ONCASPAR- pegaspargase injection, solution
Baxalta US Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ONCASPAR safely and effectively. See full prescribing information for ONCASPAR.
ONCASPAR (pegaspargase) injection, for intramuscular or intravenous use Initial U.S. Approval: 1994 RECENT MAJOR CHANGESINDICATIONS AND USAGEDOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common (>5%) grade ≥ 3 adverse reactions with ONCASPAR included hypoalbuminemia, elevated transaminase, febrile neutropenia, hypertriglyceridemia, hyperglycemia, bilirubin increased, pancreatitis, abnormal clotting studies, embolic and thrombotic events, and hypersensitivity. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Baxalta US Inc., at 1-800-999-1785 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. See 17 for PATIENT COUNSELING INFORMATION. Revised: 1/2019 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Modifications
Monitor patients at least weekly, with bilirubin, transaminases, glucose and clinical examinations until recovery from the cycle of therapy. If an adverse reaction should occur, modify treatment according to Table 1.
| Adverse Reaction | Severity* | Action |
|---|---|---|
|
||
| Infusion Reaction/ Hypersensitivity Reaction | Grade 1 |
|
| Grade 2 |
|
|
| Grade 3 to 4 |
|
|
| Hemorrhage | Grade 3 to 4 |
|
| Pancreatitis | Grades 3 to 4 |
|
| Thromboembolism | Uncomplicated deep vein thrombosis |
|
| Severe or life-threatening thrombosis |
|
|
| Hepatotoxicity | Total bilirubin more than 3 times to no more than 10 times the upper limit of normal |
|
| Total bilirubin more than 10 times the upper limit of normal |
|
|
2.3 Preparation and Administration
Administer ONCASPAR in a healthcare setting with appropriate medical support and resuscitation equipment to manage hypersensitivity reactions, should they occur [see Warnings and Precautions (5.1)].
ONCASPAR is a clear and colorless solution. Visually inspect parenteral drug products for particulate matter, cloudiness, or discoloration prior to administration. If any of these are present, discard the vial.
When ONCASPAR is administered intramuscularly:
- Limit the volume at a single injection site to 2 mL.
- If the volume to be administered is greater than 2 mL, use multiple injection sites.
When ONCASPAR is administered intravenously:
- Dilute ONCASPAR with 100 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP, using aseptic technique.
- After dilution, administer immediately into a running infusion of either 0.9% Sodium Chloride, USP or 5% Dextrose Injection, USP, respectively.
- Administer over a period of 1-2 hours.
- Do not infuse other drugs through the same intravenous line during administration of ONCASPAR.
- The diluted solution should be used immediately. If immediate use is not possible, the diluted solution should be stored refrigerated at 2°C to 8°C (36°F to 46°F) for up to 48 hours. Protect infusion bags from direct sunlight.
ONCASPAR does not contain a preservative. Use only one dose per vial; discard unused product.
3 DOSAGE FORMS AND STRENGTHS
Injection: 3,750 International Units/5 mL (750 International Units/mL) clear, colorless solution in a single-dose vial.
4 CONTRAINDICATIONS
ONCASPAR is contraindicated in patients with a:
- History of serious hypersensitivity reactions, including anaphylaxis, to ONCASPAR or to any of the excipients [see Warnings and Precautions (5.1)].
- History of serious thrombosis with prior L-asparaginase therapy [see Warnings and Precautions (5.2)].
- History of pancreatitis, including pancreatitis related to prior L-asparaginase therapy [see Warnings and Precautions (5.3)].
- History of serious hemorrhagic events with prior L-asparaginase therapy [see Warnings and Precautions (5.5)].
- Severe hepatic impairment [see Warnings and Precautions (5.6)].
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Serious Hypersensitivity Reactions
Anaphylaxis and serious hypersensitivity reactions can occur in patients receiving ONCASPAR. The risk of serious hypersensitivity reactions is higher in patients with known hypersensitivity to E. coli derived L-asparaginase formulations. Other hypersensitivity reactions can include angioedema, lip swelling, eye swelling, erythema, blood pressure decreased, bronchospasm, dyspnea, pruritus, and rash.
