- bSee Table 1 for recommended dose reductions of TAFINLAR.
3 DOSAGE FORMS AND STRENGTHS
50 mg capsules: Dark red capsule imprinted with ‘GS TEW’ and ‘50 mg’.
75 mg capsules: Dark pink capsule imprinted with ‘GS LHF’ and ‘75 mg’.
5 WARNINGS AND PRECAUTIONS
Review the Full Prescribing Information for trametinib for information on the serious risks of trametinib prior to initiation of TAFINLAR in combination with trametinib.
5.1 New Primary Malignancies
New primary malignancies, cutaneous and non-cutaneous, can occur when TAFINLAR is administered as a single agent or when used with trametinib.
Cutaneous Malignancies
TAFINLAR results in an increased incidence of cutaneous squamous cell carcinoma, keratoacanthoma, and melanoma.
In Trial 1, cutaneous squamous cell carcinomas and keratoacanthomas (cuSCC) occurred in 7% (14/187) of patients receiving TAFINLAR and in none of the patients receiving dacarbazine.
Across clinical trials of TAFINLAR (N = 586), the incidence of cuSCC was 11%. The median time to first cuSCC was 2.1 months (range: 7 days to 12.2 months). Of those patients who developed new cuSCC, approximately 33% developed one or more cuSCC with continued administration of TAFINLAR. The median time between diagnosis of the first cuSCC and the second cuSCC was 6 weeks.
In Trial 2, the incidence of basal cell carcinoma in patients receiving TAFINLAR in combination with trametinib was 3.3% (7/209) compared with 6% (13/211) of patients receiving single-agent TAFINLAR. The median time to first diagnosis of basal cell carcinoma was 5.1 months (range: 2.8 to 23.9 months) in the TAFINLAR plus trametinib arm and was 4.4 months (range: 29 days to 16.5 months) in the single-agent TAFINLAR arm. Among the 7 patients receiving TAFINLAR with trametinib who developed basal cell carcinoma, 2 (29%) experienced more than one occurrence (range: 1 to 3).
Cutaneous squamous cell carcinoma and keratoacanthoma occurred in 3% of patients receiving TAFINLAR with trametinib and 10% of patients receiving single-agent TAFINLAR. The median time to first diagnosis of cuSCC was 7.3 months (range: 1.8 to 16.8 months) in the Tafinlar plus trametinib arm and was 2 months (range: 9 days to 20.9 months) in the single-agent TAFINLAR arm.
New primary melanoma occurred in 0.5% (1/209) of patients receiving TAFINLAR with trametinib and in 1.9% (4/211) of patients receiving single-agent TAFINLAR.
Perform dermatologic evaluations prior to initiation of TAFINLAR, every 2 months while on therapy, and for up to 6 months following discontinuation of TAFINLAR. No dose modifications of TAFINLAR are required in patients who develop new primary cutaneous malignancies [see Dosage and Administration (2.3)].
Non-cutaneous Malignancies
Based on its mechanism of action, TAFINLAR may promote the growth and development of malignancies with activation of RAS through mutation or other mechanisms [see Warnings and Precautions (5.2)]. In Trial 2, non-cutaneous malignancies occurred in 1.4% (3/209) of patients receiving TAFINLAR with trametinib and in 2.8% (6/211) of patients receiving single-agent TAFINLAR.
Monitor patients receiving TAFINLAR for signs or symptoms of non-cutaneous malignancies. Permanently discontinue TAFINLAR for RAS mutation-positive non-cutaneous malignancies [see Dosage and Administration (2.3)].
5.2 Tumor Promotion in BRAF Wild-Type Melanoma
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells which are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation status prior to initiation of TAFINLAR as a single agent or in combination with trametinib [see Indications and Usage (1), Dosage and Administration (2.1)].
5.3 Hemorrhage
Hemorrhage, including major hemorrhage defined as symptomatic bleeding in a critical area or organ, can occur when TAFINLAR is administered with trametinib.
In Trial 2, the incidence of hemorrhagic events in patients receiving TAFINLAR with trametinib was 19% (40/209) compared with 15% (32/211) of patients receiving single-agent TAFINLAR. Gastrointestinal hemorrhage occurred in 6% (12/209) of patients receiving TAFINLAR with trametinib compared with 3% (6/211) of patients receiving single-agent TAFINLAR. Intracranial hemorrhage was fatal in 1.4% (3/209) of patients receiving TAFINLAR with trametinib compared with none of the patients receiving single-agent TAFINLAR.
Permanently discontinue TAFINLAR for all Grade 4 hemorrhagic events and for any persistent Grade 3 hemorrhagic events. Withhold TAFINLAR for Grade 3 hemorrhagic events; if improved, resume at the next lower dose level.
5.4 Cardiomyopathy
Cardiomyopathy can occur with TAFINLAR.
In Trial 2, all patients were required to have an echocardiogram at baseline to document normal left ventricular ejection fraction (LVEF) and serial echocardiograms at Week 4, Week 12, and every 12 weeks thereafter. Cardiomyopathy, defined as a decrease in LVEF ≥ 10% from baseline and below the institutional lower limit of normal, occurred in 6% (12/206) of patients receiving TAFINLAR with trametinib and 2.9% (6/207) of patients receiving single-agent TAFINLAR. The median time to onset of cardiomyopathy on the TAFINLAR plus trametinib arm was 8.2 months (range: 28 days to 24.9 months), and was 4.4 months (range: 28 days to 19.1 months) on the TAFINLAR arm.
In Trial 2, cardiomyopathy was identified within the first month of initiation of TAFINLAR with trametinib in 2 of 12 patients, and in 2 of 6 patients receiving single-agent TAFINLAR. Development of cardiomyopathy in patients receiving TAFINLAR and trametinib resulted in dose interruption of TAFINLAR (4.4%) or discontinuation of TAFINLAR (1.0%). In patients receiving single-agent TAFINLAR, development of cardiomyopathy resulted in dose interruption (2.4%), dose reduction (0.5%), or discontinuation (1.0%). Cardiomyopathy resolved in 10 of 12 patients receiving TAFINLAR with trametinib, and in 3 of 6 patients receiving single-agent TAFINLAR.
Assess LVEF by echocardiogram or multigated acquisition (MUGA) scan before initiation of TAFINLAR with trametinib, one month after initiation of TAFINLAR, and then at 2- to 3-month intervals while on treatment. Withhold TAFINLAR for symptomatic cardiomyopathy or asymptomatic LV dysfunction of >20% from baseline that is below institutional lower limit of normal (LLN). Resume TAFINLAR at the same dose level upon recovery of cardiac function to at least the institutional LLN for LVEF and absolute decrease ≤10% compared to baseline [see Dosage and Administration (2.3)].
5.5 Uveitis
Uveitis (including iritis and iridocyclitis) can occur with TAFINLAR.
Uveitis occurred in 1% (6/586) of patients receiving TAFINLAR across multiple clinical trials and in 2% (9/559) of patients receiving TAFINLAR with trametinib across Trials 2 and 3. Treatment employed in clinical trials included steroid and mydriatic ophthalmic drops.
Monitor patients for visual signs and symptoms of uveitis (e.g., change in vision, photophobia, eye pain). If iritis is diagnosed, administer ocular therapy and continue TAFINLAR without dose modification; for severe uveitis or iridocyclitis, interrupt TAFINLAR and treat as clinically indicated. Permanently discontinue TAFINLAR for persistent Grade 2 or greater uveitis of >6 weeks duration [see Dosage and Administration (2.3)].
5.6 Serious Febrile Reactions
Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills, dehydration, or renal failure, can occur with TAFINLAR.
The incidence and severity of pyrexia are increased when TAFINLAR is administered with trametinib compared with TAFINLAR as a single agent [see Adverse Reactions (6.1)].