Observe patients for 1 hour after administration of ONCASPAR in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis (for example, epinephrine, oxygen, intravenous steroids, antihistamines). Discontinue ONCASPAR in patients with serious hypersensitivity reactions [see Dosage and Administration (2.2)].
5.2 Thrombosis
Serious thrombotic events, including sagittal sinus thrombosis can occur in patients receiving ONCASPAR. Discontinue ONCASPAR in patients with serious thrombotic events [see Dosage and Administration (2.2)].
5.3 Pancreatitis
Pancreatitis can occur in patients receiving ONCASPAR. Hemorrhagic or necrotizing pancreatitis with fatal outcomes have been reported.
Inform patients of the signs and symptoms of pancreatitis, which, if left untreated, could be fatal. Assess serum amylase and/or lipase levels to confirm early signs of pancreatic inflammation. Discontinue ONCASPAR in patients where pancreatitis is suspected. If pancreatitis is confirmed, do not resume ONCASPAR [see Dosage and Administration (2.2)].
5.4 Glucose Intolerance
Glucose intolerance can occur in patients receiving ONCASPAR. In some cases, glucose intolerance is irreversible. Monitor serum glucose.
5.5 Hemorrhage
Increased prothrombin time, increased partial thromboplastin time, and hypofibrinogenemia can occur in patients receiving ONCASPAR. Evaluate patients with signs and symptoms of hemorrhage with coagulation parameters including PT, PTT, fibrinogen. Consider appropriate replacement therapy in patients with severe or symptomatic coagulopathy [see Dosage and Administration (2.2)].
5.6 Hepatotoxicity
Hepatotoxicity and abnormal liver function, including elevations of transaminase, bilirubin (direct and indirect), reduced serum albumin, and plasma fibrinogen can occur. Evaluate bilirubin and transaminases at least weekly during cycles of treatment that include ONCASPAR through at least 6 weeks after the last dose of ONCASPAR. In the event of serious liver toxicity, discontinue treatment with ONCASPAR and provide supportive care [see Dosage and Administration (2.2)].
6 ADVERSE REACTIONS
The following serious adverse reactions are described in greater detail in other sections of the labeling:
- Anaphylaxis and serious hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Thrombosis [see Warnings and Precautions (5.2)]
- Pancreatitis [see Warnings and Precautions (5.3)]
- Glucose intolerance [see Warnings and Precautions (5.4)]
- Hemorrhage [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
The most common grade 3 and 4 adverse reactions (>5%) included: hypoalbuminemia, elevated transaminase, febrile neutropenia, hypertriglyceridemia, hyperglycemia, bilirubin increased, pancreatitis abnormal clotting studies, embolic and thrombotic events, and hypersensitivity.
First-Line Treatment of Acute Lymphoblastic Leukemia (ALL)
Study CCG-1962 was a randomized (1:1), active-controlled study that enrolled 118 patients, with a median age of 4.7 years (1.1-9.9 years), of whom 54% were males and 65% White, 14% Hispanic, 8% Black, 8% Asian, and 6% other race. Of the 59 patients in Study 1 who were randomized to ONCASPAR, 48 patients (81%) received all 3 planned doses of ONCASPAR, 6 (10%) received 2 doses, 4 (7%) received 1 dose, and 1 patient (2%) did not receive the assigned treatment.
In Study CCG-1962, detailed safety information was collected for pre-specified adverse reactions identified as asparaginase-induced adverse reactions and for grade 3 and 4 nonhematologic adverse reactions according to the Children's Cancer Group (CCG) Toxicity and Complication Criteria. The per-patient incidence, by treatment arm, for these selected adverse reactions occurring at a severity of grade 3 or 4 are presented in Table 2 below:
| ONCASPAR (n=58) | Native E. coli L-Asparaginase (n=59) |
|
|---|---|---|
| Abnormal Liver Tests | 3 (5%) | 5 (8%) |
| Elevated Transaminases* | 2 (3%) | 4 (7%) |
| Hyperbilirubinemia | 1 (2%) | 1 (2%) |
| Hyperglycemia | 3 (5%) | 2 (3%) |
| Central Nervous System Thrombosis | 2 (3%) | 2 (3%) |
| Coagulopathy† | 1 (2%) | 3 (5%) |
| Pancreatitis | 1 (2%) | 1 (2%) |
| Allergic Reactions to Asparaginase | 1 (2%) | 0 |
The safety of ONCASPAR was investigated in Study DFCI 11-001, an open-label, randomized, active-controlled multicenter clinical trial that included 119 children and adolescents with newly-diagnosed ALL or lymphoblastic lymphoma treated with ONCASPAR in combination with the Dana Farber Cancer Institute (DFCI) ALL Consortium backbone therapy. The median age on enrollment was 4 years (range, 1-18) years. The majority of patients were male (60%) and white (75%). Most patients were considered standard risk ALL (59%) and had B-cell lineage ALL (87%).
The median number of doses of ONCASPAR during the study was 16 doses (one dose during induction therapy then administered every two weeks during post induction therapy). The median duration of exposure to ONCASPAR was 8 months. Table 3 summarizes the incidence of selected Grades ≥ 3 adverse reactions that occurred in 8 or more patients receiving ONCASPAR. Because not all grade 1 and 2 adverse reactions were collected prospectively, only grade 3 and 4 adverse reactions are presented in Table 3.
| Adverse Reaction* | ONCASPAR 2500 IU/m2 N=119 Grade ≥ 3† n (%) |
|---|---|
|
|
| Elevated transaminase* | 79 (66) |
| Febrile neutropenia | 48 (40) |
| Hypertriglyceridemia | 36 (30) |
| Hypoalbuminemia | 33 (28) |
| Bilirubin increased* | 30 (25) |
| Hyperglycemia | 29 (24) |
| Pancreatitis* | 29 (24) |
| Abnormal clotting studies* | 25 (21) |
| Embolic and thrombotic events* | 10 (8) |
| Hypersensitivity* | 8 (7) |
Previously Treated ALL
Adverse reaction information was obtained from 5 clinical trials that enrolled a total of 174 patients with relapsed ALL who received ONCASPAR as a single agent or in combination with multi-agent chemotherapy [see Clinical Studies (14.2)]. The toxicity profile of ONCASPAR in patients with previously treated relapsed ALL is similar to that reported above with the exception of clinical allergic reactions (see Table 3). The most common adverse reactions of ONCASPAR were clinical allergic reactions, elevated transaminases, hyperbilirubinemia, and coagulopathies. The most common serious adverse events due to ONCASPAR treatment were thrombosis (4%), hyperglycemia requiring insulin therapy (3%), and pancreatitis (1%).
Allergic Reactions
Allergic reactions include the following: bronchospasm, hypotension, laryngeal edema, local erythema or swelling, systemic rash, and urticaria.
Among 58 ONCASPAR-treated patients enrolled in Study CCG-1962, clinical allergic reactions were reported in 2 patients (3%). One patient experienced a grade 1 allergic reaction and the other grade 3 hives; both occurred during the first delayed intensification phase of the study (see Table 4).
Among 62 patients with relapsed ALL and prior hypersensitivity reactions to asparaginase, 35 patients (56%) had a history of clinical allergic reactions to native Escherichia (E.) coli L-asparaginase, and 27 patients (44%) had a history of clinical allergic reactions to both native E. coli and native Erwinia L-asparaginase. Twenty (32%) of these 62 patients experienced clinical allergic reactions to ONCASPAR (see Table 4).