In Trial 1, the incidence of fever (serious and non-serious) was 28% in patients receiving TAFINLAR and 10% in patients receiving dacarbazine. In patients receiving TAFINLAR, the median time to initial onset of fever (any severity) was 11 days (range: 1 day to 6.6 months) and the median duration of fever was 3 days (range: 1 day to 4.2 months). Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills occurred in 3.7% (7/187) of patients receiving TAFINLAR and in none of the 59 patients receiving dacarbazine.
In Trials 2 and 3, fever occurred in 54% (303/559) of patients receiving TAFINLAR with trametinib; the median time to onset of first occurrence of fever was 1 month (range: 1 day to 23.5 months) and the median duration of fever was 3 days (range: 1 day to 11.3 months). Approximately one-half of the patients who received TAFINLAR with trametinib and experienced pyrexia had 3 or more discrete episodes.
Serious febrile reactions or fever of any severity complicated by severe rigors/chills, hypotension, dehydration, renal failure, or syncope, occurred in 17% (93/559) of patients receiving TAFINLAR with trametinib. Fever was complicated by severe chills/rigors in 0.4% (2/559), dehydration in 1.8% (10/559), renal failure in 0.5% (3/559), and syncope in 0.7% (4/559) of patients.
Withhold TAFINLAR for fever of 101.3ºF or higher. Withhold TAFINLAR for any serious febrile reaction or fever complicated by hypotension, rigors or chills, dehydration, or renal failure and evaluate for signs and symptoms of infection. Monitor serum creatinine and other evidence of renal function during and following severe pyrexia. Refer to Table 2 for recommended dose modifications for adverse reactions [see Dosage and Administration (2.3)]. Administer antipyretics as secondary prophylaxis when resuming TAFINLAR if patient had a prior episode of severe febrile reaction or fever associated with complications. Administer corticosteroids (e.g., prednisone 10 mg daily) for at least 5 days for second or subsequent pyrexia if temperature does not return to baseline within 3 days of onset of pyrexia, or for pyrexia associated with complications such as dehydration, hypotension, renal failure or severe chills/rigors, and there is no evidence of active infection.
5.7 Serious Skin Toxicity
Serious skin toxicity can occur with TAFINLAR.
Across clinical trials of TAFINLAR administered with trametinib (N = 559), serious skin toxicity occurred in 0.7% (4/559) of patients.
Withhold TAFINLAR for intolerable or severe skin toxicity. TAFINLAR may be resumed at the next lower dose level in patients with improvement or recovery from skin toxicity within 3 weeks [see Dosage and Administration (2.3)].
5.8 Hyperglycemia
Hyperglycemia can occur with TAFINLAR.
In Trial 1, 5 of 12 patients with a history of diabetes required more intensive hypoglycemic therapy receiving TAFINLAR. The incidence of Grade 3 hyperglycemia based on laboratory values was 6% (12/187) in patients receiving TAFINLAR compared with none of the dacarbazine-treated patients.
In Trial 2, 27% (4/15) of patients with a history of diabetes receiving TAFINLAR with trametinib and 13% (2/16) of patients with a history of diabetes receiving single-agent TAFINLAR required more intensive hypoglycemic therapy. Grade 3 and Grade 4 hyperglycemia based on laboratory values occurred in 5% (11/208) and 0.5% (1/208) of patients, respectively, receiving TAFINLAR with trametinib compared with 4.3% (9/209) for Grade 3 hyperglycemia and no patients with Grade 4 hyperglycemia for patients receiving single-agent TAFINLAR.
Monitor serum glucose levels upon initiation and as clinically appropriate when TAFINLAR is administered in patients with pre-existing diabetes or hyperglycemia.
5.9 Glucose-6-Phosphate Dehydrogenase Deficiency
TAFINLAR, which contains a sulfonamide moiety, confers a potential risk of hemolytic anemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Monitor patients with G6PD deficiency for signs of hemolytic anemia while taking TAFINLAR.
5.10 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TAFINLAR can cause fetal harm when administered to a pregnant woman. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose. If TAFINLAR is used during pregnancy or if the patient becomes pregnant while taking TAFINLAR, advise the patient of the potential risk to a fetus [see Use in Specific Populations (8.1)].
Advise female patients of reproductive potential to use an effective non-hormonal method of contraception since TAFINLAR can render hormonal contraceptives ineffective, during treatment and for 2 weeks after the last dose of TAFINLAR. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR [see Drug Interactions (7.2), Use in Specific Populations (8.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in another section of the label:
- •
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- •
- Tumor Promotion in BRAF Wild-Type Melanoma [see Warnings and Precautions (5.2)]
- •
- Hemorrhage [see Warnings and Precautions (5.3)]
- •
- Cardiomyopathy [see Warnings and Precautions (5.4)]
- •
- Uveitis [see Warnings and Precautions (5.5)]
- •
- Serious Febrile Reactions [see Warnings and Precautions (5.6)]
- •
- Serious Skin Toxicity [see Warnings and Precautions (5.7)]
- •
- Hyperglycemia [see Warnings and Precautions (5.8)]
- •
- Glucose-6-Phosphate Dehydrogenase Deficiency [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the Warnings and Precautions section and below reflect exposure to TAFINLAR as a single agent and in combination with trametinib.
TAFINLAR Administered as a Single Agent
The safety of TAFINLAR as a single agent was evaluated in 586 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma, previously treated or untreated, who received TAFINLAR 150 mg orally twice daily until disease progression or unacceptable toxicity, including 181 patients treated for at least 6 months and 86 additional patients treated for more than 12 months. TAFINLAR was studied in open-label, single-arm trials and in an open-label, randomized, active-controlled trial. The median daily dose of TAFINLAR was 300 mg (range: 118 to 300 mg).
Table 3 and Table 4 present adverse drug reactions and laboratory abnormalities identified from analyses of Trial 1 [see Clinical Studies (14.1)]. Trial 1, a multicenter, international, open-label, randomized (3:1), controlled trial allocated 250 patients with unresectable or metastatic BRAF V600E mutation-positive melanoma to receive TAFINLAR 150 mg orally twice daily (n = 187) or dacarbazine 1,000 mg/m2 intravenously every 3 weeks (n = 63). The trial excluded patients with abnormal left ventricular ejection fraction or cardiac valve morphology (≥Grade 2), corrected QT interval greater than or equal to 480 milliseconds on electrocardiogram, or a known history of glucose-6-phosphate dehydrogenase deficiency. The median duration on treatment was 4.9 months for patients treated with TAFINLAR and 2.8 months for dacarbazine-treated patients. The population exposed to TAFINLAR was 60% male, 99% White, and had a median age of 53 years.
The most commonly occurring adverse reactions (≥20%) in patients treated with TAFINLAR were, in order of decreasing frequency: hyperkeratosis, headache, pyrexia, arthralgia, papilloma, alopecia, and palmar-plantar erythrodysesthesia syndrome (PPES).
The incidence of adverse events resulting in permanent discontinuation of study medication in Trial 1 was 3% for patients treated with TAFINLAR and 3% for patients treated with dacarbazine. The most frequent (≥2%) adverse reactions leading to dose reduction of TAFINLAR were pyrexia (9%), PPES (3%), chills (3%), fatigue (2%), and headache (2%).