Among 112 patients with relapsed ALL with no prior hypersensitivity reactions to asparaginase, 11 patients (10%) experienced clinical allergic reactions to ONCASPAR (see Table 4).
| Patient Status | Toxicity Grade, n (%) | Total | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| Previously Hypersensitive Patients (n=62) | 7 (11) | 8 (13) | 4 (6) | 1 (2) | 20 (32) |
| Non-Hypersensitive Patients (n=112) | 5 (4) | 4 (4) | 1 (1) | 1 (1) | 11 (10) |
| First-Line (n=58) | 1 (2) | 0 | 1 (2) | 0 | 2 (3) |
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to pegaspargase with the incidence of antibodies in other studies or to other products may be misleading.
In Study CCG-1962, ONCASPAR-treated patients were assessed for evidence of binding antibodies using an enzyme-linked immunosorbent assay (ELISA) method. The incidence of protocol-specified "high-titer" antibody formation was 2% in Induction (n=48), 10% in Delayed Intensification 1 (n=50), and 11% in Delayed Intensification 2 (n=44).
There is insufficient information to determine whether the development of antibodies is associated with an increased risk of clinical allergic reactions or altered pharmacokinetics (i.e., loss of asparaginase activity).
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ONCASPAR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Coagulopathy.
Gastrointestinal disorders: Hepatic impairment, pancreatic cyst, pancreatitis.
Immune system disorders: Anaphylactic shock, hypersensitivity reaction.
Investigations: Blood cholesterol increased.
Metabolism and nutrition disorders: Hyperglycemia, hyperammonemia.
Vascular disorders: Hemorrhage including central nervous system hemorrhage, thrombosis including superior sagittal sinus thrombosis.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk summary
There are no available data on the use of ONCASPAR in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Published literature studies in pregnant animals suggest asparagine depletion may cause harm to the animal offspring (see Data). Advise patients of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2%-4% and 15%-20%, respectively.
Data
Animal Data
Animal reproduction studies have not been conducted with ONCASPAR to evaluate its effect on reproduction and fetal development. Published literature studies in which pregnant rabbits were administered L-asparaginase or pregnant rats were deprived of dietary asparagine suggested harm to the animal offspring.
8.2 Lactation
Risk summary
There are no data on the presence of pegaspargase in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for adverse reactions in a breastfed child, advise lactating women not to breastfeed while receiving ONCASPAR and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
Based on published literature studies in pregnant animals, ONCASPAR can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential prior to starting treatment with ONCASPAR.
Contraception
Advise females of reproductive potential to avoid becoming pregnant while receiving ONCASPAR. Females should use effective contraceptive methods, including a barrier method, during treatment and for at least 3 months after the last dose of ONCASPAR. Since there is a potential for an indirect interaction between ONCASPAR and oral contraceptives, the concomitant use of ONCASPAR and oral contraceptives is not recommended. Another, non-oral contraceptive method should be used in women of childbearing potential.
8.4 Pediatric Use
The safety and effectiveness of ONCASPAR in the treatment of ALL have been established in pediatric patients. Use of ONCASPAR in these age groups is supported by evidence of efficacy as first-line treatment from one adequate and well-controlled trial, and evidence of efficacy for treatment of patients with hypersensitivity to asparaginase from four adequate and well-controlled trials [see Clinical Studies (14.1)], and safety data from 7 total trials. The pediatric patients treated with ONCASPAR 2,500 International Units/m2 on these trials included 26 infants (1 month to <2 years old), 165 children (2 years to <12 years old), and 39 adolescents (12 to 17 years old).
10 OVERDOSAGE
Three patients received 10,000 International Units/m2 of ONCASPAR as an intravenous infusion. One patient experienced a slight increase in liver enzymes. A second patient developed a rash 10 minutes after the start of the infusion, which was controlled with the administration of an antihistamine and by slowing down the infusion rate. A third patient did not experience any adverse reactions.