Table 3. Select Common Adverse Reactions Occurring in ≥10% (All Grades) or ≥2% (Grades 3 or 4) of Patients Treated with TAFINLARa
|
Primary System Organ Class Preferred Term |
TAFINLAR
|
Dacarbazine
|
||
|
All Grades (%) |
Grades 3 and 4b (%) |
All Grades (%) |
Grades 3 and 4 (%) |
|
|
Skin and subcutaneous tissue disorders | ||||
|
37 |
1 |
0 |
0 |
|
22 |
NAf |
2 |
NAf |
|
20 |
2 |
2 |
0 |
|
17 |
0 |
0 |
0 |
|
Nervous system disorders | ||||
|
32 |
0 |
8 |
0 |
|
General disorders and administration site conditions | ||||
|
28 |
3 |
10 |
0 |
|
Musculoskeletal and connective tissue disorders | ||||
|
27 |
1 |
2 |
0 |
|
12 |
3 |
7 |
0 |
|
11 |
0 |
0 |
0 |
|
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | ||||
|
27 |
0 |
2 |
0 |
|
7 |
4 |
0 |
0 |
|
Respiratory, thoracic, and mediastinal disorders | ||||
|
12 |
0 |
5 |
0 |
|
Gastrointestinal disorders | ||||
|
11 |
2 |
14 |
0 |
|
Infections and infestations | ||||
|
10 |
0 |
3 |
0 |
- aAdverse drug reactions, reported using MedDRA and graded using NCI CTCAE version 4.0 for assessment of toxicity.
- b Grade 4 adverse reactions limited to hyperkeratosis (n = 1) and constipation (n = 1).
- c Includes skin papilloma and papilloma.
- d cuSCC = cutaneous squamous cell carcinoma, includes squamous cell carcinoma of the skin and keratoacanthoma.
- e Cases of cuSCC were required to be reported as Grade 3 per protocol.
- f NA = not applicable.
Table 4. Incidence of Laboratory Abnormalities Increased from Baseline Occurring at a Higher Incidence in Patients Treated with TAFINLAR in Trial 1 [Between-Arm Difference of ≥5% (All Grades) or ≥2% (Grades 3 or 4)]a
|
Test |
TAFINLAR N = 187 |
DTIC N = 59 |
||
|
All Grades (%) |
Grades 3 and 4 (%) |
All Grades (%) |
Grades 3 and 4 (%) |
|
|
Hyperglycemia |
50 |
6 |
43 |
0 |
|
Hypophosphatemia |
37 |
6b |
14 |
2 |
|
Increased alkaline phosphatase |
19 |
0 |
14 |
2 |
|
Hyponatremia |
8 |
2 |
3 |
0 |
- a Adverse drug reactions, reported using MedDRA and graded using NCI CTCAE version 4.0 for assessment of toxicity.
- b Grade 4 laboratory abnormality limited to hypophosphatemia (n = 1).
Other clinically important adverse reactions observed in less than 10% of patients (N = 586) treated with TAFINLAR were:
Gastrointestinal Disorders: Pancreatitis.
Immune System Disorders: Hypersensitivity manifesting as bullous rash.
Renal and Urinary Disorders: Interstitial nephritis.
TAFINLAR Administered with Trametinib
The safety of TAFINLAR when administered with trametinib was evaluated in 559 patients with previously untreated, unresectable or metastatic, BRAF V600E or V600K mutation-positive melanoma who received TAFINLAR in two trials, Trial 2 (n = 209) a multicenter, double-blind, randomized (1:1), active controlled trial and Trial 3 (n = 350) a multicenter, open-label, randomized (1:1), active controlled trial. In Trials 2 and 3, patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily until disease progression or unacceptable toxicity. Both trials excluded patients with abnormal left ventricular ejection fraction, history of acute coronary syndrome within 6 months, history of Class II or greater congestive heart failure (New York Heart Association), history of RVO or RPED, QTcB interval ≥480 msec, treatment refractory hypertension, uncontrolled arrhythmias, active brain metastases, or a known history of G6PD deficiency [see Clinical Studies (14.2)].
Among these 559 patients, 199 (36%) were exposed to TAFINLAR for > 6 months to 12 months while 185 (33%) were exposed to TAFINLAR for ≥ 1 year. The median age was 55 years (range: 18 to 91), 57% were male, 98% were White, 72% had baseline ECOG performance status 0 and 28% had ECOG performance status 1, 64% had M1c stage disease, 35% had elevated LDH at baseline and 0.5% had a history of brain metastases.
The most commonly occurring adverse reactions (≥20%) for TAFINLAR in patients receiving TAFINLAR plus trametinib in Trials 2 and 3 were: pyrexia, rash, chills, headache, arthralgia, and cough.
Table 5 and Table 6 present adverse drug reactions and laboratory abnormalities, respectively, observed in Trial 2.
The demographics and baseline tumor characteristics of patients enrolled in Trial 2 are summarized in Clinical Studies [see Clinical Studies (14.2)]. Patients receiving TAFINLAR plus trametinib had a median duration of exposure of 11 months (range: 3 days to 30 months) to TAFINLAR. Among the 209 patients receiving TAFINLAR plus trametinib, 26% were exposed to TAFINLAR for >6 months to 12 months while 46% were exposed to TAFINLAR for >1 year.
In Trial 2, adverse reactions resulting in discontinuation of TAFINLAR occurred in 11% of patients receiving TAFINLAR plus trametinib; the most common was pyrexia (1.9%). Adverse reactions leading to dose reductions of TAFINLAR occurred in 26% of patients receiving TAFINLAR plus trametinib; the most common were pyrexia (14%), neutropenia (1.9%), rash (1.9%), and chills (1.9%). Adverse reactions leading to dose interruptions of Tafinlar occurred in 56% of patients receiving TAFINLAR plus trametinib; the most common were pyrexia (35%), chills (11%), vomiting (7%), nausea (5%), and decreased ejection fraction (5%).
Table 5. Select Adverse Reactions Occurring in ≥10% (All Grades) of Patients Treated with TAFINLAR in Combination with Trametinib in Trial 2a
|
Trial 2 |
||||||
|
Pooled TAFINLAR plus Trametinib N = 559 |
TAFINLAR plus Trametinib N =209 |
TAFINLAR N = 211 |
||||
|
Adverse Reactions |
All Grades (%) |
Grades 3 and 4b (%) |
All Grades (%) |
Grades 3 and 4 (%) |
All Grades (%) |
Grades 3 and 4 (%) |
|
General disorders and administrative site conditions |
||||||
|
54 |
5 |
57 |
7 |
33 |
1.9 |
|
31 |
0.5 |
31 |
0 |
17 |
0.5 |
|
Gastrointestinal disorders |
||||||
|
13 |
0.2 |
13 |
0.5 |
10 |
0 |
|
Nervous system disorders |
||||||
|
30 |
0.9 |
33 |
0.5 |
30 |
1.4 |
|
11 |
0.2 |
14 |
0 |
7 |
0 |
|
Musculoskeletal, connective tissue, and bone disorders |
||||||
|
25 |
0.9 |
26 |
0.9 |
31 |
0 |
|
15 |
0.2 |
13 |
0.5 |
13 |
0 |
|
Skin and subcutaneous tissue disorders |
||||||
|
32 |
1.1 |
42 |
0 |
27 |
1.4 |
|
10 |
0 |
12 |
0 |
16 |
0 |
|
Respiratory, thoracic, and mediastinal disorders |
||||||
|
20 |
0 |
21 |
0 |
21 |
0 |
|
Infections and infestations |
||||||
|
12 |
0 |
12 |
0 |
10 |
0 |
- aNCI CTCAE version 4
- bGrade 4 adverse reactions limited to headache (n = 1).
- cIncludes rash generalized, rash pruritic, rash erythematous, rash papular, rash vesicular, rash macular, rash maculo-papular, and rash folliculitis.
Other clinically important adverse reactions for TAFINLAR across Trials 2 and 3 (N = 559) observed in less than 10% of patients receiving TAFINLAR in combination with trametinib were:
Gastrointestinal Disorders: pancreatitis
Subcutaneous Tissue Disorders: panniculitis
Table 6. Select Treatment-Emergent Laboratory Abnormalities Occurring at ≥10% (All Grades) of Patients Receiving TAFINLAR with Trametinib in Trial 2
|
Trial 2 |
||||||
|
Pooled TAFINLAR plus Trametinib N = 559a |
TAFINLAR plus Trametinib N = 209b |
TAFINLAR N = 211b |
||||
|
Test |
All Grades (%) |
Grades 3 and 4c (%) |
All Grades (%) |
Grades 3 and 4c (%) |
All Grades (%) |
Grades 3 and 4c (%) |
|
Liver Function Tests |
||||||
|
49 |
2.7 |
50 |
1.0 |
25 |
0.5 |
|
Chemistry |
||||||
|
60 |
4.7 |
65 |
6 |
57 |
4.3 |
|
38 |
6 |
38 |
3.8 |
35 |
7 |
|
25 |
8 |
24 |
6 |
14 |
2.9 |
- aFor these laboratory tests the denominator is 556.
- bFor these laboratory tests the denominator is 208 for the combination arm, 208-209 for the TAFINLAR arm.
- cGrade 4 adverse reactions limited to hyperglycemia (n = 4), hyponatremia and hypophosphatemia (each n = 1), in the pooled combination arm; hyperglycemia (n = 1) in the Trial 2 combination arm; hypophosphatemia (n = 1) in the TAFINLAR arm.
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Dabrafenib
Dabrafenib is primarily metabolized by CYP2C8 and CYP3A4. Strong inhibitors of CYP3A4 or CYP2C8 may increase concentrations of dabrafenib and strong inducers of CYP3A4 or CYP2C8 may decrease concentrations of dabrafenib [see Clinical Pharmacology (12.3)]. Substitution of strong inhibitors or strong inducers of CYP3A4 or CYP2C8 is recommended during treatment with TAFINLAR. If concomitant use of strong inhibitors (e.g., ketoconazole, nefazodone, clarithromycin, gemfibrozil) or strong inducers (e.g., rifampin, phenytoin, carbamazepine, phenobarbital, St John’s wort) of CYP3A4 or CYP2C8 is unavoidable, monitor patients closely for adverse reactions when taking strong inhibitors or loss of efficacy when taking strong inducers.
7.2 Effects of Dabrafenib on Other Drugs
Dabrafenib induces CYP3A4 and CYP2C9. Dabrafenib decreased the systemic exposures of midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), and R-warfarin (a CYP3A4/CYP1A2 substrate) [see Clinical Pharmacology (12.3)]. Monitor international normalized ratio (INR) levels more frequently in patients receiving warfarin during initiation or discontinuation of dabrafenib. Coadministration of TAFINLAR with other substrates of these enzymes, including dexamethasone or hormonal contraceptives, can result in decreased concentrations and loss of efficacy [see Use in Specific Populations (8.1, 8.3)]. Substitute for these medications or monitor patients for loss of efficacy if use of these medications is unavoidable.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies and its mechanism of action, TAFINLAR can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There is insufficient data in pregnant women exposed to TAFINLAR to assess the risks. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose of 150 mg twice daily [see Data]. If TAFINLAR is used during pregnancy or if the patient becomes pregnant while taking TAFINLAR, advise the patient of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data: In a combined female fertility and embryo-fetal development study in rats conducted during the period of organogenesis, developmental toxicity consisted of embryo-lethality, ventricular septal defects, and variation in thymic shape at a dabrafenib dose of 300 mg/kg/day (approximately three times the human exposure at the recommended dose based on AUC). At doses of 20 mg/kg/day or greater (equivalent to the human exposure at the recommended dose based on AUC), rats demonstrated delays in skeletal development and reduced fetal body weight.
8.2 Lactation
Risk Summary
There are no data on the presence of dabrafenib in human milk, or the effects of dabrafenib on the breastfed infant, or on milk production. Because of the potential for serious adverse reactions from TAFINLAR in breastfed infants, advise women not to breastfeed during treatment with TAFINLAR and for 2 weeks following the last dose of TAFINLAR.
8.3 Females and Males of Reproductive Potential
Based on data from animal studies and its mechanism of action, TAFINLAR can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with TAFINLAR and for 2 weeks after the last dose of TAFINLAR. Counsel patients to use a non-hormonal method of contraception since TAFINLAR can render hormonal contraceptives ineffective [see Drug Interactions (7.1)]. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR.
Infertility
Females
Advise female patients of reproductive potential that TAFINLAR may impair fertility. A reduction in fertility was observed in female rats at dose exposures equivalent to the human exposure at the recommended dose. A reduction in the number of corpora lutea was noted in pregnant rats at dose exposures approximately three times the human exposure at the recommended dose [see Nonclinical Toxicology (13.1)].
Males
Advise male patients of the potential risk for impaired spermatogenesis which may be irreversible. Effects on spermatogenesis have been observed in animals treated with dabrafenib at dose exposures up to three times the human exposure at the recommended dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TAFINLAR as a single agent or with trametinib have not been established in pediatric patients.
Juvenile Animal Data
In a repeat-dose toxicity study in juvenile rats, an increased incidence of kidney cysts and tubular deposits were noted at doses as low as 0.2 times the human exposure at the recommended adult dose based on AUC. Additionally, forestomach hyperplasia, decreased bone length, and early vaginal opening were noted at doses as low as 0.8 times the human exposure at the recommended adult dose based on AUC.
8.5 Geriatric Use
One hundred and twenty-six (22%) of 586 patients in clinical trials of TAFINLAR administered as a single agent and 40 (21%) of the 187 patients receiving TAFINLAR in Trial 1 were greater than or equal to 65 years of age. No overall differences in the effectiveness or safety of TAFINLAR were observed in elderly patients as compared to younger patients in Trial 1.
Of the 559 patients randomized to receive TAFINLAR plus trametinib in Trials 2 and 3, 24% were aged 65 years and older and 6% patients aged 75 years and older. No overall differences in the effectiveness of TAFINLAR plus trametinib were observed in elderly patients as compared to younger patients. The incidences of peripheral edema (26% vs. 12%) and anorexia (21% vs. 9%) were increased in elderly patients as compared to younger patients.
8.6 Hepatic Impairment
No formal pharmacokinetic trial in patients with hepatic impairment has been conducted. Dose adjustment is not recommended for patients with mild hepatic impairment based on the results of the population pharmacokinetic analysis. As hepatic metabolism and biliary secretion are the primary routes of elimination of dabrafenib and its metabolites, patients with moderate to severe hepatic impairment may have increased exposure. An appropriate dose has not been established for patients with moderate to severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No formal pharmacokinetic trial in patients with renal impairment has been conducted. Dose adjustment is not recommended for patients with mild or moderate renal impairment based on the results of the population pharmacokinetic analysis. An appropriate dose has not been established for patients with severe renal impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no information on overdosage of TAFINLAR. Since dabrafenib is highly bound to plasma proteins, hemodialysis is likely to be ineffective in the treatment of overdose with TAFINLAR.
11 DESCRIPTION
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure.
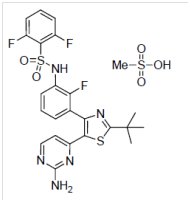
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50 mg and 75 mg capsules for oral administration. Each 50 mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75 mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dabrafenib is an inhibitor of some mutated forms of BRAF kinases with in vitro IC50 values of 0.65, 0.5, and 1.84 nM for BRAF V600E, BRAF V600K, and BRAF V600D enzymes, respectively. Dabrafenib also inhibits wild-type BRAF and CRAF kinases with IC50 values of 3.2 and 5.0 nM, respectively, and other kinases such as SIK1, NEK11, and LIMK1 at higher concentrations. Some mutations in the BRAF gene, including those that result in BRAF V600E, can result in constitutively activated BRAF kinases that may stimulate tumor cell growth [see Indications and Usage (1)]. Dabrafenib inhibits BRAF V600 mutation-positive melanoma cell growth in vitro and in vivo.