There is no specific antidote for ONCASPAR overdosage. In case of overdose, monitor patients closely for signs and symptoms of adverse reactions, and appropriately manage with symptomatic and supportive treatment.
11 DESCRIPTION
Pegaspargase is a conjugate of monomethoxypolyethylene glycol (mPEG) and L-asparaginase (L-asparagine amidohydrolase), an asparagine specific enzyme. L-asparaginase is a tetrameric enzyme that is produced endogenously by E. coli and consists of identical 34.5 kDa subunits. Approximately 69 to 82 molecules of mPEG are linked to L-asparaginase; the molecular weight of each mPEG molecule is about 5 kDa. ONCASPAR activity is expressed in International Units.
ONCASPAR (pegaspargase) injection is supplied as a clear, colorless, preservative-free, isotonic sterile solution in phosphate-buffered saline, pH 7.3, for intramuscular use or for dilution prior to intravenous infusion. Each vial of ONCASPAR contains 3,750 International Units of pegaspargase in 5 mL of solution. Each milliliter contains 750 International Units of pegaspargase, dibasic sodium phosphate, USP (5.58 mg), monobasic sodium phosphate, USP (1.20 mg), and Sodium Chloride, USP (8.50 mg) in Water for Injection, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
L-asparaginase is an enzyme that catalyzes the conversion of the amino acid L-asparagine into aspartic acid and ammonia. The pharmacological effect of ONCASPAR is thought to be based on selective killing of leukemic cells due to depletion of plasma L-asparagine. Leukemic cells with low expression of asparagine synthetase have a reduced ability to synthesize L-asparagine and therefore depend on an exogenous source of L-asparagine for survival.
12.2 Pharmacodynamics
Pharmacodynamic activity was assessed through serial measurements of asparagine in sera and cerebrospinal fluid (CSF).
In Study CCG-1962, pharmacodynamics were assessed in 57 newly diagnosed pediatric patients with standard-risk ALL who received three intramuscular doses of ONCASPAR (2,500 International Units/m2), one each during induction and two delayed intensification treatment phases [see Clinical Studies (14.1)].
In Study AALL07P4, the pharmacodynamic response of pegaspargase was assessed in 47 evaluable patients with newly diagnosed high risk B-precursor ALL. Asparagine concentrations in plasma (N=42) were maintained below the assay limit of quantification for at least 11 days following a single dose of ONCASPAR 2,500 International Units/m2 during the induction phase. CSF asparagine concentration was decreased from a mean pretreatment concentration of 0.6 µg/mL (N=20) to 0.2 µg/mL on Day 4 (N=41) and remained decreased at 0.2 µg/mL (N=39) 25 days after the administration of a single dose of ONCASPAR in the induction phase.
12.3 Pharmacokinetics
Pharmacokinetic assessments were based on an enzymatic assay measuring asparaginase activity after intramuscular (IM, CCG-1962) and intravenous (IV, AALL07P4) administration or 2,500 International Units/m2 in patients with ALL.
Absorption
The mean maximum asparaginase activity (Cmax) was reached at approximately 1 IU/mL (n=45-52) on Day 5 after a single IM injection. The mean half-life of absorption from the IM site was 1.7 days. The relative bioavailability was 82% following the first IM dose and 98% following repeat dosing.
The mean Cmax and the area under the curve (AUC0-inf) was 1.6 IU/mL and 16.6 IU/mL*day, respectively, after a single IV infusion (n=47) during the induction phase.
Distribution
The mean volume of distribution at steady state was estimated to be 1.86 L/m2 after a single IM injection and approximately 2 L after a single IV infusion based on non-compartmental analysis.