Dabrafenib and trametinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Use of dabrafenib and trametinib in combination resulted in greater growth inhibition of BRAF V600 mutation-positive melanoma cell lines in vitro and prolonged inhibition of tumor growth in BRAF V600 mutation positive melanoma xenografts compared with either drug alone.
12.2 Pharmacodynamics
Cardiac Electrophysiology
A dedicated study to evaluate the QT prolongation potential has not been conducted for TAFINLAR. In clinical trials, QTc (heart rate-corrected QT) prolongation to ≥500 ms occurred in 0.8% (2/264) of patients receiving TAFINLAR plus trametinib and in 1.5 % (4/264) of patients receiving TAFINLAR as a single agent. The QTc was increased >60 ms from baseline in 3.8% (10/264) of patients receiving TAFINLAR plus trametinib and 3% (8/264) of patients treated with TAFINLAR as a single agent.
12.3 Pharmacokinetics
Absorption
After oral administration, median time to achieve peak plasma concentration (Tmax) is 2 hours. Mean absolute bioavailability of oral dabrafenib is 95%. Following a single dose, dabrafenib exposure (Cmax and AUC) increased in a dose-proportional manner across the dose range of 12 to 300 mg, but the increase was less than dose-proportional after repeat twice-daily dosing. After repeat twice-daily dosing of 150 mg, the mean accumulation ratio was 0.73 and the inter-subject variability (CV%) of AUC at steady-state was 38%.
Administration of dabrafenib with a high-fat meal decreased Cmax by 51%, decreased AUC by 31%, and delayed median Tmax by 3.6 hours as compared with the fasted state [see Dosage and Administration (2.2)].
Distribution
Dabrafenib is 99.7% bound to human plasma proteins. The apparent volume of distribution (Vc/F) is 70.3 L.
Metabolism
The metabolism of dabrafenib is primarily mediated by CYP2C8 and CYP3A4 to form hydroxy-dabrafenib. Hydroxy-dabrafenib is further oxidized via CYP3A4 to form carboxy-dabrafenib and subsequently excreted in bile and urine. Carboxy-dabrafenib is decarboxylated to form desmethyl-dabrafenib; desmethyl-dabrafenib may be reabsorbed from the gut. Desmethyl-dabrafenib is further metabolized by CYP3A4 to oxidative metabolites. Mean metabolite-to-parent AUC ratios following repeat-dose administration are 0.9, 11, and 0.7 for hydroxy-, carboxy-, and desmethyl-dabrafenib, respectively. Based on systemic exposure, relative potency, and pharmacokinetic properties, both hydroxy- and desmethyl-dabrafenib are likely to contribute to the clinical activity of dabrafenib.
Elimination
The mean terminal half-life of dabrafenib is 8 hours after oral administration. Hydroxy-dabrafenib terminal half-life (10 hours) parallels that of dabrafenib while the carboxy- and desmethyl-dabrafenib metabolites exhibit longer half-lives (21 to 22 hours). The apparent clearance of dabrafenib is 17.0 L/h after single dosing and 34.4 L/h after 2 weeks of twice-daily dosing.
Fecal excretion is the major route of elimination accounting for 71% of radioactive dose while urinary excretion accounted for 23% of total radioactivity as metabolites only.
Specific Populations
Age, Body Weight, and Gender: Based on the population pharmacokinetics analysis, age has no effect on dabrafenib pharmacokinetics. Pharmacokinetic differences based on gender and on weight are not clinically relevant.
Pediatric: Pharmacokinetics of dabrafenib has not been studied in pediatric patients.
Renal Impairment: No formal pharmacokinetic trial in patients with renal impairment has been conducted. The pharmacokinetics of dabrafenib were evaluated using a population analysis in 233 patients with mild renal impairment (GFR 60 to 89 mL/min/1.73 m2) and 30 patients with moderate renal impairment (GFR 30 to 59 mL/min/1.73 m2) enrolled in clinical trials. Mild or moderate renal impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with severe renal impairment.
Hepatic Impairment: No formal pharmacokinetic trial in patients with hepatic impairment has been conducted. The pharmacokinetics of dabrafenib was evaluated using a population analysis in 65 patients with mild hepatic impairment enrolled in clinical trials. Mild hepatic impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with moderate to severe hepatic impairment.
Drug Interactions
Effect of Strong Inhibitors of CYP3A4 or CYP2C8 on Dabrafenib: In vitro studies show that dabrafenib is a substrate of CYP3A4 and CYP2C8 while hydroxy-dabrafenib and desmethyl-dabrafenib are CYP3A4 substrates. Coadministration of dabrafenib 75 mg twice daily and ketoconazole 400 mg once daily (a strong CYP3A4 inhibitor) for 4 days increased dabrafenib AUC by 71%, hydroxy-dabrafenib AUC by 82%, and desmethyl-dabrafenib AUC by 68%. Coadministration of dabrafenib 75 mg twice daily and gemfibrozil 600 mg twice daily (a strong CYP2C8 inhibitor) for 4 days increased dabrafenib AUC by 47%, with no change in the AUC of dabrafenib metabolites.
Effect of Dabrafenib on CYP Substrates: In vitro data demonstrate that dabrafenib is an inducer of CYP3A4 and CYP2B6 via activation of the pregnane X receptor (PXR) and constitutive androstane receptor (CAR) nuclear receptors. Dabrafenib may also induce CYP2C enzymes via the same mechanism. Coadministration of dabrafenib 150 mg twice daily for 15 days and a single dose of midazolam 3 mg (a CYP3A4 substrate) decreased midazolam AUC by 74%. Coadministration of dabrafenib 150 mg twice daily for 15 days and a single dose of warfarin 15 mg decreased the AUC of S-warfarin (a CYP2C9 substrate) by 37% and the AUC of R-warfarin (a CYP3A4/CYP1A2 substrate) by 33% [see Drug Interactions (7.2)].
Effect of Transporters on Dabrafenib: Dabrafenib is a substrate of human P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but is not a substrate of organic cation transporter (OCT1) or organic anion transporting polypeptide (OATP1B1, OATP1B3, OATP2B1) in vitro. Hydroxy-dabrafenib and desmethyl-dabrafenib are not substrates of OATP1B1 or OATP1B3 in vitro.
Effect of Dabrafenib on Transporters: Dabrafenib and its metabolites, hydroxy-dabrafenib, carboxy-dabrafenib, and desmethyl-dabrafenib, are inhibitors of OATP1B1, OATP1B3 and organic anion transporter (OAT1 and OAT3) in vitro. Dabrafenib and desmethyl-dabrafenib are inhibitors of BCRP in vitro.
Effect of Trametinib on Dabrafenib: Coadministration of trametinib 2 mg daily with dabrafenib 150 mg twice daily resulted in a 23% increase in AUC of dabrafenib, a 33% increase in AUC of desmethyl-dabrafenib, and no change in AUC of hydroxy-dabrafenib as compared with administration of dabrafenib.
Effect of Acid Reducing Agents on Dabrafenib: Drugs that alter the pH of the upper GI tract (e.g., proton pump inhibitors, H2-receptor antagonists, antacids) may alter the solubility of dabrafenib and reduce its bioavailability. However, no formal clinical trial has been conducted to evaluate the effect of gastric pH-altering agents on the systemic exposure of dabrafenib. When TAFINLAR is coadministered with a proton pump inhibitor, H2-receptor antagonist, or antacid, systemic exposure of dabrafenib may be decreased and the effect on efficacy of TAFINLAR is unknown.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with dabrafenib have not been conducted. TAFINLAR increased the risk of cutaneous squamous cell carcinomas in patients in clinical trials.
Dabrafenib was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test) or the mouse lymphoma assay, and was not clastogenic in an in vivo rat bone marrow micronucleus test.
In a combined female fertility and embryo-fetal development study in rats, a reduction in fertility was noted at doses greater than or equal to 20 mg/kg/day (equivalent to the human exposure at the recommended dose based on AUC). A reduction in the number of ovarian corpora lutea was noted in pregnant females at 300 mg/kg/day (which is approximately three times the human exposure at the recommended dose based on AUC).
Male fertility studies with dabrafenib have not been conducted; however, in repeat-dose studies, testicular degeneration/depletion was seen in rats and dogs at doses equivalent to and three times the human exposure at the recommended dose based on AUC, respectively.
13.2 Animal Toxicology and/or Pharmacology
Adverse cardiovascular effects were noted in dogs at dabrafenib doses of 50 mg/kg/day (approximately five times the human exposure at the recommended dose based on AUC) or greater, when administered for up to 4 weeks. Adverse effects consisted of coronary arterial degeneration/necrosis and hemorrhage, as well as cardiac atrioventricular valve hypertrophy/hemorrhage.
14 CLINICAL STUDIES
14.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma – TAFINLAR Administered as a Single Agent
In Trial 1, the safety and efficacy of TAFINLAR as a single agent were demonstrated in an international, multicenter, randomized (3:1), open-label, active-controlled trial conducted in 250 patients with previously untreated BRAF V600E mutation-positive, unresectable or metastatic melanoma. Patients with any prior use of BRAF inhibitors or MEK inhibitors were excluded. Patients were randomized to receive TAFINLAR 150 mg orally twice daily (n = 187) or dacarbazine 1,000 mg/m2 intravenously every 3 weeks (n = 63). Randomization was stratified by disease stage at baseline [unresectable Stage III (regional nodal or in-transit metastases), M1a (distant skin, subcutaneous, or nodal metastases), or M1b (lung metastases) versus M1c melanoma (all other visceral metastases or elevated serum LDH)]. The main efficacy outcome measure was progression-free survival (PFS) as assessed by the investigator. In addition, an independent radiology review committee (IRRC) assessed the following efficacy outcome measures in pre-specified supportive analyses: PFS, confirmed objective response rate (ORR), and duration of response.
The median age of patients in Trial 1 was 52 years. The majority of the trial population was male (60%), White (99%), had an ECOG performance status of 0 (67%), M1c disease (66%), and normal LDH (62%). All patients had tumor tissue with mutations in BRAF V600E as determined by a clinical trial assay at a centralized testing site. Tumor samples from 243 patients (97%) were tested retrospectively, using an FDA-approved companion diagnostic test, THxID™-BRAF assay.
The median durations of follow-up prior to initiation of alternative treatment in patients randomized to receive TAFINLAR was 5.1 months and in the dacarbazine arm was 3.5 months. Twenty-eight (44%) patients crossed over from the dacarbazine arm at the time of disease progression to receive TAFINLAR.
Trial 1 demonstrated a statistically significant increase in progression-free survival in the patients treated with TAFINLAR. Table 7 and Figure 1 summarize the PFS results.
|
Investigator-Assessed Endpoints† |
TAFINLAR N = 187 |
Dacarbazine N = 63 |
|
Progression-Free Survival | ||
|
78 (42%) |
41 (65%) |
|
76 |
41 |
|
2 |
0 |
|
5.1 (4.9, 6.9) |
2.7 (1.5, 3.2) |
|
0.33 (0.20, 0.54) |
|
|
P <0.0001 |
|
|
Confirmed Tumor Responses | ||
|
52% |
17% |
|
(44, 59) |
(9, 29) |
|
6 (3%) |
0 |
|
91 (48%) |
11 (17%) |
| ||
|
5.6 (5.4, NR) |
NR (5.0, NR) |
- † CI = Confidence interval; HR = Hazard ratio; CR = Complete response; PR = Partial response; NR = Not reached.
- a Pike estimator, stratified by disease state.
- b Stratified log-rank test.
Figure 1. Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival in Trial 1
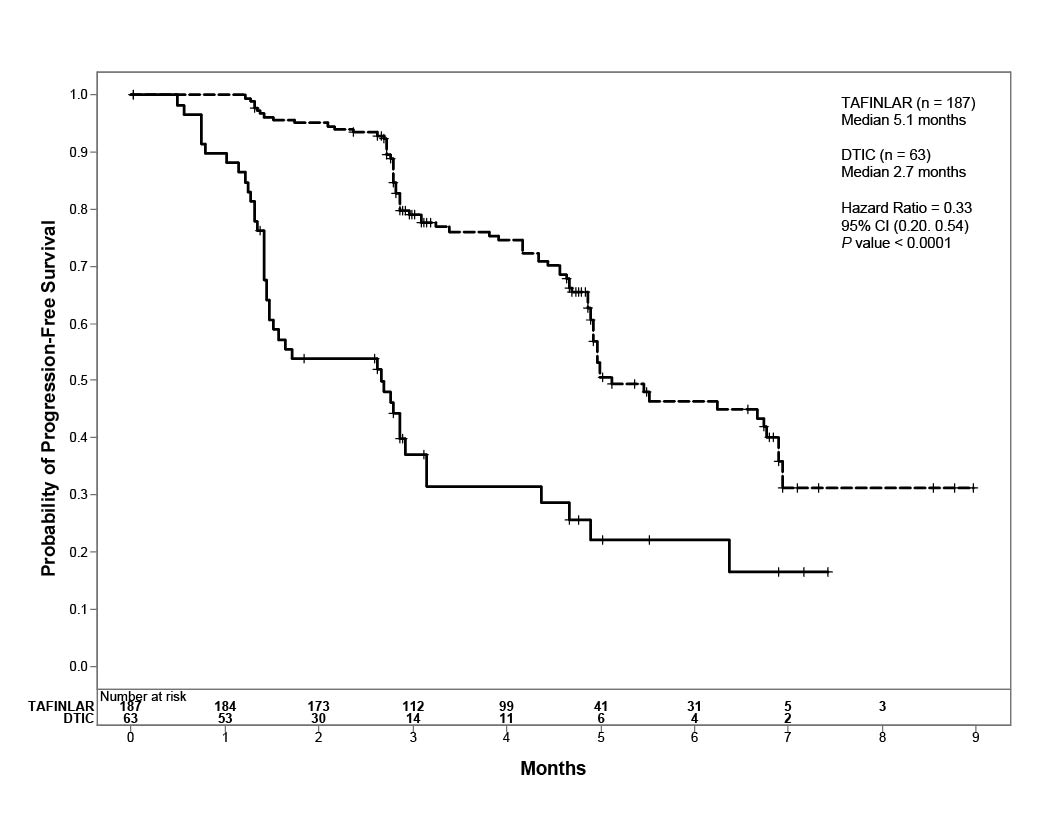
In supportive analyses based on IRRC assessment and in an exploratory subgroup analysis of patients with retrospectively confirmed V600E mutation-positive melanoma with the THxID™-BRAF assay, the PFS results were consistent with those of the primary efficacy analysis.
The activity of TAFINLAR for the treatment of BRAF V600E mutation-positive melanoma, metastatic to the brain was evaluated in a single-arm, open-label, two-cohort multicenter trial. All patients received TAFINLAR 150 mg twice daily. Patients in Cohort A (n = 74) had received no prior local therapy for brain metastases, while patients in Cohort B (n = 65) had received at least one local therapy for brain metastases, including, but not limited to, surgical resection, whole brain radiotherapy, or stereotactic radiosurgery such as gamma knife, linear-accelerated-based radiosurgery, or charged particles. In addition, patients in Cohort B were required to have evidence of disease progression in a previously treated lesion or an untreated lesion. Additional eligibility criteria were at least one measurable lesion of 0.5 cm or greater in largest diameter on contrast-enhanced MRI, stable or decreasing corticosteroid dose, and no more than two prior systemic regimens for treatment of metastatic disease. The primary outcome measure was estimation of the overall intracranial response rate (OIRR) in each cohort.