14 CLINICAL STUDIES
14.1 First-Line Treatment of ALL
Study CCG-1962
The safety and effectiveness of ONCASPAR was evaluated in an open-label, multicenter, randomized, active-controlled study (Study CCG-1962). In this study, 118 pediatric patients aged 1 to 9 years with previously untreated standard-risk ALL were randomized 1:1 to ONCASPAR or native E. coli L-asparaginase as part of combination therapy. ONCASPAR was administered intramuscularly at a dose of 2,500 International Units/m2 on Day 3 of the 4-week induction phase and on Day 3 of each of two 8-week delayed intensification phases. Native E. coli L-asparaginase was administered intramuscularly at a dose of 6,000 International Units/m2 three times weekly for 9 doses during induction and for 6 doses during each delayed intensification phase.
The primary determination of effectiveness was based on demonstration of similar asparagine depletion (magnitude and duration) in the ONCASPAR and native E. coli L-asparaginase arms. The protocol-specified goal was achievement of asparagine depletion to a serum concentration of ≤1 μM. The proportion of patients with this level of depletion was similar between the 2 study arms during all 3 phases of treatment at the protocol-specified time points.
In all phases of treatment, serum asparagine concentrations decreased within 4 days of the first dose of asparaginase in the treatment phase and remained low for approximately 3 weeks for both ONCASPAR and native E. coli L-asparaginase arms. Serum asparagine concentrations during the induction phase are shown in Figure 1. The patterns of serum asparagine depletion in the 2 delayed intensification phases are similar to the pattern of serum asparagine depletion in the induction phase.
| Note: ONCASPAR (2,500 International Units/m2 intramuscular) was administered on Day 3 of the 4-week induction phase. Native E. coli L-asparaginase (6,000 International Units/m2 intramuscular) was administered 3 times weekly for 9 doses during induction. |
| Figure 1. Mean (± Standard Error) Serum Asparagine Concentrations During Study CCG-1962 Induction Phase |
|
|
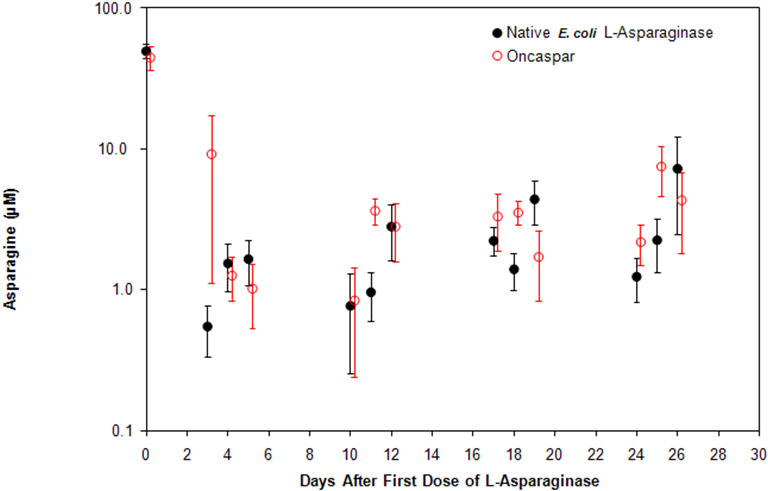
CSF asparagine concentrations were determined in 50 patients during the induction phase. CSF asparagine decreased from a mean pre-treatment concentration of 3.1 µM to 1.7 µM on Day 4±1 and 1.5 µM at 25±1 days after administration of ONCASPAR. These findings were similar to those observed in the native E. coli L-asparaginase treatment arm.
Concentrations of asparaginase activities greater than 0.1 International Units/mL were observed in over 90% of the samples from patients treated with ONCASPAR during Induction, Delayed Intensification 1, and Delayed Intensification 2 for approximately 20 days.
While the 3-year Event-Free Survival (EFS) for the ONCASPAR and native E. coli L-asparaginase study arms were similar and in the range of 80%, Study CCG-1962 was not designed to evaluate for differences in EFS rates.