The median age of patients in Cohort A was 50 years, 72% were male, 100% were White, 59% had a pre-treatment ECOG performance status of 0, and 57% had an elevated LDH value at baseline. The median age of patients in Cohort B was 51 years, 63% were male, 98% were White, 66% had a pre-treatment ECOG performance status of 0, and 54% had an elevated LDH value at baseline. Efficacy results as determined by an independent radiology review committee, masked to investigator response assessments, are provided in Table 8.
|
IRRC-assessed Endpoints |
Cohort A n = 74 |
Cohort B n = 65 |
|
Overall Intracranial Response Rate (OIRR) | ||
|
18 (9.7, 28.2) |
18 (9.9, 30.0) |
|
Duration of OIRR |
(n = 13) |
(n = 12) |
|
Median, months (95% CI) |
4.6 (2.8, NR) |
4.6 (1.9, 4.6) |
IRRC = Independent radiology review committee; CI = Confidence interval; NR = Not reached.
14.2 BRAF V600E or V600K Unresectable or Metastatic Melanoma – TAFINLAR Administered with Trametinib
The safety and efficacy of TAFINLAR administered with trametinib were evaluated in two international, randomized, active-controlled trials: one double-blind trial (Trial 2) and one open-label trial (Trial 3).
Trial 2 compared TAFINLAR and trametinib to TAFINLAR and placebo as first-line therapy for patients with unresectable (Stage IIIC) or metastatic (Stage IV) BRAF V600E or V600K mutation-positive cutaneous melanoma. Patients were randomized (1:1) to receive TAFINLAR 150 mg twice daily and trametinib 2 mg once daily or TAFINLAR 150 mg twice daily plus matching placebo. Randomization was stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) vs. ≤ULN) and BRAF mutation subtype (V600E vs. V600K). The major efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST v1.1 with additional efficacy outcome measures of overall survival (OS) and confirmed overall response rate (ORR).
Trial 3 compared TAFINLAR and trametinib to vemurafenib as first-line treatment therapy for patients with unresectable (Stage IIIc) or metastatic (Stage IV) BRAF V600E or V600K mutation-positive cutaneous melanoma. Patients were randomized (1:1) to receive TAFINLAR 150 mg twice daily and trametinib 2 mg once daily or vemurafenib 960 mg twice daily. Randomization was stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) vs. ≤ULN) and BRAF mutation subtype (V600E vs. V600K). The major efficacy outcome measure was overall survival. Additional efficacy outcome measures were PFS and ORR as assessed by investigator per RECIST v1.1.
In Trial 2, 423 patients were randomized to TAFINLAR plus trametinib (n = 211) or TAFINLAR plus placebo (n = 212). The median age was 56 years (range: 22 to 89 years), 53% were male, >99% were White, 72% had ECOG performance status of 0, 4% had Stage IIIc, 66% had M1c disease, 65% had a normal LDH, and 2 patients had a history of brain metastases. All patients had tumor containing BRAF V600E or V600K mutations as determined by centralized testing, 85% with BRAF V600E mutations and 15% with BRAF V600K mutations.
In Trial 3, 704 patients were randomized to TAFINLAR plus trametinib (n = 352) or single-agent vemurafenib (n = 352). The median age was 55 years (range: 18 to 91 years), 96% were White, and 55% were male, 6% percent of patients had Stage IIIC, 61% had M1c disease, 67% had a normal LDH, 70% had ECOG performance status of 0, 89% had BRAF V600E mutation-positive melanoma, and one patient had a history of brain metastases.
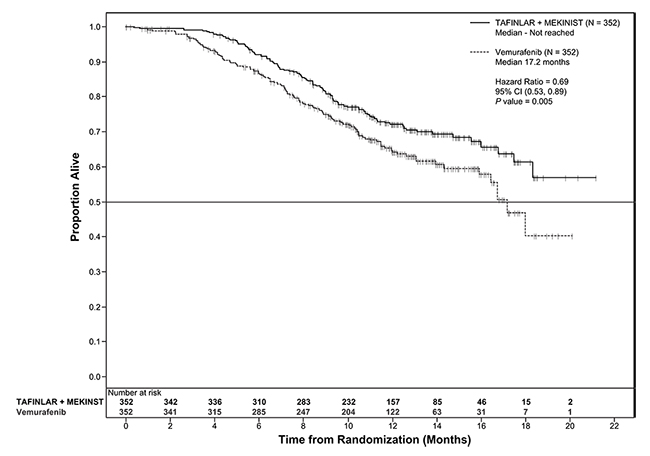
Trial 2 and Trial 3 demonstrated statistically significant improvements in OS and PFS (see Table 9 and Figures 2 and 3).
Table 9. Efficacy Results in Patients with BRAF V600E or V600K Melanomaa
|
Endpoint† |
Trial 2 |
Trial 3 |
||
|
TAFINLAR plus Trametinib N=211 |
TAFINLAR plus Placebo N=212 |
TAFINLAR plus Trametinib N=352 |
Vemurafenib N=352 |
|
|
Overall Survival |
||||
|
99 (47%) |
123 (58%) |
100 (28%) |
122 (35%) |
|
25.1 (19.2, NR) |
18.7 (15.2, 23.1) |
NR (18.3, NR) |
17.2 (16.4, NR) |
|
0.71 (0.55, 0.92) |
0.69 (0.53, 0.89) |
||
|
0.01 |
0.005a |
||
|
Progression-Free Survival (PFS)b |
||||
|
102 (48%) |
109 (51%) |
166 (47%) |
217 (62%) |
|
9.3 (7.7, 11.1) |
8.8 (5.9, 10.9) |
11.4 (9.9, 14.9) |
7.3 (5.8, 7.8) |
|
0.75 (0.57, 0.99) |
0.56 (0.46, 0.69) |
||
|
0.035 |
<0.001 |
||
|
Overall Response Rate (ORR)b |
||||
|
66 (60, 73) |
51 (44, 58) |
64 (59, 69) |
51 (46, 56) |
|
<0.001 |
<0.001 |
||
|
10 |
8 |
13 |
8 |
|
56 |
42 |
51 |
43 |
|
9.2 (7.4, NR) |
10.2 (7.5, NR) |
13.8 (11.0, NR) |
7.5 (7.3, 9.3) |
- †CI = Confidence interval; HR = Hazard ratio; CR = Complete response; PR = Partial response; NR = Not reached.
- a P-value is comparing with the allocated alpha of 0.021 for the interim analysis based on 77% information.
bPFS and ORR were assessed by investigator.
Figure 2. Kaplan-Meier Curves for Overall Survival in Trial 2
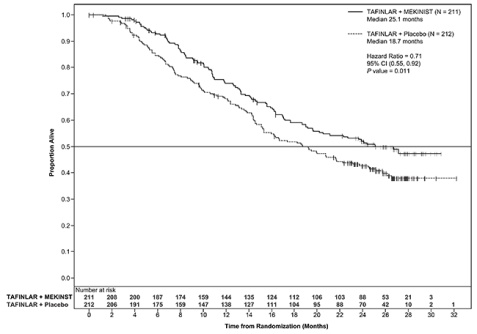
Figure 3. Kaplan-Meier Curves for Overall Survival in Trial 3
16 HOW SUPPLIED/STORAGE AND HANDLING
50 mg capsules: Dark red capsule imprinted with ‘GS TEW’ and ‘50 mg’ available in bottles of 120 (NDC 0173-0846-08). Each bottle contains a silica gel desiccant.
75 mg capsules: Dark pink capsule imprinted with ‘GS LHF’ and ‘75 mg’ available in bottles of 120 (NDC 0173-0847-08). Each bottle contains a silica gel desiccant.
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the following:
Confirmation of BRAF V600E or V600K mutation
- •
- TAFINLAR as a single agent: Evidence of BRAF V600E mutation in the tumor specimen using an FDA-approved test is necessary to identify patients for whom treatment is indicated [see Dosage and Administration (2.1)].
- •
- TAFINLAR with trametinib: Evidence of BRAF V600 mutation in tumor specimens using an FDA-approved test is necessary to identify patients for whom treatment is indicated [see Dosage and Administration (2.1)].
New cutaneous and non-cutaneous malignancies
- TAFINLAR increases the risk of developing new primary cutaneous and non-cutaneous malignancies. Advise patients to contact their healthcare provider immediately for any new lesions, changes to existing lesions on their skin, or signs and symptoms of other malignancies [see Warnings and Precautions (5.1)].
Hemorrhage
- TAFINLAR when administered with trametinib increases the risk of intracranial and gastrointestinal hemorrhage. Advise patients to contact their healthcare provider to seek immediate medical attention for signs or symptoms of unusual bleeding or hemorrhage [see Warnings and Precautions (5.3)].
Cardiomyopathy
- TAFINLAR can cause cardiomyopathy. Advise patients to immediately report any signs or symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.4)].
Uveitis
- TAFINLAR can cause uveitis, including iritis and iridocyclitis. Advise patients to contact their healthcare provider if they experience any changes in their vision [see Warnings and Precautions (5.5)].
Serious febrile reactions
- TAFINLAR can cause pyrexia including serious febrile reactions. Inform patients that the incidence and severity of pyrexia are increased when TAFINLAR is given in combination with trametinib. Instruct patients to contact their healthcare provider if they develop fever while taking TAFINLAR [see Warnings and Precautions (5.6)].
Serious skin toxicities
- TAFINLAR can cause serious skin toxicities. Advise patients to contact their healthcare provider for progressive or intolerable rash [see Warnings and Precautions (5.7)].
Hyperglycemia
- TAFINLAR can impair glucose control in diabetic patients resulting in the need for more intensive hypoglycemic treatment. Advise patients to contact their healthcare provider to report symptoms of severe hyperglycemia [see Warnings and Precautions (5.8)].
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
- TAFINLAR may cause hemolytic anemia in patients with G6PD deficiency. Advise patients with known G6PD deficiency to contact their healthcare provider to report signs or symptoms of anemia or hemolysis [see Warnings and Precautions (5.9)].
Embryo-fetal toxicity
- TAFINLAR can cause fetal harm if taken during pregnancy. Advise a pregnant woman of the potential risk to a fetus [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Females and males of reproductive potential
- Instruct females of reproductive potential to use non-hormonal, effective non-hormonal contraception during treatment and for 2 weeks after discontinuation of treatment with TAFINLAR. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Infertility
- Advise males and females of reproductive potential of the potential risk for impaired fertility with TAFINLAR [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment with TAFINLAR and for 2 weeks after the last dose of TAFINLAR [see Use in Specific Populations (8.2)].
Instructions for taking Tafinlar
- Instruct patients to take TAFINLAR at least 1 hour before or at least 2 hours after a meal [see Dosage and Administration (2.2)].
TAFINLAR is a registered trademark of the GSK group of companies.
THxID™ is a trademark of bioMérieux.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2015, GSK group of companies. All rights reserved.
TFR:4PI
|
MEDICATION GUIDE TAFINLAR® (TAFF-in-lar) (dabrafenib) capsules |
|
If your healthcare provider prescribes TAFINLAR for you to be taken with trametinib, also read the Patient Information leaflet that comes with trametinib. |
|
What is the most important information I should know about TAFINLAR? TAFINLAR may cause serious side effects, including the risk of new cancers: TAFINLAR, when used alone or with trametinib, may cause a type of skin cancer, called cutaneous squamous cell carcinoma (cuSCC). New melanoma lesions may happen in people who take TAFINLAR alone or with trametinib. TAFINLAR with trametinib, may cause new cancers including basal cell carcinoma. Talk with your healthcare provider about your risk for these cancers. Check your skin and tell your healthcare provider right away about any skin changes including a:
Your healthcare provider should check your skin before treatment with TAFINLAR, every two months during treatment with TAFINLAR, and for up to 6 months after you stop taking TAFINLAR to look for any new skin cancers. Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that develop during treatment with TAFINLAR. See "What are the possible side effects of TAFINLAR?" for more information about side effects. |
|
What is TAFINLAR? TAFINLAR is a prescription medicine used alone or with a medicine called trametinib, to treat people with a type of skin cancer called melanoma:
Your healthcare provider will perform a test to make sure that TAFINLAR is right for you. TAFINLAR alone or with trametinib is not used to treat people with a type of skin cancer called wild-type BRAF melanoma. It is not known if TAFINLAR alone or TAFINLAR with trametinib is safe and effective in children. |
|
What should I tell my healthcare provider before taking TAFINLAR? Before you take TAFINLAR, tell your healthcare provider if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. TAFINLAR and certain other medicines can affect each other, causing side effects. TAFINLAR may affect the way other medicines work, and other medicines may affect how TAFINLAR works. You can ask your pharmacist for a list of medicines that may interact with TAFINLAR. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
How should I take TAFINLAR?
|
|
What are the possible side effects of TAFINLAR? TAFINLAR may cause serious side effects, including:
The most common side effects of TAFINLAR alone or with trametinib include:
TAFINLAR may cause fertility problems in females. This could affect your ability to become pregnant. Talk to your healthcare provider if this is a concern for you. TAFINLAR may cause lower sperm counts in males. This could affect the ability to father a child. Talk to your healthcare provider if this is a concern for you. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of TAFINLAR. For more information about side effects, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store TAFINLAR?
|
|
General information about the safe and effective use of TAFINLAR Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TAFINLAR for a condition for which it was not prescribed. Do not give TAFINLAR to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TAFINLAR that is written for health professionals. |
|
What are the ingredients in TAFINLAR? Active ingredient: dabrafenib Inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose Capsule shells: hypromellose, red iron oxide (E172), titanium dioxide (E171). GlaxoSmithKline Research Triangle Park, NC 27709 Revised: November 2015 TAFINLAR is a registered trademark of the GSK group of companies. ©2015, GSK group of companies. All rights reserved. TFR:4MG |
PRINCIPAL DISPLAY PANEL
NDC 0173-0846-08
Tafinlar®
(Dabrafenib) Capsules
50 mg
Rx only
120 Capsules
Federal Law requires dispensing of TAFINLAR® with the Medication Guide provided with this bottle.
Each capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg dabrafenib.
Dosage: See accompanying prescribing information.
Store at 25°C (77°F); excursions permitted 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Do not use if printed safety seal under cap is broken or missing.
GlaxoSmithKline
RTP, NC 27709
- 10000000124205 Rev. 3/14

PRINCIPAL DISPLAY PANEL
NDC 0173-0847-08
Tafinlar®
(Dabrafenib) Capsules
75 mg
Rx only
120 Capsules
Federal Law requires dispensing of TAFINLAR® with the Medication Guide provided with this bottle.
Each capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg dabrafenib.
Dosage: See accompanying prescribing information.
Store at 25°C (77°F); excursions permitted 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Do not use if printed safety seal under cap is broken or missing.
GlaxoSmithKline
RTP, NC 27709
- 10000000124207 Rev. 3/14

| TAFINLAR
dabrafenib capsule |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| TAFINLAR
dabrafenib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - GlaxoSmithKline LLC (167380711) |