14.2 Patients with ALL Hypersensitive to Asparaginase
The safety and effectiveness of ONCASPAR was evaluated in 4 open-label studies enrolling a total of 42 patients with multiply-relapsed, acute leukemia [39 (93%) with ALL] with a history of prior clinical allergic reaction to asparaginase. Hypersensitivity to asparaginase was defined by a history of systemic rash, urticaria, bronchospasm, laryngeal edema, hypotension, or local erythema, urticaria, or swelling, greater than 2 centimeters, for at least 10 minutes following administration of any form of native E. coli L-asparaginase. All patients received ONCASPAR at a dose of 2,000 or 2,500 International Units/m2 administered intramuscularly or intravenously every 14 days. Patients received ONCASPAR as a single agent or in combination with multi-agent chemotherapy. The re-induction response rate was 50% (95% confidence interval: 35%, 65%), based upon 36% complete remissions and 14% partial remissions. These results were similar to the overall response rates reported for patients with ALL receiving second-line, native E. coli L-asparaginase-containing re-induction chemotherapy. Anti-tumor activity was also observed with single-agent ONCASPAR. Three responses (1 complete remission and 2 partial remissions) were observed in 9 adult and pediatric patients with relapsed ALL and hypersensitivity to native E. coli L-asparaginase.
16 HOW SUPPLIED/STORAGE AND HANDLING
ONCASPAR (pegaspargase) Injection is supplied as a sterile, clear, colorless, preservative-free solution in Type I single-dose vials containing 3,750 International Units of pegaspargase per 5 mL (750 International Units per mL) solution (NDC 0944-3810-01).
17 PATIENT COUNSELING INFORMATION
Advise Patients/Caregivers of the following risks of ONCASPAR:
Anaphylaxis and Serious Hypersensitivity Reactions
Inform patients of the possibility of serious allergic reactions, including anaphylaxis, and to seek immediate medical care for any swellings or difficulty breathing [see Warnings and Precautions (5.1)].
Thrombosis
Instruct patients on the risk of thrombosis and hemorrhage and to seek immediate medical attention if they experience severe headache, arm or leg swelling, shortness of breath, or chest pain [see Warnings and Precautions (5.2)].
Pancreatitis
Instruct patients on the signs and symptoms of pancreatitis and to seek immediate medical attention if they experience severe abdominal pain [see Warnings and Precautions (5.3)].
Glucose Intolerance
Instruct patients on the risk of hyperglycemia and glucose intolerance. Advise patients to immediately report excessive thirst or increase in the volume or frequency of urination [see Warnings and Precautions (5.4)].
Hemorrhage
Advise patients to report any unusual bleeding or bruising to their physician [see Warnings and Precautions (5.5)].
Hepatotoxicity
Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding [see Warnings and Precautions (5.6)].
Pregnancy and Lactation
Advise female patients of reproductive potential to use effective contraceptive methods while receiving ONCASPAR and for at least 3 months after the last dose. Advise patients to notify their healthcare provider immediately in the event of a pregnancy, or if pregnancy is suspected during ONCASPAR treatment. Since there is a potential for an indirect interaction between ONCASPAR and oral contraceptives, the concomitant use of ONCASPAR and oral contraceptives is not recommended [see Use in Specific Populations (8.3)].
Advise lactating women not to breastfeed during treatment with ONCASPAR and for at least 3 months after the last dose [see Use in Specific Populations (8.2)].
BAXALTA® is a trademark of Baxalta Incorporated, a wholly-owned, indirect subsidiary of Shire plc. ONCASPAR® is a registered trademark of Servier IP UK LTD, a wholly owned, indirect subsidiary of Les Laboratoires Servier.
SHIRE and the Shire Logo are trademarks or registered trademarks of Shire Pharmaceutical Holdings Ireland Limited or its affiliates. Servier and the Servier logo are trademarks of Les Laboratoires Servier.
I-301-21-US-E
Manufactured by: Baxalta US Inc., Lexington, MA 02421
U.S. License No. 2020


| ONCASPAR
pegaspargase injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Baxalta US Inc. (079887619) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Exelead, Inc | 961822389 | MANUFACTURE(0944-3810) | |